
Molecular Dynamic Simulations of Bromodomain and Extra-Terminal Protein 4 Bonded to Potent Inhibitors




Abstract
:1. Introduction
2. Methods
2.1. Protein Prepare
2.2. Molecular Dynamics Simulations
2.3. Principal Components Analysis (PCA) and Cross-Correlation Matrix
2.4. MM/GBSA Calculations
3. Result and Discussion
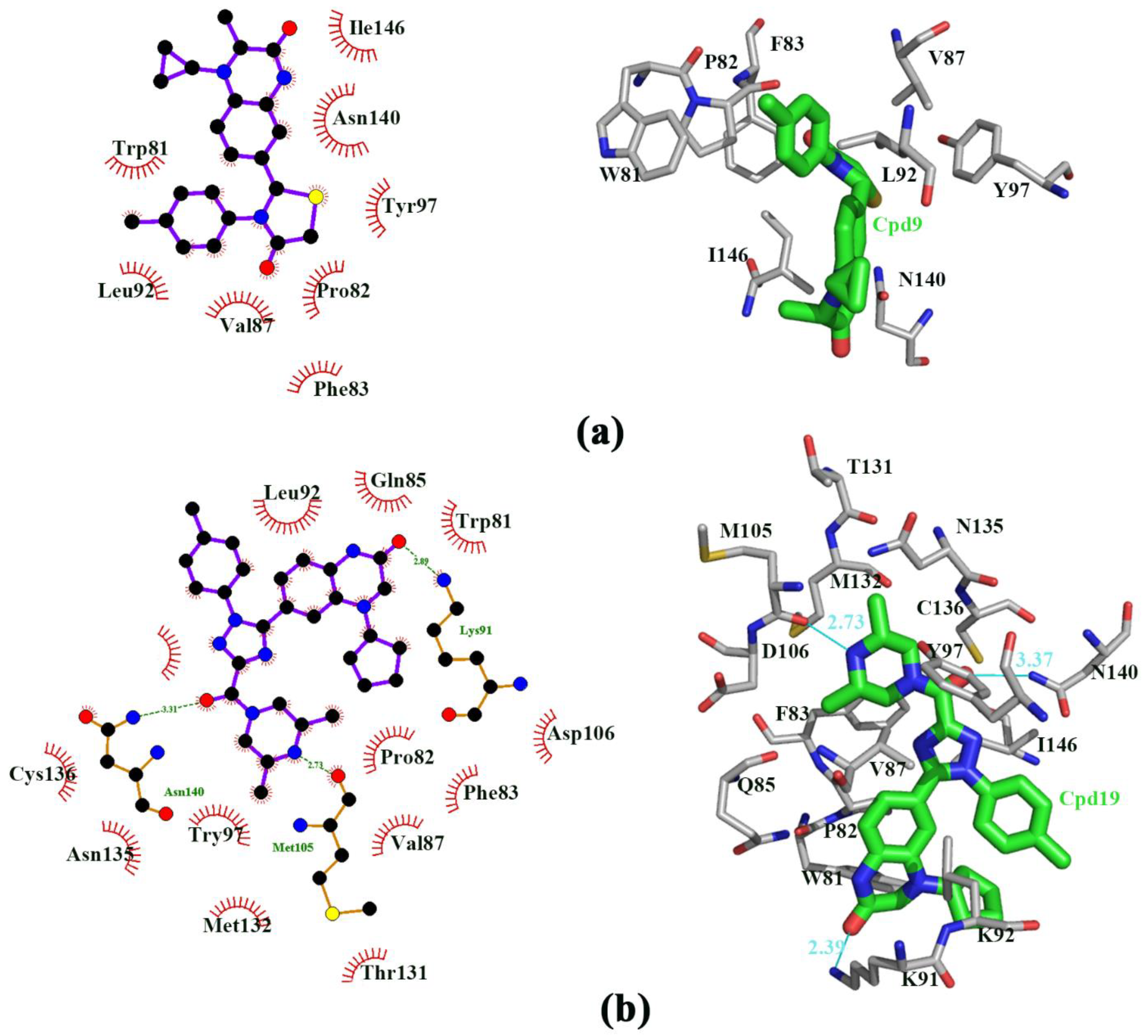
3.1. Docking Study
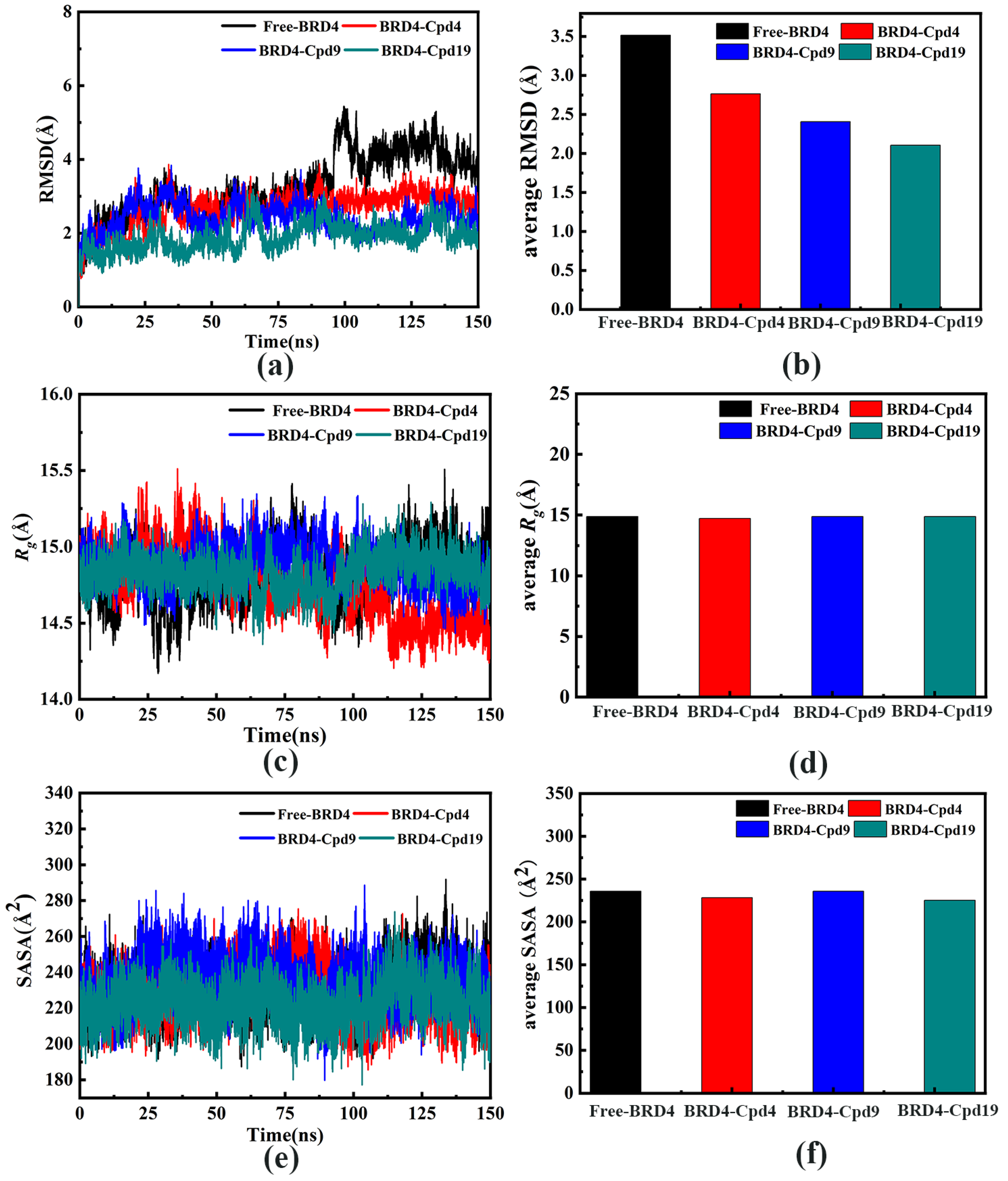
3.2. The Stability of the Four Systems
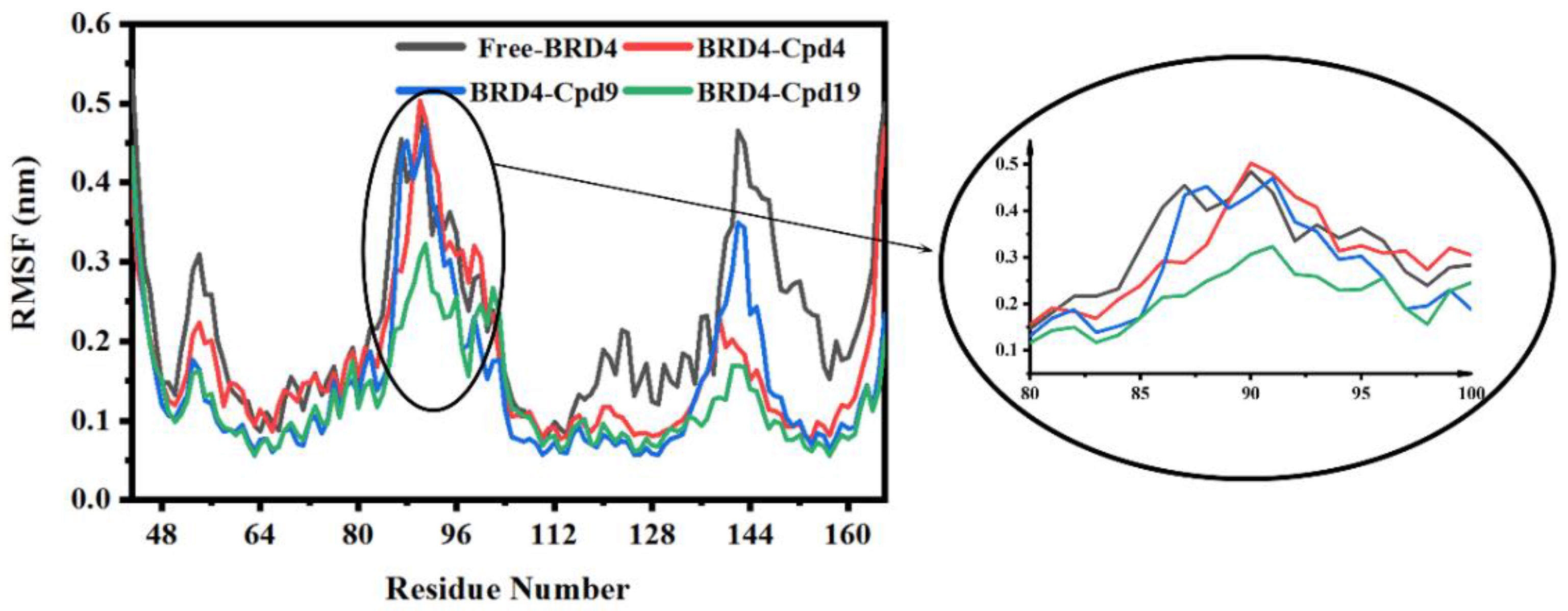
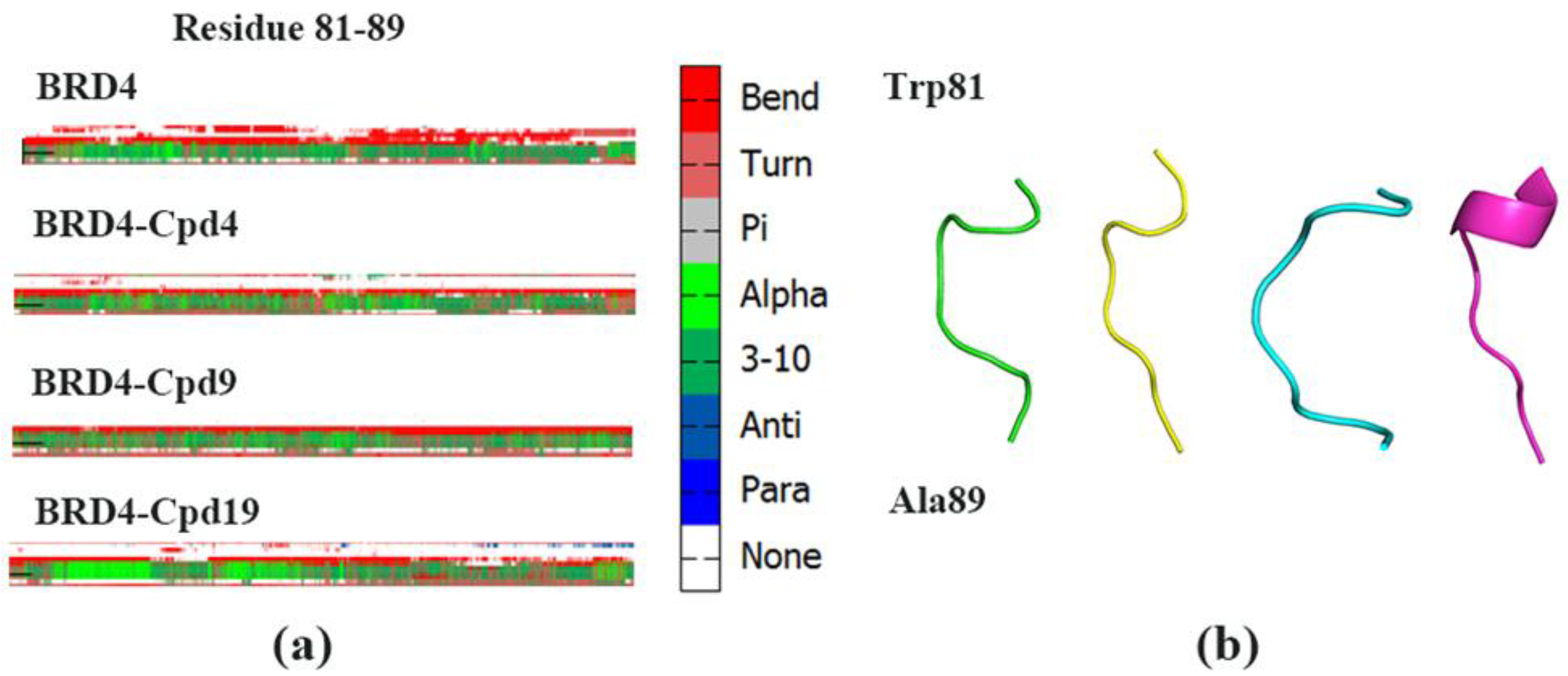
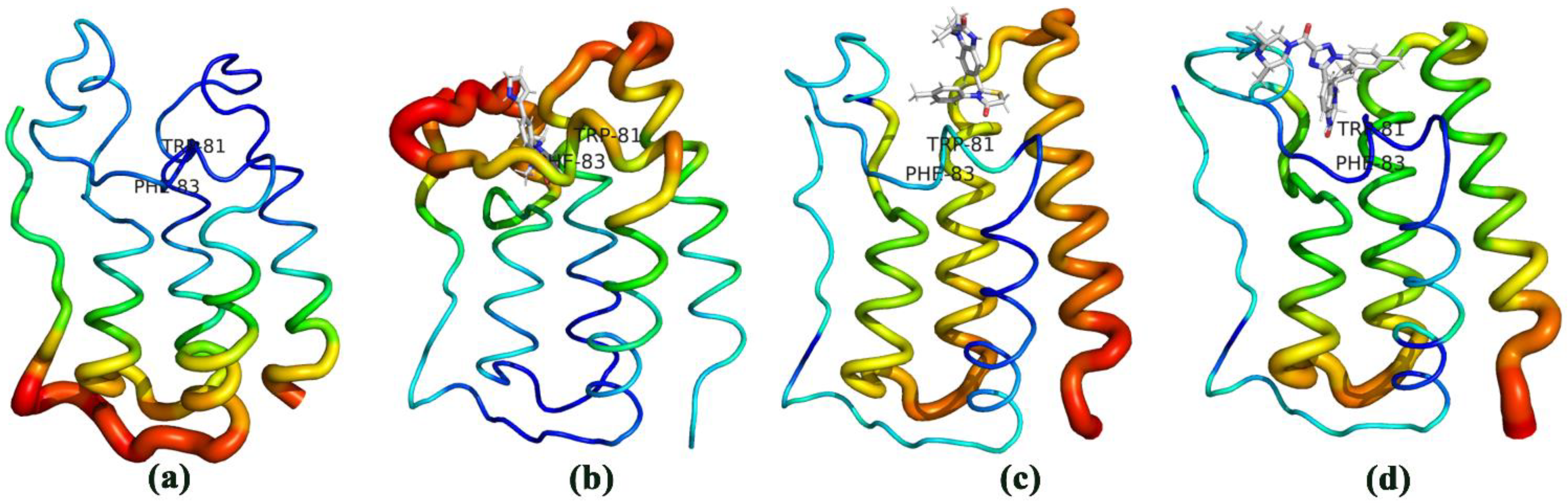
3.3. The Conformational Changes for Three Inhibitors Binding
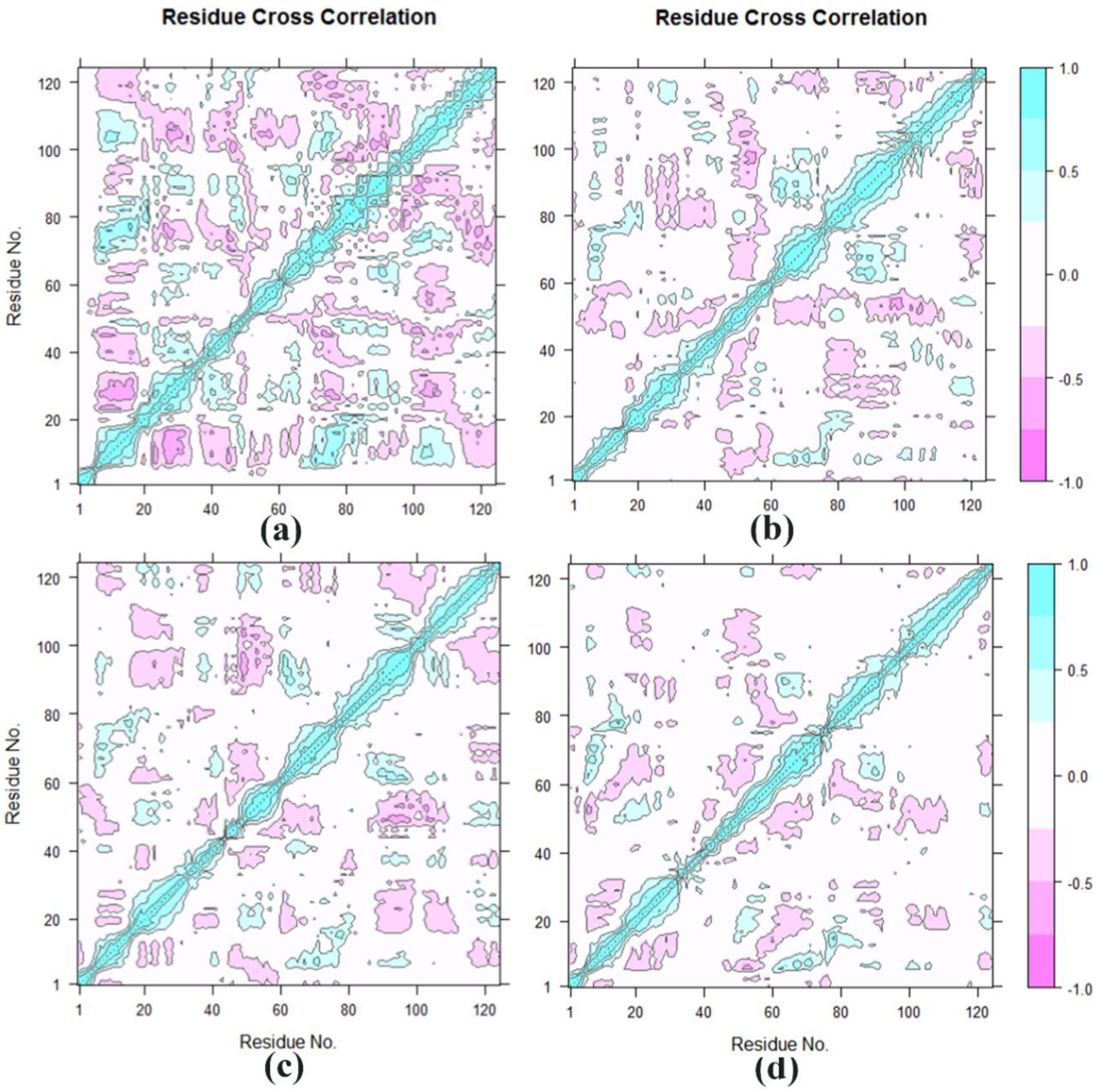
3.4. Cross-Correlation Analysis
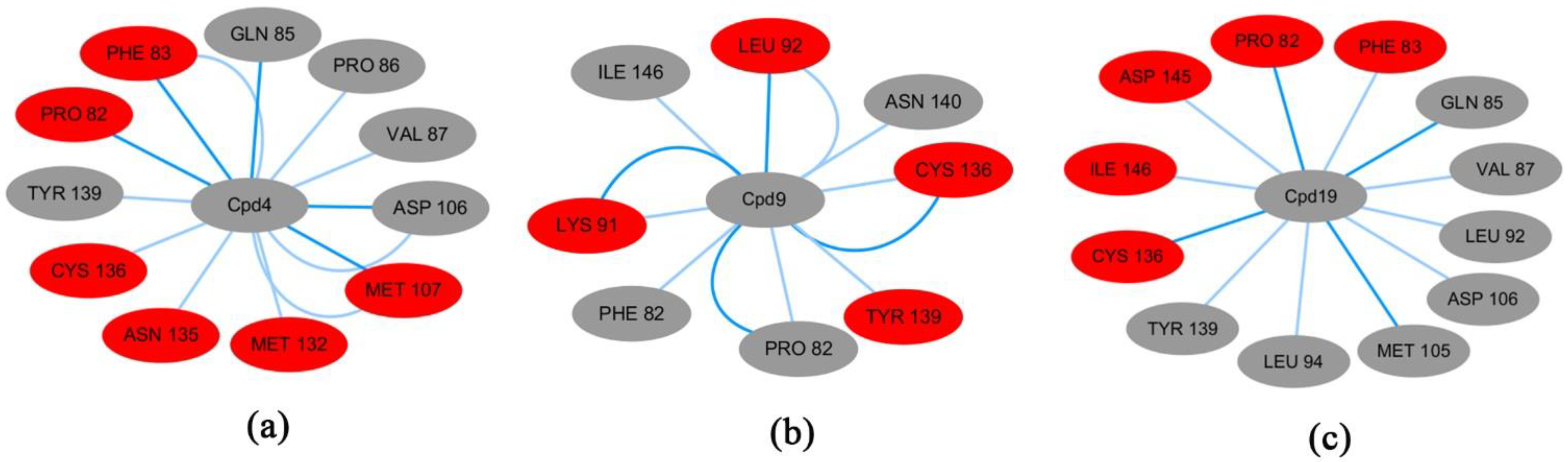
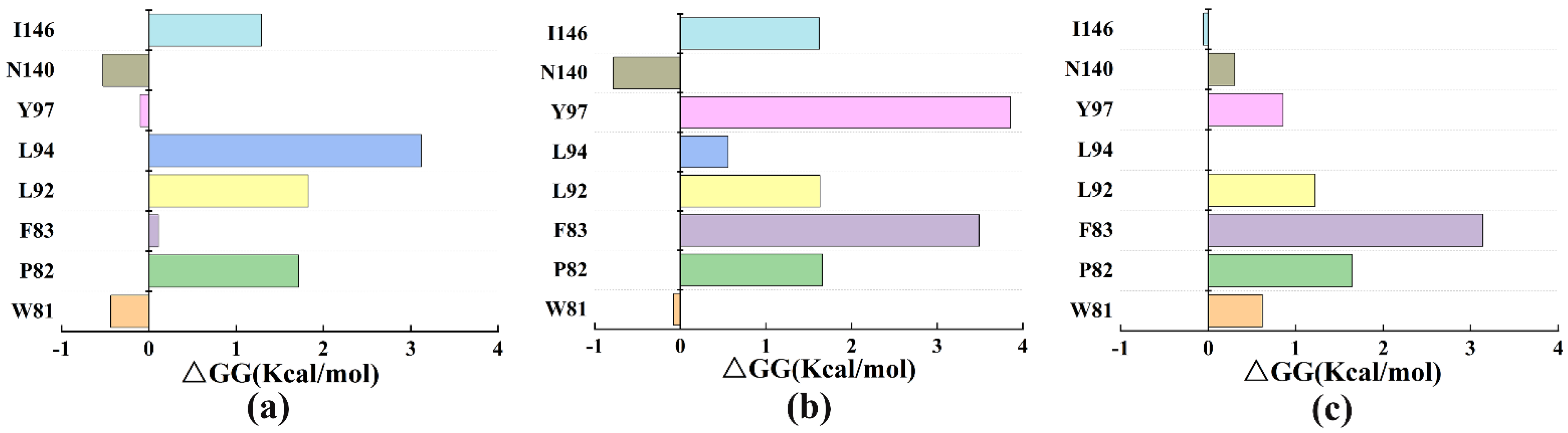
3.5. The Free Energy Binding of Three Systems
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Urbanucci, A.; Mills, I.G. Bromodomain-containing proteins in prostate cancer. Mol. Cell. Endocrinol. 2018, 462, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Huang, J.; Zhang, C.; Zhao, F.; Kim, W.; Tu, X.; Zhang, Y.; Nowsheen, S.; Zhu, Q.; Deng, M.; et al. The bromodomain containing protein BRD-9 orchestrates RAD51-RAD54 complex formation and regulates homologous recombination-mediated repair. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Lee, C.-F.; Lai, H.-T.; Yu, C.-T.; Lee, J.-E.; Zuo, H.; Tsai, S.Y.; Tsai, M.-J.; Ge, K.; Wan, Y.; et al. Opposing functions of BRD4 isoforms in breast cancer. Mol. Cell 2020, 78, 1114–1132. [Google Scholar] [CrossRef] [PubMed]
- Wen, N.; Guo, B.; Zheng, H.; Xu, L.; Liang, H.; Wang, Q.; Wang, D.; Chen, X.; Zhang, S.; Li, Y.; et al. Bromodomain inhibitor jq1 induces cell cycle arrest and apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling pathway. Int. J. Oncol. 2019, 55, 879–895. [Google Scholar] [CrossRef]
- Wang, D.; Kon, N.; Lasso, G.; Jiang, L.; Leng, W.; Zhu, W.-G.; Qin, J.; Hong, B.; Gu, W. Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature 2016, 538, 118–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Michelle, C.B. Bromodomain histone readers and cancer. J. Mol. Biol. 2017, 429, 2003–2010. [Google Scholar] [CrossRef]
- Lu, L.; Chen, Z.; Lin, X.; Tian, L.; Su, Q.; An, P.; Li, W.; Wu, Y.; Du, J.; Shan, H.; et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2020, 27, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Zhang, Y.; Cai, S.; Dong, L.; Hu, X.; Chen, M.-B.; Zhu, Y.-H. MicroRNA-4651 targets bromodomain-containing protein 4 to inhibit non-small cell lung cancer cell progression. Cancer Lett. 2020, 476, 129–139. [Google Scholar] [CrossRef]
- Sanchez, R.; Meslamani, J.; Zhou, M.-M. The bromodomain: From epigenome reader to druggable target. Biochim. et Biophys. Acta 2014, 1839, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Tian, C.-Q.; Damaneh, M.S.; Li, Y.; Cao, D.; Lv, K.; Yu, T.; Meng, T.; Chen, D.; Wang, X.; et al. Structure-based discovery and development of a series of potent and selective bromodomain and extra-terminal protein inhibitors. J. Med. Chem. 2019, 62, 8642–8663. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Cai, J.; Ma, S.; Fang, Y.; Huang, H.; Lin, T.; Rao, H.; Li, M.; Xia, Z.; Kang, T.; et al. BRD2 induces drug resistance through activation of the RasGRP1/Ras/ERK signaling pathway in adult T-cell lymphoblastic lymphoma. Cancer Commun. 2020, 40, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Daneshvar, K.; Ardehali, M.B.; Klein, I.A.; Hsieh, F.-K.; Kratkiewicz, A.J.; Mahpour, A.; Cancelliere, S.O.L.; Zhou, C.; Cook, B.M.; Li, W.; et al. lncRNA DIGIT and BRD3 protein form phase-separated condensates to regulate endoderm differentiation. Nat. Cell Biol. 2020, 22, 1211–1222. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cai, S.; Wang, J.-M.; Huai, Y.-Y.; Lu, P.-H.; Chu, Q. BRDT promotes ovarian cancer cell growth. Cell Death Dis. 2020, 11, 1021. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Stefan, K. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.-C.; Eckert, F.; Heinrich, V.; Bamberg, M.; Brucker, S.; Hehr, T. Resurgery and chest wall re-irradiation for recurrent breast cancer: A second curative approach. BMC Cancer 2011, 11, 197. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noel, J.K.; Iwata, K.; Ooike, S.; Sugahara, K.; Nakamura, H.; Daibata, M. Abstract C244: Development of the BET bromodomain inhibitor OTX015. Mol. Cancer Ther. 2013, 12, C244. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Côté, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- Bui, M.H.; Lin, X.; Albert, D.H.; Li, L.; Lam, L.T.; Faivre, E.J.; Warder, S.E.; Huang, X.; Wilcox, D.; Donawho, C.K.; et al. Preclinical characterization of BET family bromodomain inhibitor ABBV-075 suggests combination therapeutic strategies. Cancer Res. 2017, 77, 2976–2989. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Wang, Y.; Li, Y.; Cao, D.; Xu, L.; Song, S.; Damaneh, M.S.; Li, J.; Chen, Y.; Wang, X.; et al. Structure-based optimization of a series of selective BET inhibitors containing aniline or indoline groups. Eur. J. Med. Chem. 2018, 150, 156–175. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, Y.; Li, Y.; Xu, L.; Cao, D.; Song, S.; Damaneh, M.S.; Wang, X.; Meng, T.; Chen, Y.-L.; et al. Discovery of a series of dihydroquinoxalin-2(1H)-ones as selective BET inhibitors from a dual PLK1-BRD4 inhibitor. Eur. J. Med. Chem. 2017, 137, 176–195. [Google Scholar] [CrossRef]
- Watts, E.; Heidenreich, D.; Tucker, E.; Raab, M.; Strebhardt, K.; Chesler, L.; Knapp, S.; Bellenie, B.; Hoelder, S. Designing dual inhibitors of anaplastic lymphoma kinase (ALK) and bromodomain-4 (BRD4) by tuning kinase selectivity. J. Med. Chem. 2019, 62, 2618–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Yosief, H.O.; Dai, L.; Huang, H.; Dhawan, G.; Zhang, X.; Muthengi, A.M.; Roberts, J.; Buckley, D.L.; Perry, J.A.; et al. Structure-guided design and development of potent and selective dual bromodomain 4 (BRD4)/polo-like kinase 1 (PLK1) inhibitors. J. Med. Chem. 2018, 61, 7785–7795. [Google Scholar] [CrossRef]
- Prieto-Martínez, F.D.; Medina-Franco, J.L. Flavonoids as Putative Epi-Modulators: Insight into Their Binding Mode with BRD4 Bromodomains Using Molecular Docking and Dynamics. Biomolecules 2018, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Kadir, M.A.; Abdul Razak, F.I.; Haris, N.S.H. Experimental and DFT data of p-chlorocalix[4]arene as drugs receptor. Data Brief 2020, 32, 106263. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Watanabe, A.; Yutani, M.; Yamano, Y.; Sagara, M.; Nagai, S.; Saito, K.; Yamashita, M.; Ihara, M.; Iida, A. STAT3 inhibitory activity of naphthoquinones isolated from Tabebuia avellanedae. Bioorg. Med. Chem. 2020, 28, 115347. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Liu, X.; Zhang, S.; Yan, F.; Zhang, Q.; Chen, J. Insight into selective mechanism of class of I-BRD9 inhibitors toward BRD9 based on molecular dynamics simulations. Chem. Biol. Drug Des. 2019, 93, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Liu, X.; Zhang, S.; Yan, F.; Zhang, Q.; Chen, J. A computational insight into binding modes of inhibitors XD29, XD35, and XD28 to bromodomain-containing protein 4 based on molecular dynamics simulations. J. Biomol. Struct. Dyn. 2018, 36, 1212–1224. [Google Scholar] [CrossRef]
- Wu, S.L.; Wang, L.F.; Sun, H.B.; Wang, W.; Yu, Y.X. Probing molecular mechanism of inhibitor bindings to bromodomain-containing protein 4 based on molecular dynamics simulations and principal component analysis. SAR QSAR Environ. Res. 2020, 31, 547–570. [Google Scholar] [CrossRef] [PubMed]
- Tumdam, R.; Kumar, A.; Subbarao, N.; Balaji, B.S. In silico study directed towards identification of novel high-affinity inhibitors targeting an oncogenic protein: BRD4-BD1. SAR QSAR Environ. Res. 2018, 29, 975–996. [Google Scholar] [CrossRef]
- Zhu, M.; Song, L.T.; Liu, R.R.; Zhai, H.L.; Meng, Y.J.; Ren, C.L. Selective inhibition mechanism of nitroxoline to the BET family: Insight from molecular simulations. Life Sci. 2021, 270, 119141. [Google Scholar] [CrossRef]
- Zhong, H.; Wang, Z.; Wang, X.; Liu, H.; Li, D.; Liu, H.; Yao, X.; Hou, T. Importance of a crystalline water network in docking-based virtual screening: A case study of BRD4. Phys. Chem. Chem. Phys. 2019, 21, 25276–25289. [Google Scholar] [CrossRef]
- Mathur, S.; Deshmukh, P.; Tripathi, S.; Marimuthu, P.; Padmanabhan, B. Insights into the crystal structure of BRD2-BD2—Phenanthridinone complex and theoretical studies on phenanthridinone analogs. J. Biomol. Struct. Dyn. 2018, 36, 2342–2360. [Google Scholar] [CrossRef]
- Weber, O.C.; Uversky, V.N. How accurate are your simulations? Effects of confined aqueous volume and AMBER FF99SB and CHARMM22/CMAP force field parameters on structural ensembles of intrinsically disordered proteins: Amyloid-β42 in water. Intrinsically Disord. Proteins 2017, 5, e1377813. [Google Scholar] [CrossRef] [Green Version]
- Janowski, P.A.; Liu, C.; Deckman, J.; Case, D.A. Molecular dynamics simulation of triclinic lysozyme in a crystal lattice. Protein Sci. A Publ. Protein Soc. 2016, 25, 87–102. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.T.; Pabit, S.A.; Pollack, L.; Case, D.A. Extracting water and ion distributions from solution x-ray scattering experiments. J. Chem. Phys. 2016, 144, 214105. [Google Scholar] [CrossRef] [Green Version]
- Linse, B.; Linse, P. Tuning the smooth particle mesh Ewald sum: Application on ionic solutions and dipolar fluids. J. Chem. Phys. 2014, 141, 184114. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. Absolute and relative binding free energy calculations of the interaction of biotin and its analogs with streptavidin using molecular dynamics/free energy perturbation approaches. Proteins 1993, 16, 226–245. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, M.; Wilkins, D.M.; Ceriotti, M. Fast-forward Langevin dynamics with momentum flips. J. Chem. Phys. 2018, 148, 184109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.T.; Shaikh, N.; Kumar, S.U.; Doss, C.G.P.; Zayed, H. Structure-Based Virtual Screening to Identify Novel Potential Compound as an Alternative to Remdesivir to Overcome the RdRp Protein Mutations in SARS-CoV-2. Front. Mol. Biosci. 2021, 8, 645216. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, Z.; Zhu, J.; Wang, S.; Xu, D.; Han, W. Why Is a High Temperature Needed by Thermus thermophilus Argonaute During mRNA Silencing: A Theoretical Study. Front. Chem. 2018, 6, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhu, J.; Guo, X.; Huang, T.; Han, J.; Gao, J.; Xu, D.; Han, W. How oncogenic mutations activate human MAP kinase 1 (MEK1): A molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2020, 38, 3942–3958. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Rahnamoun, A.; Kaymak, M.C.; Manathunga, M.; Götz, A.W.; van Duin, A.C.T.; Merz, K.M., Jr.; Aktulga, H.M. ReaxFF/AMBER-A Framework for Hybrid Reactive/Nonreactive Force Field Molecular Dynamics Simulations. J. Chem. Theory Comput. 2020, 16, 7645–7654. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Protein | Ligand |
|---|---|---|
| Free-BRD4-BD1 | BRD4-BD1 | None |
| BRD4-BD1-Cpd4 | BRD4-BD1 |  |
| BRD4-BD1-Cpd9 | BRD4-BD1 |  |
| BRD4-BD1-Cpd19 | BRD4-BD1 |  |
| Residue No. | Free-BRD4 | BRD4-Cpd4 | BRD4-Cpd9 | BRD4-Cpd19 |
|---|---|---|---|---|
| W81 | 14.94 | 14.09 | 13.20 | 33.25 |
| P82 | 14.94 | 14.09 | 13.20 | 33.25 |
| F83 | 14.94 | 14.09 | 13.20 | 33.25 |
| System | BRD4-Cpd4 | BRD4-Cpd9 | BRD4-Cpd19 |
|---|---|---|---|
| △Gvdw | −32.94 ± 5.14 | −38.17 ± 4.10 | −46.04 ± 6.40 |
| △Gele | −12.47 ± 6.03 | −13.72 ± 5.40 | −9.58 ± 8.74 |
| △Gpolar | 24.22 ± 4.87 | 27.73 ± 4.97 | 28.64 ± 8.62 |
| △Gnonpolar | −4.12 ± 0.48 | −4.69 ± 0.47 | −5.48 ± 0.76 |
| △Ggas | −45.41 ± 9.18 | −51.89 ± 7.81 | −55.62 ± 11.58 |
| △Gsolv | 20.11 ± 4.63 | 23.04 ± 4.74 | 23.15 ± 8.40 |
| △Gtotal | −25.31 ± 5.48 | −28.85 ± 4.23 | −32.46 ± 5.60 |
| IC50 | 303.0 nM | 142.0 ± 3.0 nM | 5.3 ± 0.4 nM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; He, Y.; Geng, Y.; Wang, Z.; Han, L.; Han, W. Molecular Dynamic Simulations of Bromodomain and Extra-Terminal Protein 4 Bonded to Potent Inhibitors. Molecules 2022, 27, 118. https://doi.org/10.3390/molecules27010118
Chen S, He Y, Geng Y, Wang Z, Han L, Han W. Molecular Dynamic Simulations of Bromodomain and Extra-Terminal Protein 4 Bonded to Potent Inhibitors. Molecules. 2022; 27(1):118. https://doi.org/10.3390/molecules27010118
Chicago/Turabian StyleChen, Siao, Yi He, Yajiao Geng, Zhi Wang, Lu Han, and Weiwei Han. 2022. "Molecular Dynamic Simulations of Bromodomain and Extra-Terminal Protein 4 Bonded to Potent Inhibitors" Molecules 27, no. 1: 118. https://doi.org/10.3390/molecules27010118
APA StyleChen, S., He, Y., Geng, Y., Wang, Z., Han, L., & Han, W. (2022). Molecular Dynamic Simulations of Bromodomain and Extra-Terminal Protein 4 Bonded to Potent Inhibitors. Molecules, 27(1), 118. https://doi.org/10.3390/molecules27010118