
Stable Isotope Geochemistry of the Organic Elements within Shales and Crude Oils: A Comprehensive Review



Abstract
:1. Introduction
2. Standards and Notation
3. Stable Isotope Fractionation
Kinetic Isotope Effect (KIE)
4. Experimental and Instrumental Methods in Stable Isotopes (C, H, and S)
4.1. Stable Isotopic Analyses
4.1.1. Fractionation of Sediment Extracts and Crude Oils
4.1.2. Molecular Sieving
4.1.3. Elemental Analysis–Isotope Ratio Mass Spectrometry (Bulk δ13C Analysis)
4.1.4. Gas Chromatography–Isotope Ratio Mass Spectrometry (GC-IRMS)
4.1.5. Methods of Stable Sulfur Isotope Analysis
Bulk Stable Sulfur Isotope Analysis
Individual Sulfur Compounds Isotope Analysis
5. Applications of Stable Isotopes (C, H, S) in Shales and Crude Oils
5.1. Stable Carbon Isotope
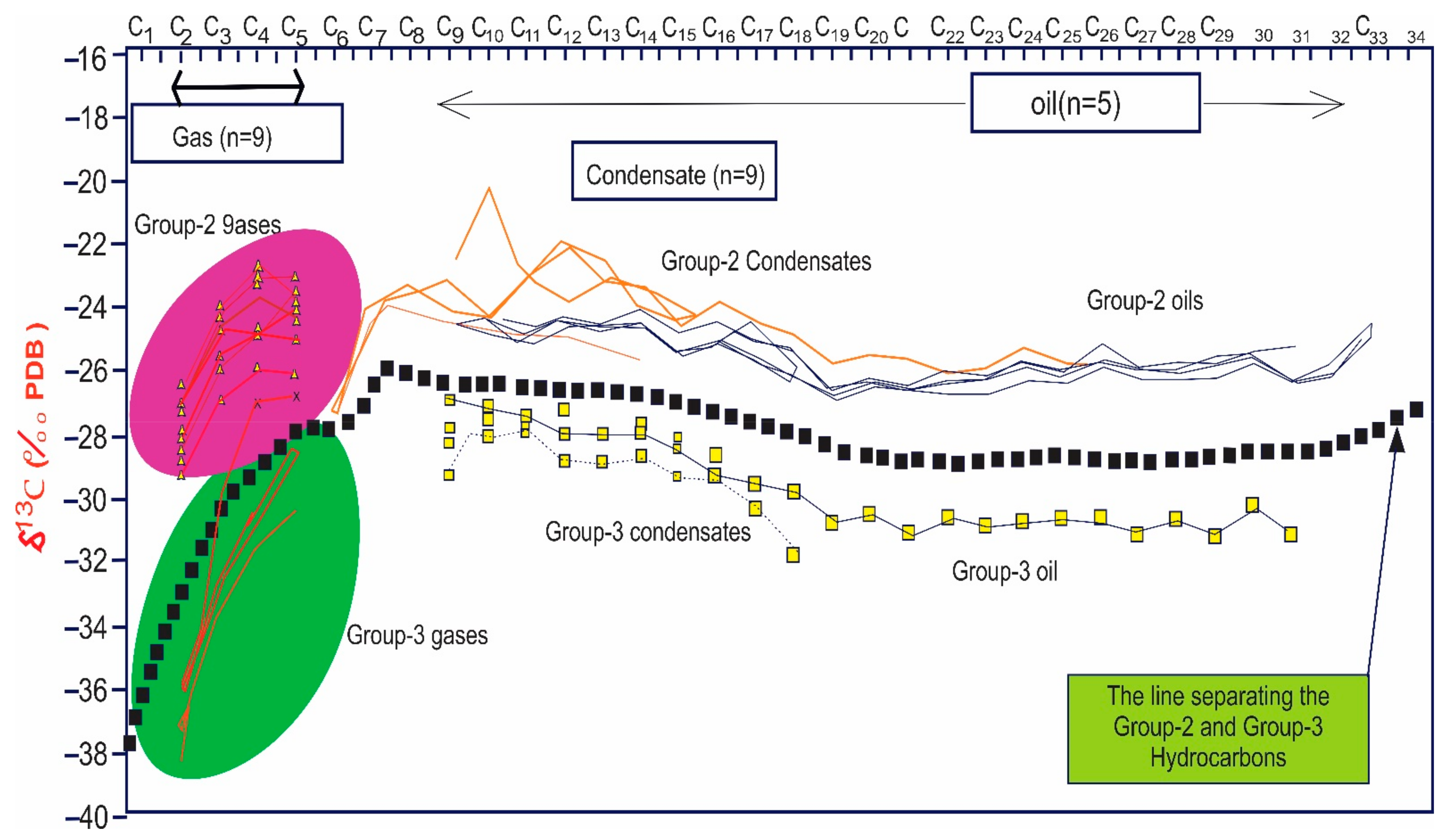
5.1.1. Source and Depositional Facies
5.1.2. Biodegradation
5.1.3. Maturity
5.1.4. Age Dating
5.1.5. Carbon CSIA
5.1.6. Application of Stable Carbon Isotopes to Gas Exploration
5.2. Stable Hydrogen Isotope
5.2.1. Hydrogen CSIA
5.2.2. Application of Hydrogen Isotope to Gas Exploration
5.3. Stable Sulfur Isotopes
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sofer, Z. Stable Isotopes in Petroleum Exploration Source and Migration Process and Evaluation Techniques; Amoco Production: Tulsa, OK, USA, 1991; pp. 103–106. [Google Scholar]
- Silverman, S.R.; Epstein, S. Carbon isotopic composition of petroleums and other sedimentary organic materials. Am. Assoc. Pet. Geol. Bull. 1958, 42, 998–1012. [Google Scholar]
- Silverman, S.R. Investigations of Petroleum Origin and Evolution Mechanisms by Carbon Isotope Studies. In Isotopic and Cosmic Chemistry; Craid, H., Miller, S.L., Wesserburg, G.J., Eds.; North-Holland Publishing Company: North-Holland, Amsterdam, 1964; pp. 92–102. [Google Scholar]
- Kvenvolden, K.A.; Squires, R.M. Carbon isotopic composition of crude oils from Ellenburger group (Lower Ordovician), Permian basin West Texas and Eastern New Mexico. Am. Assoc. Pet. Geol. Bull. 1967, 5, 1293–1303. [Google Scholar]
- Eckelman, W.R.; Broecker, W.S.; Whitlock, D.W.; Allsup, J.R. Implications of carbon isotopic composition of total organic carbon of some recent sediments and ancient oils. Am. Assoc. Pet. Geol. Bull. 1962, 46, 699–704. [Google Scholar]
- Alexeyev, F.A.; Lebedev, V.S.; Krylova, T.A.; Ovsyannikov, V.M.; Grachev, A.V. Carbon Isotopic Composition of Natural Hydrocarbons and the Problems of Their Origin; ONTI VNIYaGG: Moscow, Russia, 1967. [Google Scholar]
- Anderson, T.F.; Pratt, L.M. Isotopic Evidence for the Origin of Organic Sulfur and Elemental Sulfur in Marine Sediments. ACS Symp. Ser. 1995, 612, 378–396. [Google Scholar]
- Habicht, K.S.; Canfield, D.E. Isotope fractionation by sulfate-reducing natural populations and the isotopic composition of sulfide in marine sediments. Geology 2001, 29, 555–558. [Google Scholar] [CrossRef]
- Dawson, D.; Grice, K.; Alexander, R. Effect of maturation on the indigenous delta D signatures of individual hydrocarbons in sediments and crude oils from the Perth Basin (Western Australia). Org. Geochem. 2005, 36, 95–104. [Google Scholar] [CrossRef]
- Dawson, D.; Grice, K.; Alexander, R.; Edwards, D. The effect of source and maturity on the stable isotopic compositions of individual hydrocarbons in sediments and crude oils from the Vulcan Sub-basin, Timor Sea, Northern Australia. Org. Geochem. 2007, 38, 1015–1038. [Google Scholar] [CrossRef]
- Cai, C.; Zhang, C.; Worden, R.H.; Wang, T.; Li, H.; Jiang, L.; Huang, S.; Zhang, B. Application of sulfur and carbon isotopes to oil–source rock correlation: A case study from the Tazhong area, Tarim Basin, China. Org. Geochem. 2015, 83–84, 140–152. [Google Scholar] [CrossRef]
- Cai, C.; Li, K.; Anlai, M.; Zhang, C.; Xu, Z.; Worden, R.H.; Wu, G.; Zhang, B.; Chen, L. Distinguishing Cambrian from Upper Ordovician source rocks: Evidence from sulfur isotopes and biomarkers in the Tarim Basin. Org. Geochem. 2009, 40, 755–768. [Google Scholar] [CrossRef]
- Sofer, Z. Stable Carbon Isotope Compositions of Crude Oils: Application to Source Depositional Environments and Petroleum Alteration. AAPG Bull. 1984, 68, 31–49. [Google Scholar]
- Radke, J.; Bechtel, A.; Gaupp, R.; Pu¨ttmann, W.; Schwark, L.; Sachse, D.; Gleixner, G. Correlation between hydrogen isotope ratios of lipid biomarkers and sediment maturity. Geochim. Cosmochim. Acta 2005, 69, 5517–5530. [Google Scholar] [CrossRef]
- Xie, S.; Nott, C.; Avsejs, L.; Volders, F.; Maddy, D.; Chambers, F.; Gledhill, A.; Carter, J.; Evershed, R. Palaeoclimate records in compound-specific δD values of a lipid biomarker in ombrotrophic peat. Org. Geochem. 2000, 31, 1053–1057. [Google Scholar] [CrossRef]
- Sauer, P.; Eglinton, T.; Hayes, J.M.; Schimmelmann, A.; Sessions, A. Compound-specific D/H ratios of lipid biomarkers from sediments as a proxy for environmental and climatic conditions. Geochim. Cosmochim. Acta 2001, 65, 213–222. [Google Scholar] [CrossRef]
- Dawson, D.; Grice, K.; Wang, S.X.; Alexander, R.; Radke, J. Stable hydrogen isotopic composition of hydrocarbons in torbanites (Late Carboniferous to Late Permian) deposited under various climatic conditions. Org. Geochem. 2004, 35, 189–197. [Google Scholar] [CrossRef]
- Asif, M.; Fazeelat, T.; Grice, K. Petroleum geochemistry of the Potwar Basin, Pakistan: 1-Oil–oil correlation from biomarkers and δ13C/δD. Org. Geochem. 2011, 42, 1226–1240. [Google Scholar] [CrossRef]
- Rigby, D.; Batts, B.; Smith, J. The effect of maturation on the isotopic composition of fossil fuels. Org. Geochem. 1981, 3, 29–36. [Google Scholar] [CrossRef]
- Hoefs, J. Stable Isotope Geochemistry, 8th ed.Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Philp, R.P.; Monaco, G.L. Applications of Stable Isotopes in Hydrocarbon Exploration and Environmental Forensics. In Advances in Isotope Geochemistry; Mark, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 639–677. [Google Scholar]
- Tissot, B.T.; Welte, D.H. Petroleum Formation and Occurrences: A New Approach to Oil and Gas Exploration; Springer: Berlin/Heidelberg, Germany, 1978. [Google Scholar]
- Frank, D.; Sackett, W. Kinetic isotope effects in the thermal cracking of neopentane. Geochim. Cosmochim. Acta 1969, 33, 811–820. [Google Scholar] [CrossRef]
- Park, R.; Dunning, H. Stable carbon isotope studies of crude oils and their porphyrin aggregates. Geochim. Cosmochim. Acta 1961, 22, 99–105. [Google Scholar] [CrossRef]
- Muller, P.; Wienholz, R. Bestimmung der natürlichen Variationen der Kohlenstoffisotope in Erdöl-und Erdgaskomponenten und ihre Beziehung zur Genese. Z. Angew. Geol. 1967, 13, 427–450. [Google Scholar]
- May, F.; Freund, W.; Mu¨ller, P. Modellversuche über Isotopenfraktionierung von Erdgaskomponenten während der Migration. Z. Angew. Geol. 1968, 14, 376. [Google Scholar]
- Colombo, U.; Gazzarrini, F.; Sironi, G.; Gonfiantini, R.; Tongiorgi, E. Carbon isotope composition of individual hydrocarbons from italian natural gases. Nature 1965, 205, 1303–1304. [Google Scholar] [CrossRef]
- Welte, D.H. Der 13C-Isotopengehalt von geradzahligen und ungerazahligen höheren n-parafinen Paraffinen aus Erdöl. Erdöl und Kohle 1969, 22, 150–162. [Google Scholar]
- Botneva, T.A.; Müller, P.; Maas, I. On carbon isotopic composition of oils and their fractions. Geol. Nefti Gaza 1969, 7, 33–39. [Google Scholar]
- Lebedev, V.S. Isotope composition of oil and gas. Geokhimiya 1964, 11, 1128–1137. [Google Scholar]
- Galimov, E.M. 13C enrichment of methane during passage through the rocks. Geochem. Int. 1967, 4, 1180–1181. [Google Scholar]
- Galimov, E.M. Geochemistry of Carbon Stable Isotopes; Nedra: Moscow, Russia, 1968; p. 224. [Google Scholar]
- Galimov, E. Isotopic composition of carbon in gases of the crust. Int. Geol. Rev. 1969, 11, 1092–1104. [Google Scholar] [CrossRef]
- Galimov, E.M. Die Isotopenzusammensetzung des Koh-lenstoffs in den Gasen der Erdkruste. Z. Angew. Geol. 1969, 15, 69–81. [Google Scholar]
- Galimov, E.M. On the relationship of the fractionation coefficient of isotopes to the equilibrium constants of the isotope exchange reactions of carbon in hydrocarbon systems. Zh. Fiz. Khim. 1971, 45, 1187–1191. [Google Scholar]
- Galimov, E.M.; Kuznetsova, N.G.; P’yankov, N.A.; Vinnikov-ski, S.A. Genetic types of the Permian Prikam’e oils on the basis of their carbon isotope composition. Geol. Nefti Gaza 1972, 1, 33–39. [Google Scholar]
- Galimov, E.M.; Posyagin, V.I.; Prokhorov, V.S. Experimental study of carbon isotope fractionation in the CH4–C2H6–C3H8–C4H10 system at different temperatures. Geokhimiya 1972, 8, 977–987. [Google Scholar]
- Galimov, E.M.; Migdisov, A.A.; Ronov, A.B. Controlling factors of carbon isotope composition in the Precambrian and Phanerozoic. In Litologiya Osadochnaya Geologiya Dokembriya (Precambrian Lithology and Sedimentary Geology); Sidorenko, A.V., Ed.; Akademiya Nauk USSR: Moscow, Russia, 1973; pp. 279–281. [Google Scholar]
- Galimov, E.M.; Teplinskiy, G.I.; Tabassaranskiy, Z.A.; Gavrilov, Y.Y. On the conditions of formation of gas deposits in the eastern part of the Turan Plate as revealed by carbon isotopic composition of the gases. Geochem. Int. 1973, 10, 1259–1271. [Google Scholar]
- Galimov, E.M. Carbon Isotopes in Oil and Gas Geology; Nedra Press: Moscow, Russia, 1975; p. 385. [Google Scholar]
- Lowenstein, T.K.; Timofeeff, M.N.; Kovalevych, V.M.; Horita, J. The major-ion composition of Permian seawater. Geochim. Cosmochim. Acta 2005, 69, 1701–1719. [Google Scholar] [CrossRef]
- Zerkle, A.L.; Kamyshny, A.; Kump, L.R.; Farquhar, J.; Oduro, H.; Arthur, M.A. Sulfur cycling in a stratified euxinic lake with moderately high sulfate: Constraints from quadruple S isotopes. Geochim. Cosmochim. Acta 2010, 74, 4953–4970. [Google Scholar] [CrossRef]
- Johnston, D.T. Multiple sulfur isotopes and the evolution of Earth’s surface sulfur cycle. Earth Sci. Rev. 2011, 106, 161–183. [Google Scholar] [CrossRef]
- Canfield, D.E. Biogeochemistry of sulfur isotopes. In Stable Isotope Geochemistry; Valley, J.W., Cole, D., Eds.; Mineralogical Society of America and Geochemical Society, Reviews of Mineralogy and Geochemistry: Washington, DC, USA, 2001; Volume 43, pp. 607–636. [Google Scholar]
- Hurtgen, M.T.; Arthur, M.A.; Suits, N.S.; Kaufman, A.J. The sulfur isotopic composition of Neoproterozoic seawater sulfate: Implications for asnowball Earth? Earth Planet. Sci. Lett. 2002, 203, 413–430. [Google Scholar] [CrossRef]
- Berner, R.A.; Raisswell, R. Burial of organic carbon and pyrite sulfur in sediments over Phanerozoic time: A new theory. Geochim. Cosmochim. Acta 1983, 47, 855–862. [Google Scholar] [CrossRef]
- Kleeberg, A. Interactions between bentic phosphorus release and sulfur cycling in Lake Scharmützelsee (Germany). Water Air Soil Pollut. 1997, 99, 391–399. [Google Scholar] [CrossRef]
- Sim, M.S.; Ono, S.; Donovan, K.; Templer, S.P.; Bosak, T. Effect of electron donors on the fractionation of sulfur isotopes by a marine Desulfovibriosp. Geochim. Cosmochim. Acta 2011, 75, 4244–4259. [Google Scholar] [CrossRef]
- Urey, H.C. The thermodynamic properties of isotopic substances. J. Chem. Soc. 1947, 1, 562–581. [Google Scholar] [CrossRef]
- Stahl, W.J. Carbon and nitrogen isotopes in hydrocarbon re-search and exploration. Chem. Geol. 1977, 20, 121–149. [Google Scholar] [CrossRef]
- Fuex, A. The use of stable carbon isotopes in hydrocarbon exploration. J. Geochem. Explor. 1977, 7, 155–188. [Google Scholar] [CrossRef]
- Schoell, M. Genetic characterization of natural gases. Am. Assoc. Pet. Geol. Bull. 1983, 67, 2225–2238. [Google Scholar]
- Schoell, M. Recent advances in petroleum isotope geochemistry. Org. Geochem. 1984, 6, 645–663. [Google Scholar] [CrossRef]
- Schoell, M. Stable isotopes in petroleum research. In Advances in Petroleum Geochemistry; Brooks, J., Welte, D., Eds.; Academic Press: London, UK, 1984; Volume 1, pp. 215–245. [Google Scholar]
- Elsner, M.; Zwank, L.; Hunkeler, D.; Schwarzenbach, R.P. A New Concept Linking Observable Stable Isotope Fractionation to Transformation Pathways of Organic Pollutants. Environ. Sci. Technol. 2005, 39, 6896–6916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherwood Lollar, B.; Slater, G.F.; Sleep, B.; Witt, M.; Kledka, G.M.; Harkness, M.; Spivack, J. Stable carbon isotope evidence for intrinsic bioremediation of tetrachloroethene and trichloroethene at area 6, Dover Air Force Base. Environ. Sci. Technol. 2001, 35, 261–269. [Google Scholar] [CrossRef]
- George, S.C.; Boreham, C.J.; Minifie, S.A.; Teerman, S.C. The effect of minor to moderate biodegradation on C5 to C9 hydrocarbons in crude oils. Org. Geochem. 2002, 33, 1293–1317. [Google Scholar] [CrossRef]
- Schüth, C.; Taubald, H.; Bolaño, N.; Maciejczyk, K. Carbon and hydrogen isotope effects during sorption of organic contaminants on carbonaceous materials. J. Contam. Hydrol. 2003, 64, 269–281. [Google Scholar] [CrossRef]
- Kopinke, F.-D.; Georgi, A.; Voskamp, M.; Richnow, H.H. Carbon Isotope Fractionation of Organic Contaminants Due to Retardation on Humic Substances: Implications for Natural Attenuation Studies in Aquifers. Environ. Sci. Technol. 2005, 39, 6052–6062. [Google Scholar] [CrossRef]
- Kuder, T.; Philp, P.; Allen, J. Effects of Volatilization on Carbon and Hydrogen Isotope Ratios of MTBE. Environ. Sci. Technol. 2009, 43, 1763–1768. [Google Scholar] [CrossRef]
- Onojake, M.C.; Osuji, L.C.; Oforka, N.C. Bulk Geochemical Characteristics of Crude Oils From the Umutu/Bomu Fields, Niger Delta, Nigeria. Pet. Sci. Technol. 2014, 32, 618–624. [Google Scholar] [CrossRef]
- Murphy, M.T.K. Analytical methods. In Organic Geochemistry; Eglinton, G., Murphy, M.T.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1969; pp. 74–88. [Google Scholar]
- Hertelendi, E.; Gál, J.; Paál, A.; Fekete, S.; Giurgiu, M.; Gál, I.; Kertész, Z.; Nagy, S. Stable isotope mass spectrometer. In Proceedings of the Fourth Working Meeting Isotopes in Nature, Leipzig, Germany, 22–26 September 1986; pp. 323–334. [Google Scholar]
- Amrani, A.; Sessions, A.L.; Adkins, J.F. Compound-Specific δ34S Analysis of Volatile Organics by Coupled GC/Multicollector-ICPMS. Anal. Chem. 2009, 81, 9027–9034. [Google Scholar] [CrossRef] [PubMed]
- Amrani, A.; Deev, A.; Sessions, A.; Tang, Y.; Adkins, J.F.; Hill, R.J.; Moldowan, J.; Wei, Z. The sulfur-isotopic compositions of benzothiophenes and dibenzothiophenes as a proxy for thermochemical sulfate reduction. Geochim. Cosmochim. Acta 2012, 84, 152–164. [Google Scholar] [CrossRef]
- Said-Ahmad, W.; Amrani, A. A sensitive method for the sulfur isotope Analysis of dimethyl sulfide and dimethyl sulfoniopropionate in seawater. Rapid Commun. Mass Spectrom. 2013, 27, 2789–2796. [Google Scholar] [CrossRef]
- Giesemann, A.; Jäger, H.A.; Norman, A.L.; Krouse, H.R.; Brand, W.A. On-line sulphur isotope determination using an elemental analyzer coupled to a mass spectrometer. Anal. Chem. 1994, 66, 2816–2819. [Google Scholar] [CrossRef]
- Puchelt, H.; Sabels, B.R.; Hoering, T.C. Preparation of sulfur hexafluoride for isotope geochemical analysis. Geochim. Cosmochim. Acta 1971, 35, 625–628. [Google Scholar] [CrossRef]
- Rees, C. Sulphur isotope measurements using SO2 and SF6. Geochim. Cosmochim. Acta 1978, 42, 383–389. [Google Scholar] [CrossRef]
- Beaudoin, G.; Taylor, B.E. High precision and spatial resolution sulfur isotope analysis using MILES laser microprobe. Geochim. Cosmochim. Acta 1994, 58, 5055–5063. [Google Scholar] [CrossRef]
- Hu, G.X.; Rumble, D.; Wang, P.L. An ultraviolet laser microprobe for the in-situ analysis of multisulfur isotopes and its use in measuring Archean sulphur isotope mass-independent anomalies. Geochim. Cosmochim. Acta 2003, 67, 3101–3118. [Google Scholar] [CrossRef]
- Kelley, K.D.; Wilkinson, J.J.; Chapman, J.B.; Crowther, H.L.; Weiss, D. Zinc isotopes in sphalerite from base metal deposits in the red dog district, northern alaska. Econ. Geol. 2009, 104, 767–773. [Google Scholar] [CrossRef]
- Crowe, D.; Valley, J.W.; Baker, K.L. Micro-analysis of sulfur-isotope ratios and zonation by laser microprobe. Geochim. Cosmochim. Acta 1990, 54, 2075–2092. [Google Scholar] [CrossRef]
- Ono, S.; Wing, B.A.; Johnston, D.; Farquhar, J.; Rumble, D. Mass-dependent fractionation of quadruple sulphur isotope system as a new tracer of sulphur biogeochemical cycles. Geochim. Cosmochim. Acta 2006, 70, 2238–2252. [Google Scholar] [CrossRef]
- Chaussidon, M.; Albarede, F.; Sheppard, S.M.F. Sulphur isotope heterogeneity in the mantle from ion microprobe measurements of sulphide inclusions in diamonds. Nature 1987, 330, 242–244. [Google Scholar] [CrossRef]
- Chaussidon, M.; Albarede, F.; Sheppard, S.M.F. Sulphur isotope variations in the mantle from ion microprobe analysis of microsulphide inclusions. Earth Planet. Sci. Lett. 1989, 92, 144–156. [Google Scholar] [CrossRef]
- Eldridge, C.S.; Compston, W.; Williams, I.S.; Both, R.A.; Walshe, J.L.; Ohmoto, H. Sulfur isotope variability in sediment hosted massive sulfide deposits as determined using the ion microprobe SHRIMP. I. An example from the Rammelsberg ore body. Econ. Geol. 1988, 83, 443–449. [Google Scholar] [CrossRef]
- Eldridge, C.S.; Williams, I.S.; Walshe, J.L. Sulfur isotope variability in sediment hosted massive sulfide deposits as determined using the ion microprobe SHRIMP. II. A study of the H.Y.C. deposit at McArthur River, Northern Territory, Australia. Econ. Geol. 1993, 88, 1–26. [Google Scholar] [CrossRef]
- Kozdon, R.; Kita, R.N.; Huberty, J.M.; Fournelle, J.H.; Johnson, C.A.; Valley, J.W. In situ sulfur isotope analysis of sulfide minerals by SIMS: Precision and accuracy with application to thermometry of similar to 3.5 Ga Pilbara cherts. Chem. Geol. 2010, 275, 243–253. [Google Scholar] [CrossRef]
- Hauri, E.H.; Papineau, D.; Wang, J.; Hillion, F. High-precision analysis of multiple sulfur isotopes using NanoSIMS. Chem. Geol. 2016, 420, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Bendall, C.; Lahaye, Y.; Fiebig, J.; Weyer, S.; Brey, G.P. In situ sulfur isotope analysis by laser ablation MC-ICPMS. Appl. Geochem. 2006, 21, 782–787. [Google Scholar] [CrossRef]
- Craddock, P.R.; Rouxel, O.J.; Ball, L.A.; Bach, W. Sulfur isotope measurement of sulfate and sulfide by high-resolution MC-ICP-MS. Chem. Geol. 2008, 253, 102–113. [Google Scholar] [CrossRef]
- Paris, G.; Sessions, A.; Subhas, A.V.; Adkins, J.F. MC-ICP-MS measurement of δ34 S and ∆33S in small amounts of dissolved sulphate. Chem. Geol. 2013, 345, 50–61. [Google Scholar] [CrossRef]
- Grotheer, H.; Greenwood, P.; McCulloch, M.; Böttcher, M.E.; Grice, K. δ34S character of organosulfur compounds in kerogen and bitumen fractions of sedimentary rocks. Org. Geochem. 2017, 110, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Peters, K.E.; Fowler, M.G. Applications of petroleum geochemistry to exploration and reservoir management. Org. Geochem. 2002, 33, 5–36. [Google Scholar] [CrossRef]
- Craig, H. The geochemistry of the stable carbon isotopes. Geochim. Cosmochim. Acta 1953, 3, 53–92. [Google Scholar] [CrossRef]
- Silverman, S.R. Migration and segregation of oil and gas fluids in subsurface environments. Am. Assoc. Pet. Geol. Memoir. 1965, 4, 54–65. [Google Scholar]
- Andrusevich, V.E.; Engel, M.H.; Zumberge, J.E.; Brothers, L.A. Secular, episodic changes in stable carbon isotope composi-tion of crude oils. Chem. Geol. 1998, 152, 59–72. [Google Scholar] [CrossRef]
- Odden, W.; Barth, T.; Talbot, M. Compound-specific carbon isotope analysis of natural and artificially generated hydro-carbons in source rocks and petroleum fluids from offshore Mid-Norway. Org. Geochem. 2002, 33, 47–65. [Google Scholar] [CrossRef]
- Welte, D.; Hagemann, H.; Hollerbach, A.; Leythauser, D.; Stahl, W. Correlation between petroleum and source rock. In Proceedings of Ninth World Petroleum Congress; Applied Science Publishers II: London, UK, 1975; pp. 179–191. [Google Scholar]
- Philp, R.P.; Jarde, E. Application of stable and radioiso-topes. In Introduction to Environmental Forensics; Murphy, B., Morrison, R., Eds.; Elsevier: New York, NY, USA, 2007; pp. 455–512. [Google Scholar]
- Stahl, W. Source rock-crude oil correlation by isotopic type-curves. Geochim. Cosmochim. Acta 1978, 42, 1573–1577. [Google Scholar] [CrossRef]
- Chung, H.; Brand, S.W.; Grizzle, P.L. Carbon isotope geochemistry of Paleozoic oils from Big Horn Basin. Geochim. Cosmochim. Acta 1981, 45, 1803–1815. [Google Scholar] [CrossRef]
- Palmer, S.E. Effect of biodegradation and water-washing on crude oil composition. In Organic Geochemistry; Engel, M.H., Macko, S.E., Eds.; Plenum: New York, NY, USA, 1993; pp. 511–534. [Google Scholar]
- Stahl, W. Compositional changes and 13C/12C fractionations during the degradation of hydrocarbons by bacteria. Geochim. Cosmochim. Acta 1980, 44, 1903–1907. [Google Scholar] [CrossRef]
- Clayton, C. Carbon isotope fractionation during natural gas generation from kerogen. Mar. Pet. Geol. 1991, 8, 232–240. [Google Scholar] [CrossRef]
- Clayton, C. Effect of maturity on carbon isotope ratios of oils and condensates. Org. Geochem. 1991, 17, 887–899. [Google Scholar] [CrossRef]
- Hunt, J.M. Petroleum Geochemistry and Geology; W.H. Freeman: New York, NY, USA, 1996. [Google Scholar]
- Park, R.; Epstein, S. Carbon isotope fractionation during photosynthesis. Geochim. Cosmochim. Acta 1960, 21, 110–126. [Google Scholar] [CrossRef]
- O’Leary, M.H. Carbon isotope fractionation in plants. Phytochemistry 1981, 20, 553–567. [Google Scholar] [CrossRef]
- Stuermer, D.H.; Peters, K.; Kaplan, I. Source indicators of humic substances and proto-kerogen. Stable isotope ratios, elemental compositions and electron spin resonance spectra. Geochim. Cosmochim. Acta 1978, 42, 989–997. [Google Scholar] [CrossRef]
- Aizenshtat, Z.; Baedecker, M.; Kaplan, I. Distribution and diagenesis of organic compounds in JOIDES sediment from Gulf of Mexico and western Atlantic. Geochim. Cosmochim. Acta 1973, 37, 1881–1898. [Google Scholar] [CrossRef]
- Sackett, W.M.; Eadie, B.J.; Exner, M.E. Stable isotope com-position of organic carbon in recent Antarctic sediments. In Advances in Organic Geochemistry; Tissot, B., Bienner, F., Eds.; Technip: Paris, France, 1974; pp. 661–671. [Google Scholar]
- Gormley, J.R.; Sackett, W.M. Carbon isotope evidence for the maturation of marine lipids. In Advances in Organic Geochemistry; Campos, R., Goni, J., Eds.; Enadisma: Madrid, Spain, 1977; pp. 321–339. [Google Scholar]
- Brown, F.; Baedecker, M.; Nissenbaum, A.; Kaplan, I. Early diagenesis in a reducing fjord, Saanich Inlet, British Columbia—III. Changes in organic constituents of sediment. Geochim. Cosmochim. Acta 1972, 36, 1185–1203. [Google Scholar] [CrossRef]
- Peters, K.E.; Rohrback, B.G.; Kaplan, I.R. Carbon and hydro-gen stable isotope variations in kerogen during laboratory-simulated thermal maturation. Am. Assoc. Pet. Geol. Bull. 1981, 65, 501–508. [Google Scholar]
- Hatcher, P.G.; Spiker, E.C.; Szeverenyi, N.M.; Maciel, G.E. Selective preservation and origin of petroleum-forming aquatic kerogen. Nature 1983, 305, 498–501. [Google Scholar] [CrossRef]
- Hunt, J. The significance of carbon isotope variations in marine sediments. In Advances in Organic Geochemistry; Hobson, G.D., Speers, G.C., Eds.; Pergamon: Oxford, UK, 1970. [Google Scholar]
- Mello, M.R.; Telnaes, N.; Gaglianone, P.C.; Chicarelli, M.I.; Brassell, S.C.; Maxwell, J.R. Organic geochemical characterization of depositional palaeoenvironments of source rocks and oils in Brazilian marginal basins. Org. Geochem. 1988, 13, 31–45. [Google Scholar] [CrossRef]
- Tissot, B.; Welte, D. Petroleum Formation and Occurrence, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 1984. [Google Scholar]
- Sun, Y.; Chen, Z.; Xu, S.; Cai, S. Stable carbon and hydrogen isotopic fractionation of individual n-alkanes accompanying biodegradation: Evidence from a group of progressively bio-degraded oils. Org. Geochem. 2005, 36, 225–238. [Google Scholar] [CrossRef]
- Peters, K.E.; Walters, C.C.; Moldowan, J.M. The Biomarker Guide; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Boreham, C.J.; Dowling, L.M.; Murray, A.P. Biodegradation and maturity influences on n-alkane isotopic profiles in terrigenous sequences. In Proceedings of the 17th International Meeting on Organic Geochemistry, San Sebastian, Spain, 4–8 September 1995; pp. 539–541. [Google Scholar]
- Mansuy, L.; Philp, R.P.; Allen, J. Source identification of oil spills based on the isotopic compositin of individual compo-nents in weathered oil samples. Environ. Sci. Technol. 1997, 31, 3417–3425. [Google Scholar] [CrossRef]
- Huang, Y.; Eglinton, G.; Ineson, P.; Latter, P.; Bol, R.; Harkness, D. Absence of carbon isotope fractionation of individual n-alkanes in a 23-year field decomposition experiment with Calluna vulgaris. Org. Geochem. 1997, 26, 497–501. [Google Scholar] [CrossRef]
- Wilkes, H.; Boreham, C.; Harms, G.; Zengler, K.; Rabus, R. Anaerobic degradation and carbon isotopic fractionation of alkylbenzenes in crude oil by sulphate-reducing bacteria. Org. Geochem. 2000, 31, 101–115. [Google Scholar] [CrossRef]
- Hunkeler, D.; Andersen, N.; Aravena, R.; Bernasconi, S.M.; Butler, B.J. Hydrogen and Carbon Isotope Fractionation during Aerobic Biodegradation of Benzene. Environ. Sci. Technol. 2001, 35, 3462–3467. [Google Scholar] [CrossRef] [Green Version]
- Masterson, W.D.; Dzou, L.I.P.; Holba, A.G.; Fincannon, A.L.; Ellis, L. Evidence for biodegradation and evaporative frac-tionation in West Sak, Kuparuk and Prudhoe Bay field areas, North Slope, Alaska. Org. Geochem. 2001, 32, 411–441. [Google Scholar] [CrossRef]
- Mazeas, L.; Budzinski, H.; Raymond, N. Absence of stable carbon isotopic fractionation of saturated and polycyclic aromatic hydrocarbons during aerobic bacterial biodegrada-tion. Org. Geochem. 2002, 33, 1259–1272. [Google Scholar] [CrossRef]
- Pond, K.L.; Huang, Y.; Wang, Y.; Kulpa, C.F. Hydrogen isotopic composition of individual n-alkanes as an intrinsic tracer for bioremediation and source identification of petro-leum contamination. Environ. Sci. Technol. 2002, 36, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Mancini, S.A.; Ulrich, A.C.; Lacrampe-Couloume, G.; Sleep, B.; Edwards, E.A.; Lollar, B.S. Carbon and Hydrogen Isotopic Fractionation during Anaerobic Biodegradation of Benzene. Appl. Environ. Microbiol. 2003, 69, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallasser, R.J. Recognizing biodegradation in gas/oil accu-mulations through the δ13C compositions of gas components. Org. Geochem. 2000, 31, 1363–1373. [Google Scholar] [CrossRef]
- Hoeffs, J.; Frey, M. The isotopic composition of carbona-ceous matter in metamorphic profile from the Swiss Alps. Geochim. Cosmochim. Acta 1976, 40, 945–951. [Google Scholar] [CrossRef]
- Lewan, M. Effects of thermal maturation on stable organic carbon isotopes as determined by hydrous pyrolysis of Woodford Shale. Geochim. Cosmochim. Acta 1983, 47, 1471–1479. [Google Scholar] [CrossRef]
- Lewan, M.; Winters, J.; McDonald, J. Generation of oil-like pyrolysates from organic-rich shales. Science 1979, 203, 896–899. [Google Scholar] [CrossRef]
- Lewan, M.; Williams, J. Evaluation of petroleum generation from resinites by hydrous pyrolysis. Am. Assoc. Pet. Geol. Bull. 1987, 71, 207–214. [Google Scholar]
- Moldowan, J.M.; Dahl, J.E.; Huizinga, B.J.; Jacobson, S.R.; Taylor, D.W. The relationship of angiosperms and oleanane in petroleum through geologic time. Am. Assoc. Pet. Geol. Bull. 1993, 77, 334–335. [Google Scholar]
- Chung, H.M.; Rooney, M.A.; Toon, M.B.; Claypool, G.E. Carbon isotope composition of marine crude oils. Bull. Am. Assoc. Pet. Geol. 1992, 76, 1000–1007. [Google Scholar]
- Knoll, A.H. End of the Proterozoic Eon. Sci. Am. 1991, 265, 64–73. [Google Scholar] [CrossRef]
- Matthews, D.E.; Hayes, J.M. Isotope-ratio-monitoring gas chromatography-mass spectrometry. Anal. Chem. 1978, 50, 1465–1473. [Google Scholar] [CrossRef]
- Hayes, J.M.; Takigiku, R.; Ocampo, R.; Callot, H.J.; Albrecht, P. Isotopic composition and probable origins of organic molecules in the Eocene Messel Shale. Nature 1987, 329, 48–51. [Google Scholar] [CrossRef]
- Hayes, J.M.; Freeman, K.H.; Popp, B.N.; Hoham, C.H. Compound-specific isotopic analysis: A novel tool for reconstruction of ancient biogeochemical processes. Org. Geochem. 1990, 16, 1115–1128. [Google Scholar] [CrossRef]
- Freeman, K.; Hayes, J.M.; Trendel, J.-M.; Albrecht, P. Evidence from carbon isotope measurements for diverse origins of sedimentary hydrocarbons. Nature 1990, 343, 254–256. [Google Scholar] [CrossRef]
- Philp, R.P. Reservoir Geochemistry; The University of Oklahoma: Norman, OK, USA, 1996. [Google Scholar]
- Rincon, J.; Jaramillo, J.; Philp, R.; Allen, J. Biomar-kers and compound-specific stable carbon isotope of n-alkanes in crude oils from Eastern Llanos Basin, Colombia. J. S. Am. Earth Sci. 2010, 29, 198–213. [Google Scholar]
- Liu, J.; Geng, A.; Xiong, Y. The application of stable carbon and hydrogen isotopic compositions of individual n-alkanes to Paleozoic oil/source rock correlation enigmas in the Huanghua depression, China. J. Pet. Sci. Eng. 2006, 54, 70–78. [Google Scholar] [CrossRef]
- Bjorøy, M.; Hall, K.; Gillyon, P.; Jumeau, J. Carbon isotope variations in n-alkanes and isoprenoids of whole oils. Chem. Geol. 1991, 93, 13–20. [Google Scholar] [CrossRef]
- Karlsen, D.A.; Nyland, B.; Flood, B.; Ohm, S.E.; Brekke, T.; Olsen, S.; Backer-Owe, K. Petroleum geochemistry of the Haltenbanken, Norwegian continental shelf. In The Geochemistry of Reservoirs Geological Society; Cubbit, J.M., England, W.A., Eds.; Special Publication: London, UK, 1995; pp. 203–256. [Google Scholar]
- Stoddart, D.P.; Hall, P.B.; Larter, S.R.; Brasher, J.; Li, M.; Bjorøy, M. The reservoir geochemistry of the Eldfisk Field, Norwegian North Sea. In The Geochemistry of Reservoirs Geological Society; Cubitt, J.M., England, W.A., Eds.; Special Publication: London, UK, 1995; pp. 257–279. [Google Scholar]
- Schoell, M.; Hwang, R.; Carlson, R.; Welton, J. Carbon isotopic composition of individual biomarkers in gilsonites (Utah). Org. Geochem. 1994, 21, 673–683. [Google Scholar] [CrossRef]
- Rooney, M.; Vuletich, A.; Griffith, C. Compound-specific isotope analysis as a tool for characterizing mixed oils: An example from the West of Shetlands area. Org. Geochem. 1998, 29, 241–254. [Google Scholar] [CrossRef]
- Inaba, T.; Suzuki, N. Gel permeation chromatography for fractionation and isotope ratio analysis of steranes and tri-terpanes in oils. Org. Geochem. 2003, 34, 635–641. [Google Scholar] [CrossRef]
- Riediger, C.; Fowler, M.; Snowdon, L. Organic geochemis-try of the Lower Cretaceous Ostracode Zone, a brackish/non-marine source for some lower Manville oils in southeastern Alberta. Can. Soc. Pet. Geol. Mem. 1997, 18, 93–102. [Google Scholar]
- Schaeffer, P.; Poinsot, J.; Hauke, V.; Adam, P.; Wehrung, P.; Trendel, J.M.; Albrecht, P.; Dessort, D.; Connan, J. Novel optically active hydrocarbons in sediments: Evidence for an extensive biological cyclization of higher regular polyphenols. Angew. Chem. Int. 1994, 33, 1166–1169. [Google Scholar] [CrossRef]
- Li, M.; Riediger, C.; Fowler, M.; Snowdon, L. Unusual polycyclic aromatic hydrocarbons in Lower Cretaceous Ostracode Zone sediments and related oils of the Western Canada sedimentary basin. Org. Geochem. 1997, 27, 439–448. [Google Scholar] [CrossRef]
- Cuishan, Z.; Hong, Z.; Peirong, W.; Zebo, Z.; Anding, C. The distribution and carbon isotopic composition of unusual polycyclic alkanes in the Cretaceous Lengshuiwu Formation, China. Org. Geochem. 2003, 34, 1027–1035. [Google Scholar] [CrossRef]
- Li, M.; Huang, Y.S.; Obermajer, M.; Jiang, C.Q.; Snowdon, L.R.; Fowler, M.G. Hydrogen isotopic compositions of individual alkanes as a new approach to petroleum correlation:case studies from the Western Canada Sedimentary Basin. Org. Geochem. 2001, 32, 1387–1399. [Google Scholar] [CrossRef]
- Xiong, Y.; Geng, A. Carbon isotopic composition of indi-vidual n-alkanes in asphaltene pyrolysates of biodegraded crude oils from the Liaohe basin, China. Org. Geochem. 2000, 31, 1441–1449. [Google Scholar] [CrossRef]
- Liao, Y.; Geng, A.; Xiong, Y.; Liu, D.; Lu, J.; Liu, J.; Zhang, H.; Geng, X. The influence of hydrocarbon expulsion on carbon isotopic compositions of individual n-alkanes in pyrolysates of selected terrestrial kerogens. Org. Geochem. 2004, 35, 1479–1488. [Google Scholar] [CrossRef]
- Xiong, Y.; Geng, A.; Pan, C.; Liu, D.; Peng, P. Characterization of the hydrogen isotopic composition of individual n-alkanes in terrestrial source rocks. Appl. Geochem. 2005, 20, 455–464. [Google Scholar] [CrossRef]
- Li, M.; Larter, S.; Mei, B.; Bjorøy, M. Compound specific isotopic compositions for end members of crude oils and related source rocks from the Liaohe Basin: Paleoenvironmental implications. In Organic Geochemistry: Developments and Application to Energy, Climate and Human History, Proceedings of the 17th International Meeting on Organic Geochemistry, San Sebastián, Spain, 4–8 September 1995; Grimalt, J.O., Dorronsoro, C., Eds.; A.I.G.O.A.: San Sebastián, Spain, 1995; pp. 38–40. [Google Scholar]
- Xiong, Y.; Geng, A.; Wang, C.; Sheng, G.; Fu, J. The origin of crude oils from the Shuguang-Huanxiling Buried Hills in the Liaohe Basin, China: Evidence from chemical and isotopic compositions. Appl. Geochem. 2003, 18, 445–456. [Google Scholar] [CrossRef]
- Bernard, B.; Brooks, J.; Sackett, W. Natural gas seepage in the Gulf of Mexico. Earth Planet. Sci. Lett. 1976, 31, 48–54. [Google Scholar] [CrossRef]
- Gurgey, K.; Philp, P.; Clayton, C.; Emiroglu, H.; Siyako, M. Geochemical and isotopic approach to maturity/source/mixing estimations for natural gas and associated condensates in the Thrace Basin, NW Turkey. Appl. Geochem. 2005, 20, 2017–2037. [Google Scholar] [CrossRef]
- Tuo, J.; Zhang, M.; Wang, X.; Zhang, C. Hydrogen isotope ratios of aliphatic and diterpenoid hydrocarbons in coals and carbonaceous mudstones from the Liaohe Basin, China. Org. Geochem. 2006, 37, 165–176. [Google Scholar] [CrossRef]
- Alexander, R.; Kagi, R.I.; Larcher, A.V. Clay catalysis of alkyl hydrogen exchange reactions—reaction mechanisms. Org. Geochem. 1984, 6, 755–760. [Google Scholar] [CrossRef]
- Schimmelmann, A.; Lewan, M.D.; Wintsch, R.P. D/H isotope ratios of kerogen, bitumen, oil, and water in hydrous pyrolysis of source rocks containing kerogen types I.; II, IIS, and III. Geochim. Cosmochim. Acta 1999, 63, 3751–3766. [Google Scholar] [CrossRef]
- Schimmelmann, A.; Boudou, J.-P.; Lewan, M.D.; Wintsch, R.P. Experimental controls on D/H and 13C/12C ratios of kerogen, bitumen and oil during hydrous pyrolysis. Org. Geochem. 2001, 32, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- Leif, R.N.; Simoneit, B.R. The role of alkenes produced during hydrous pyrolysis of a shale. Org. Geochem. 2000, 31, 1189–1208. [Google Scholar] [CrossRef]
- Sessions, A.L. Hydrogen Isotope Ratios of Individual Organic Compounds. Ph.D. Thesis, Indiana University, Bloomington, Indiana, 2001; p. 149. [Google Scholar]
- Sessions, A.L.; Sylva, S.P.; Summons, R.E.; Hayes, J.M. Isotopic exchange of carbon-bound hydrogen over geologic timescales. Geochim. Cosmochim. Acta 2004, 68, 1545–1559. [Google Scholar] [CrossRef]
- Pedentchouk, N.; Freeman, K.H.; Harris, N.B. Different response of δD values of n-alkanes, isoprenoids, and kerogen during thermal maturation. Geochim. Cosmochim. Acta 2006, 70, 2063–2072. [Google Scholar] [CrossRef]
- Stalker, L.; Larter, S.R.; Farrimond, P. Biomarker binding into keogens: Evidence from hydrous pyrolysis using heavy water (D2O). Org. Geochem. 1998, 28, 239–253. [Google Scholar] [CrossRef]
- Burgoyne, T.W.; Hayes, J.M. Quantitative Production of H2 by Pyrolysis of Gas Chromatographic Effluents. Anal. Chem. 1998, 70, 5136–5141. [Google Scholar] [CrossRef]
- Hilkert, A.W.; Douthitt, C.B.; Schlüter, H.J.; Brand, W.A. Isotope ratio monitoring gas chromatography/Mass spectrometry of D/H by high temperature conversion isotope ratio mass spectrometry. Rapid Commun. Mass Spectrom. 1999, 13, 1226–1230. [Google Scholar] [CrossRef]
- Sessions, A.; Burgoyne, T.W.; Schimmelmann, A.; Hayes, J.M. Fractionation of hydrogen isotopes in lipid biosynthesis. Org. Geochem. 1999, 30, 1193–1200. [Google Scholar] [CrossRef]
- Anderson, K.B.; Muntean, J.V. The nature and fate of natural resins in the geosphere. Part, X. Structural characteristics of the macromolecular constituents of modern dammar resin and Class II ambers. Geochem. Trans. 2000, 1, 1–9. [Google Scholar] [CrossRef]
- Maslen, E.; Grice, K.; Le Métayer, P.; Dawson, D.; Edwards, D. Stable carbon isotopic compositions of individual aromatic hydrocarbons as source and age indicators in oils from western Australian basins. Org. Geochem. 2011, 42, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Andersen, N.; Paul, H.A.; Bernasconi, S.; McKenzie, J.A.; Behrens, A.; Schaeffer, P.; Albrecht, P. Large and rapid climate variability during the Messinian salinity crisis: Evidence from deuterium concentrations of individual biomarkers. Geology 2001, 29, 799–802. [Google Scholar] [CrossRef]
- Yang, H.; Huang, Y. Preservation of lipid hydrogen isotope ratios in Miocene lacustrine sediments and plant fossils at Clarkia, northern Idaho, USA. Org. Geochem. 2003, 34, 413–423. [Google Scholar] [CrossRef]
- Dawson, D.; Grice, K.; Alexander, R. Stable hydrogen isotopic composition of hydrocarbons in crude oils and source rocks from the Perth Basin (Western Australia). In Proceedings of the 21st International Meeting on Organic Geochemistry, Krakow, Poland, 8–12 September 2003. [Google Scholar]
- Dansgaard, W. Stable isotopes in precipitation. Tellus 1964, 16, 436–468. [Google Scholar] [CrossRef]
- Kehew, A.E. Applied Chemical Hydrogeology; Prentice Hall: Upper Saddle River, NJ, USA, 2001. [Google Scholar]
- Schoell, M. The hydrogen and carbon isotopic composition of methane from natural gases of various origins. Geochim. Cosmochim. Acta 1980, 44, 649–661. [Google Scholar] [CrossRef]
- Schoell, M. Multiple origins of methane in the earth. Chem. Geol. 1988, 71, 1–10. [Google Scholar] [CrossRef]
- Dai, J.; Pei, X.; Qi, H. China Natural Gas Geology; Petroleum Industry Press: Beijing, China, 1992; Volume 1, pp. 35–86. [Google Scholar]
- Dai, J.X.; Xia, X.Y.; Li, Z.S. Inter-laboratory calibration of natural gas round robins for δ2H and δ13C using offline and online techniques. Chem. Geol. 2012, 310–311, 49–55. [Google Scholar] [CrossRef]
- Dai, J.X.; Ni, Y.Y.; Gong, D.Y. Geochemical characteristics of gases of from the largest tight sand field (Sulige) and shale gas field (Fuling) in China. Mar. Pet. Geol. 2017, 79, 426–438. [Google Scholar] [CrossRef]
- Whiticar, M.J.; Faber, E.; Schoell, M. Biogenic methane formation in marine and freshwater environments: CO2 reduction vs. acetate fermentation-isotope evidence. Geochim. Cosmochim. Acta 1986, 50, 693–709. [Google Scholar] [CrossRef]
- Whiticar, M.J. Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem. Geol. 1999, 161, 291–314. [Google Scholar] [CrossRef]
- Shen, P.; Xu, Y.C. Isotopic compositional characteristics of terrigenous natural gases in China. Chin. J. Geochem. 1993, 12, 14–24. [Google Scholar] [CrossRef]
- Xu, Y. Origin Theory and Application of Natural Gases; Science Press: Beijing, China, 1994. [Google Scholar]
- Galimov, E.M. Isotope organic geochemistry. Org. Geochem. 2006, 37, 1200–1262. [Google Scholar] [CrossRef]
- Kinnaman, F.S.; Valentine, D.L.; Tyler, S.C. Carbon and hydrogen isotope fractionation associated with aerobic microbial oxidation of methane, ethane, propane and butane. Geochim. Cosmochim. Acta 2007, 71, 271–283. [Google Scholar] [CrossRef]
- Ni, Y.Y.; Ma, Q.S.; Geoffrey, S.E. Fundamental studies on kinetic isotope effect (KIE) of hydrogen isotope fractionation in natural gas systems. Geochim. Cosmochim. Acta 2011, 75, 2696–2707. [Google Scholar] [CrossRef]
- Ni, Y.Y.; Dai, J.X.; Zhu, G.Y. Stable hydrogen and carbon isotopic ratios of coal-derived an oil-derived gases: A case study in the Tarim Basin, NW China. Int. J. Coal. Geol. 2013, 116–117, 302–313. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Worden, R.H.; Jin, Z.J. Thermochemical sulphate reduction(TSR) versus maturation and their effects on hydrogen stable isotopes of very dry alkane gases. Geochim. Cosmochim. Acta 2014, 137, 208–220. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Jin, Z.J.; Meng, Q.Q. Genetic types of natural gas and filling patterns in Daniudi gas field, Ordos Basin. China J. Asian Earth Sci. 2015, 107, 1–11. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Li, Z. The hydrogen isotopic characteristics of the Upper Paleozoic natural gas in Ordos Basin. Org. Geochem. 2014, 74, 66–75. [Google Scholar] [CrossRef]
- Wang, Y.P.; Dai, J.X.; Zhao, C.Y. Genetic origin of Mesozoic natural gases in the Ordos Basin (China): Comparison of carbon and hydrogen isotopes and pyrolytic results. Org. Geochem. 2010, 41, 1045–1048. [Google Scholar] [CrossRef]
- Dai, J.; Ni, Y.; Hu, G. Stable carbon and hydrogen isotopes of gases from the large tight gas fields in China. Sci. China Earth Sci. 2014, 57, 88–103. [Google Scholar] [CrossRef]
- Hu, G.Y.; Li, J.; Shan, X.Q. The origin of natural gas and the hydrocarbon charging history of the Yulin Gas field in the Ordos Basin, China. Int. J. Coal Geol. 2010, 81, 381–391. [Google Scholar]
- Hu, G.Y.; Yu, C.; Ni, Y.Y. Comparative study of stable carbon and hydrogen isotopes of alkane gases sourced from the Longtan and Xujiahe coal-bearing measures in the Sichuan Basin, China. Int. J. Coal Geol. 2014, 116–117, 293–301. [Google Scholar] [CrossRef]
- Wu, X.; Wang, P.; Liu, Q. The source of natural gas reservoired in the 5th member of the Upper Trias-sic Xujiahe Formation in Xinchang Gasfield, the Western Sichuan Depression and its implication. Nat. Gas Geosci. 2016, 27, 1409–1418. [Google Scholar]
- Huang, S.P.; Fang, X.; Liu, D. Natural gas genesis and sources in the Zizhou gas field, Ordos Basin, China. Int. J. Coal Geol. 2015, 152, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, J.; Li, Z. Characteristics and genetic types of the lower Paleozoic natural gas, Ordos Basin. Mar. Pet. Geol. 2018, 89, 106–119. [Google Scholar] [CrossRef]
- Wang, X.F.; Liu, W.H.; Shi, B.G. Hydrogen isotope characteristics of thermogenic methane in Chinese sedimentary basins. Org. Geochem. 2015, 83–84, 178–189. [Google Scholar] [CrossRef]
- Feng, Z.Q.; Liu, D.; Huang, S.P. Geochemical characteristics and genesis of natural gas in the Yan’an gas field, Ordos Basin, China. Org. Geochem. 2016, 102, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.Q.; Tao, X.W.; Hu, G.Y. Geochemical characteristics and source of natural gases from southeast depression of the Tarim Basin, NW China. Org. Geochem. 2014, 74, 106–115. [Google Scholar] [CrossRef]
- Peng, W.; Hu, G.; Huang, S. Natural gas geochemical characteristics and genetic analysis: A case study of the Dongsheng gas field in the Ordos Basin of China. J. China Univ. Min. Technol. 2017, 46, 74–84. [Google Scholar]
- Huang, S.; Duan, S.; Wang, Z.; Jiang, Q.; Jiang, H.; Su, W.; Feng, Q.; Huang, T.; Yuan, M.; Ren, M.; et al. Affecting factors and application of the stable hydrogen isotopes of alkane gases. Pet. Explor. Dev. 2019, 46, 518–530. [Google Scholar] [CrossRef]
- Thode, H.G. Sulfur isotope ratios in petroleum research and exploration: 202 Williston basin. Am. Associ. Pet. Geol. Bull. 1981, 65, 1527–1535. [Google Scholar]
- Orr, W.L. Kerogen/asphaltene/sulphur relationships in sulphur-rich Montereyoils. In Advances in Organic Geochemistry; By, D., Leythaeuser, J., Eds.; Rullkotter Rullkotter Pergamon Press: Oxford, UK, 1986; pp. 499–516. [Google Scholar]
- Cai, C.F.; Zhang, C.M.; Cai, L.L.; Wu, G.H.; Jiang, L.; Xu, Z.M.; Li, K.K.; Ma, A.L.; Chen, L.X. Origins of Palaeozoic oils in the Tarim Basin: Evidence from sulfur isotopes and biomarkers. Chem. Geol. 2009, 268, 197–210. [Google Scholar] [CrossRef]
- Yan, D.T.; Chen, D.Z.; Wang, Q.C.; Wang, J.G. Predominance of stratified anoxic Yangtze Sea interrupted by short-term oxygenation during the Ordo-Silurian transition. Chem. Geol. 2012, 291, 69–78. [Google Scholar] [CrossRef]
- Wei, H.Y.; Wei, X.M.; Qiu, Z.; Song, H.Y.; Shi, G. Redox conditions across the G-L boundary in South China: Evidence from pyrite morphology and sulfur isotopic compositions. Chem. Geol. 2016, 440, 1–14. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, D.; Jin, Z.; Meng, Q.; Li, S. Influence of volcanic activities on redox chemistry changes linked to the enhancement of the ancient Sinian source rocks in the Yangtze craton. Precambrian Res. 2019, 327, 1–13. [Google Scholar] [CrossRef]
- Chen, G.; Chang, X.; Gang, W.; Wang, N.; Zhang, P.; Cao, Q.; Xu, J. Anomalous positive pyrite sulfur isotope in lacustrine black shale of the Yanchang Formation, Ordos Basin: Triggered by paleoredox chemistry changes. Mar. Pet. Geol. 2020, 104587. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Notation | Ratio | Standard | Absolute Ratio |
|---|---|---|---|---|
| Hydrogen | δD | D/H(2H/1H) | SMOW | 1.557 × 10−4 |
| >Carbon | δ13C | 13C/12C | PDB | 1.122 × 10−2 |
| Nitrogen | δ15N | 15N/14N | Atmosphere | 3.613 × 10−3 |
| Oxygen | δ18O | 18O/16O | SMOW, PDB | 2.0052 × 10−3 |
| Chlorine | δ37Cl | 37Cl/35Cl | seawater | −0.31978 |
| Sulfur | δ34S | 34S/32S | CDT | 4.43 × 10−2 |
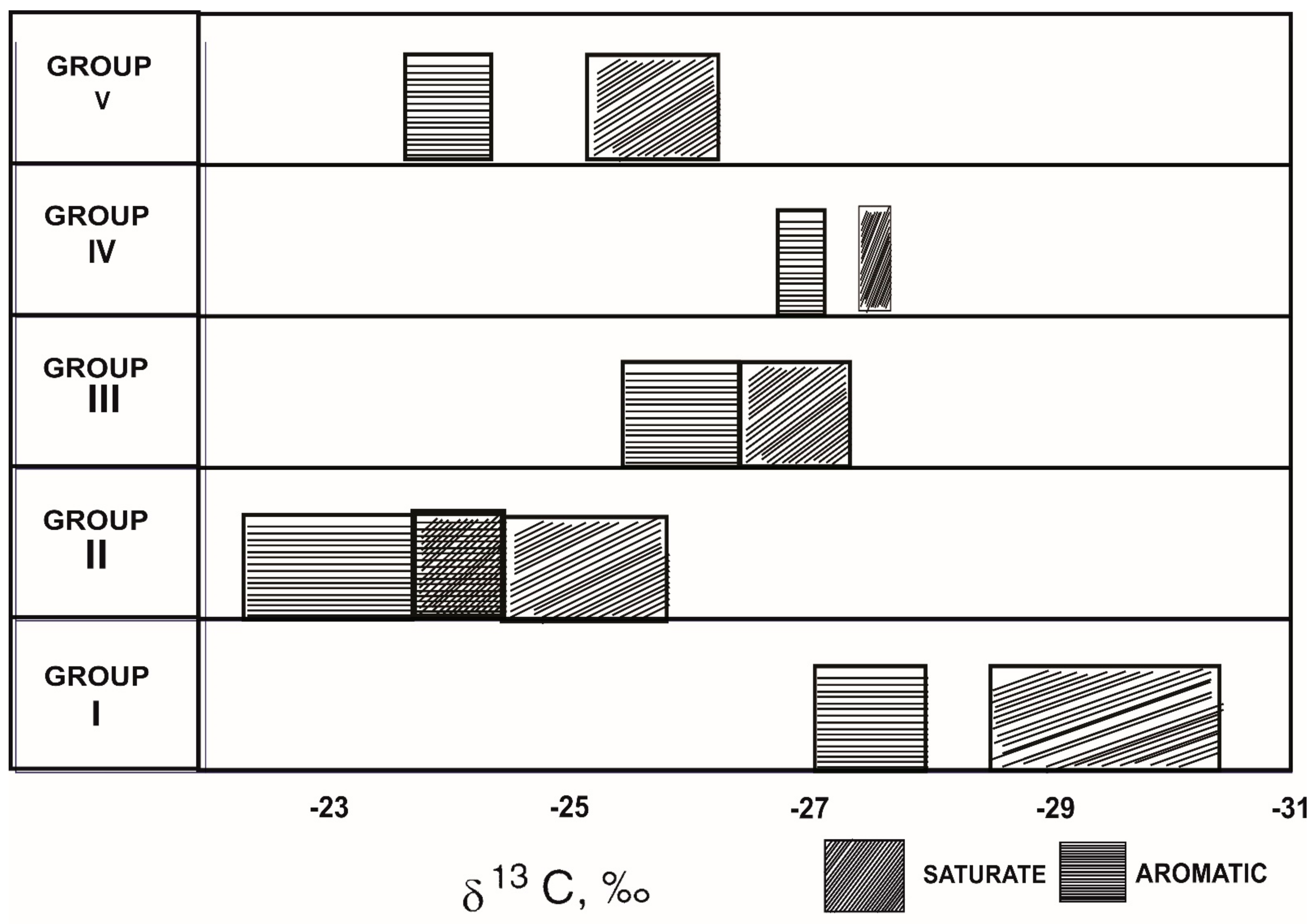
| Property | Group I | Group II | Group III | Group IV | Group V |
|---|---|---|---|---|---|
| δ13C‰ | < −28.0 | −23.0 to −25.6 | −25.4 to −26.6 | −26.8 to −27.6 | −24.4, −25.1 |
| % Sulphur | Low | Medium | High | High | Medium |
| V/Ni | Low | High/medium | Medium | High | High |
| %Saturates | High | High | Low/medium | Low/medium | High |
| n-Alkanes’ dominance | C23-C25 | C17-C21 | C17-C19 | C18-C20 | C20-C22 |
| Odd/even | High | High | Low | Low | Low |
| Pristane/Phytane | High | High | Low | Low | Low |
| Inferred depositional environment | Lacustrine/freshwater | Lacustrine/saline water | Marine evaporitic | Marine carbonate | Marine deltaic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogbesejana, A.B.; Liu, B.; Ostadhassan, M. Stable Isotope Geochemistry of the Organic Elements within Shales and Crude Oils: A Comprehensive Review. Molecules 2022, 27, 34. https://doi.org/10.3390/molecules27010034
Ogbesejana AB, Liu B, Ostadhassan M. Stable Isotope Geochemistry of the Organic Elements within Shales and Crude Oils: A Comprehensive Review. Molecules. 2022; 27(1):34. https://doi.org/10.3390/molecules27010034
Chicago/Turabian StyleOgbesejana, Abiodun Busuyi, Bo Liu, and Mehdi Ostadhassan. 2022. "Stable Isotope Geochemistry of the Organic Elements within Shales and Crude Oils: A Comprehensive Review" Molecules 27, no. 1: 34. https://doi.org/10.3390/molecules27010034
APA StyleOgbesejana, A. B., Liu, B., & Ostadhassan, M. (2022). Stable Isotope Geochemistry of the Organic Elements within Shales and Crude Oils: A Comprehensive Review. Molecules, 27(1), 34. https://doi.org/10.3390/molecules27010034