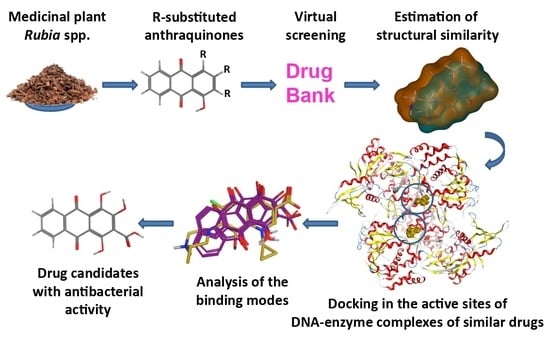
1. Introduction
The diverse pharmacological effects of plant polyphenols in parallel with their constantly increasing use in diets and as ethnomedicines justify the efforts for identification of the molecular mechanisms of their therapeutic action as well as finding their potential molecular targets. Recently, we have reported a study estimating structural similarity between plant-derived phenols and drug compounds from the DrugBank database [
1] (
https://www.drugbank.ca, last accessed on the 1 March 2022). A representative dataset of 75 phenols selected from the literature and a virtual library of more than 7000 drug compounds extracted from the DrugBank have been used [
2]. Performing structural similarity-based virtual screening (OpenEye scientific software platform,
https://www.eyesopen.com, last accessed on the 1 March 2022) of the DrugBank database, we have demonstrated that pseudopurpurin, a hydroxyanthraquinone from
Rubia spp., was structurally similar to gatifloxacin, a synthetic broad-spectrum antibacterial agent.
Gatifloxacin is a representative of the fourth-generation fluoroquinolone family. It is known to work by inhibiting the bacterial enzymes DNA gyrase and DNA topoisomerase IV [
3,
4]. These enzymes have been considered attractive targets for discovery and design of biologically active compounds with antibacterial activity, due to their essential role for the maintenance of a proper DNA topology during transcription and replication [
5]. In parallel, anthraquinones have been known as inhibitors of bacterial topoisomerases I and II [
6], antibacterial effects [
7], and biological activities against
Staphylococcus aureus and
Bacillus subtilis have been reported for
Rubia cordifolia L. [
8].
The structural similarity of pseudopurpurin to gatifloxacin pointed to both DNA-related enzymes as potential molecular targets of other hydroxyanthraquinones from Rubia spp. and motivated our further interest in investigating these natural compounds as potential inhibitors of the DNA gyrase and DNA topoisomerase IV enzymes.
In the present study, a homologous series of plant-derived hydroxyanthraquinones previously reported among the secondary metabolites typically found in
Rubia spp. [
9,
10] was comparatively explored with regard to similarity with synthetic drugs shown to interact with DNA gyrase and DNA topoisomerase IV, namely, the synthetic fluoroquinolones (S)-gatifloxacin and (S)-levofloxacin. Furthermore, docking simulations of the compounds in the two bacterial enzymes were carried out to predict potential interactions and to map molecular modes of their possible antibacterial action. Overall, the carboxyl and hydroxyl groups in the structures of hydroxyanthraquinones were outlined as significant features predisposing to higher variability of the ligands’ behavior in the binding site, and stronger binding, thus suggesting the possible use of these hydroxyanthraquinones as appropriate lead structures.
3. Discussion
DNA gyrase and DNA topoisomerase IV have been classified as type IIA topoisomerases. Their mechanism of action involves formation of a transient covalent bond to the 5ʹDNA-phosphate of both strands of the DNA duplex and to function via a strand-passage mechanism [
11]. The inhibitory effect of quinolones has been associated with binding to the enzyme-DNA complexes, thus trapping the enzyme targets on bacterial DNA as ternary drug-enzyme-DNA complexes [
12].
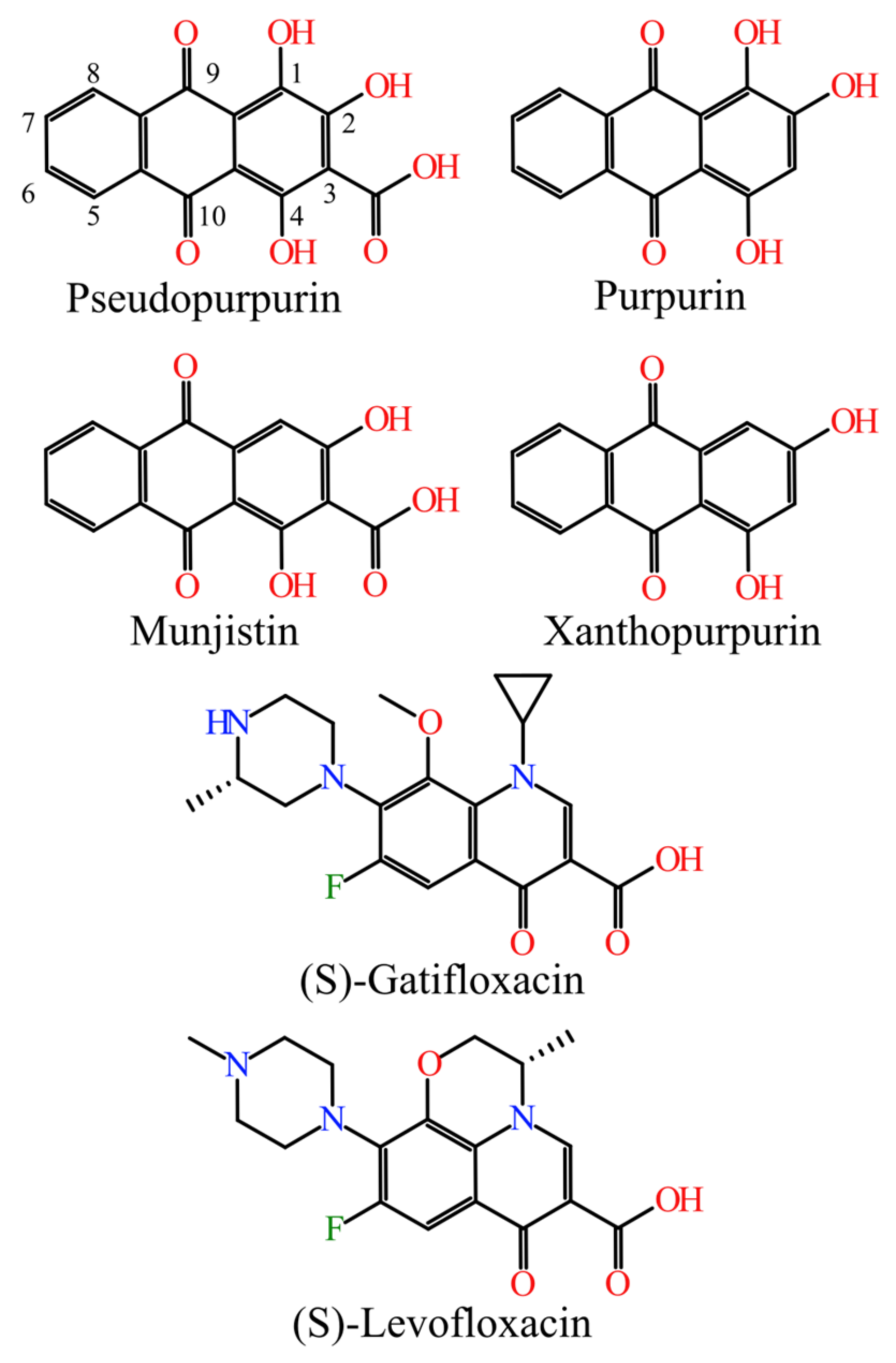
In the current study, munjistin, purpurin, and xanthopurpurin were selected based on the common structural scaffold they share with the previously highlighted hit compound pseudopurpurin [
2]. The scores of structural similarity bewteen the four investigated hydroxyanthraquinones to the referent fluoroquinolones were quite close, those of the carboxylated pseudopurpurin and munjistin being slightly higher than the ones of the non-carboxylated purpurin and xanthopurpurin. Similar trends were outlined also for (S)-levofloxacin. Overall, slightly higher similarity was reported to (S)-levofloxacin compared to (S)-gatifloxacin.
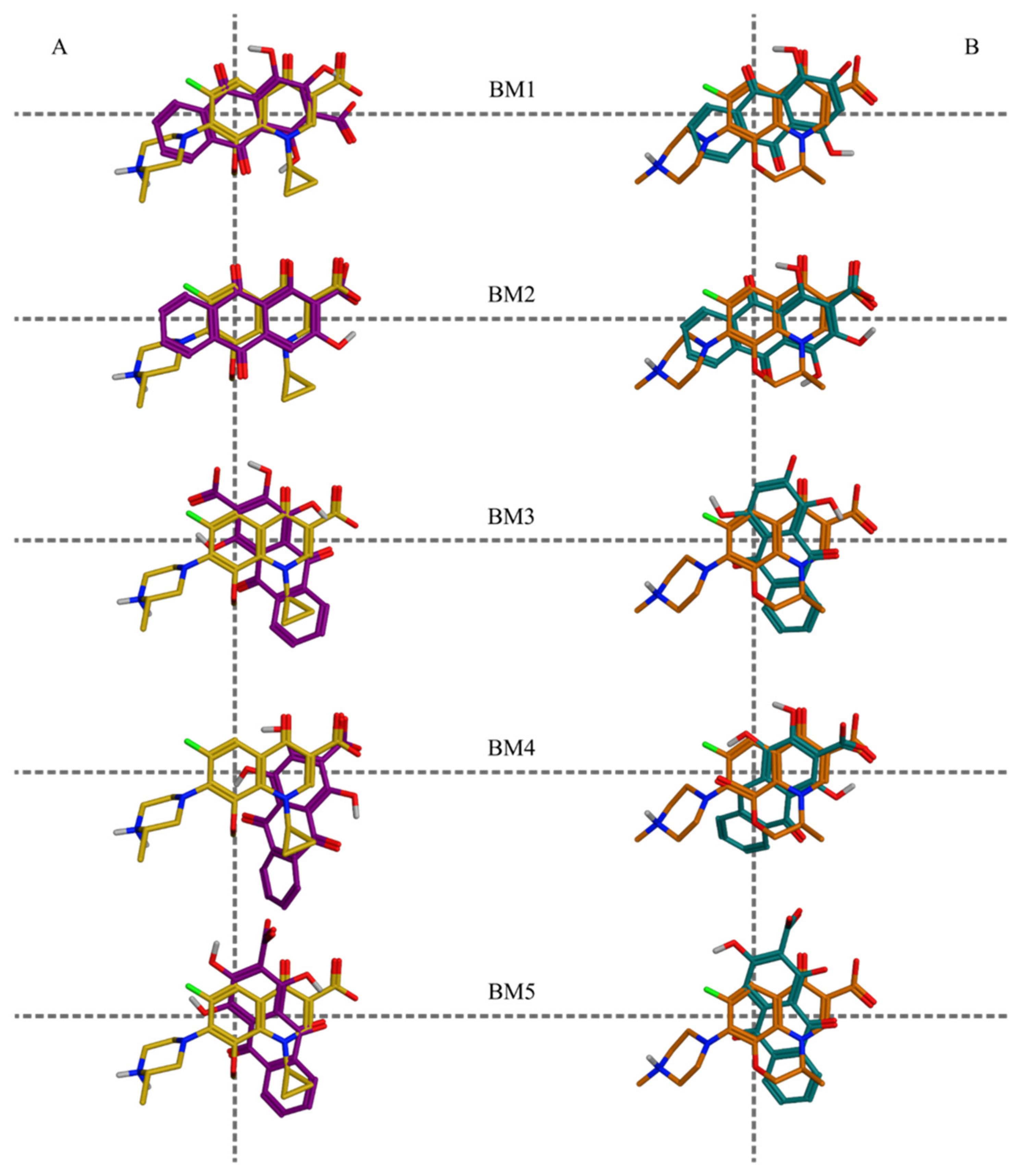
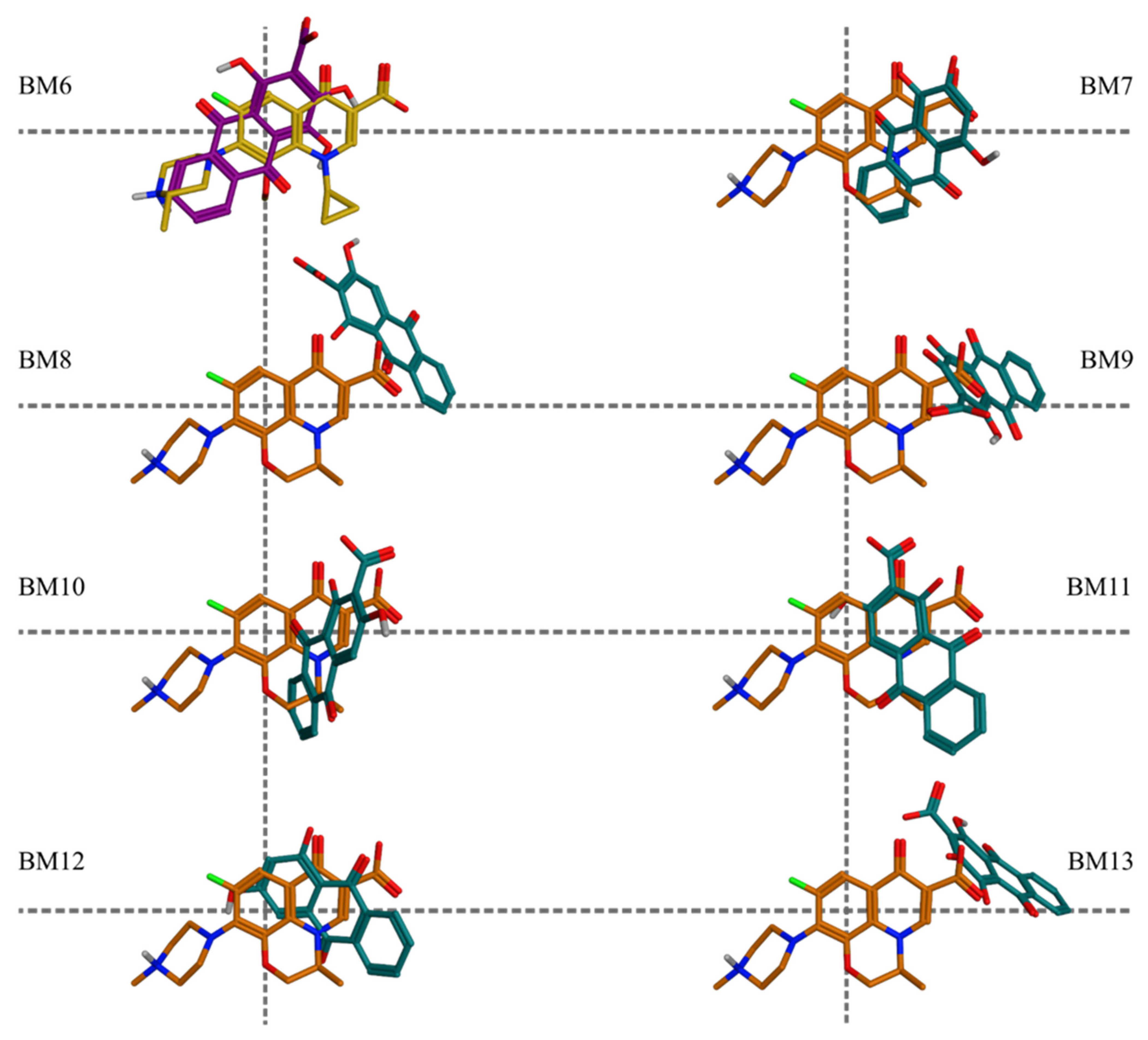
The molecular docking simulations of the structures with the highly probable ionization states in the protein-DNA-drug complexes of DNA gyrase and DNA topoisomerase IV supported the initial hypothesis about the potential mechanism(s) of the antibacterial effects of Rubia spp. Pseudopurpurin and purpurin appeared most frequently among the best scored docking poses. In particular, for both targets pseudopurpurin appeared above the median of the docking scores and within the best-scored quartiles (the 75-th percentile) in all ionization states, while purpurin appeared among the top-ranked poses representative for the highly probable ionization states, underlining the relevance of the 1-OH substituent for the affinities to the targets. The most negatively scored BMs were BM1, BM2, BM3, and BM4, respectively. According to the results, pseudopurpurin and munjistin have the highest number of different BMs in DNA gyrase suggesting higher probability of binding to this enzyme.
The variability of the molecular features of the four hydroxyanthraquinones, allowed for a deeper analysis of the relevance of the 3-COOH and the 1-OH substituents to the molecular modes of action of pseudopurpurin (
Figure 1). This was clearly outlined in the detailed PLIF analysis of the binding modes in the active sites of the two DNA-enzyme complexes and the ranking of the binding modes by their average docking scores (
Tables S3 and S4). Such complementary approach combining knowledge for the chemical domain of the studied series of compounds, the predicted patterns of ligand-receptor interactions, and the quantitative approximations of ligands’ affinities has been successfully applied in our previous investigations focused on prediction and classification of potential molecular modes of action of multiple Phase I metabolites of a triterpenoid [
13].
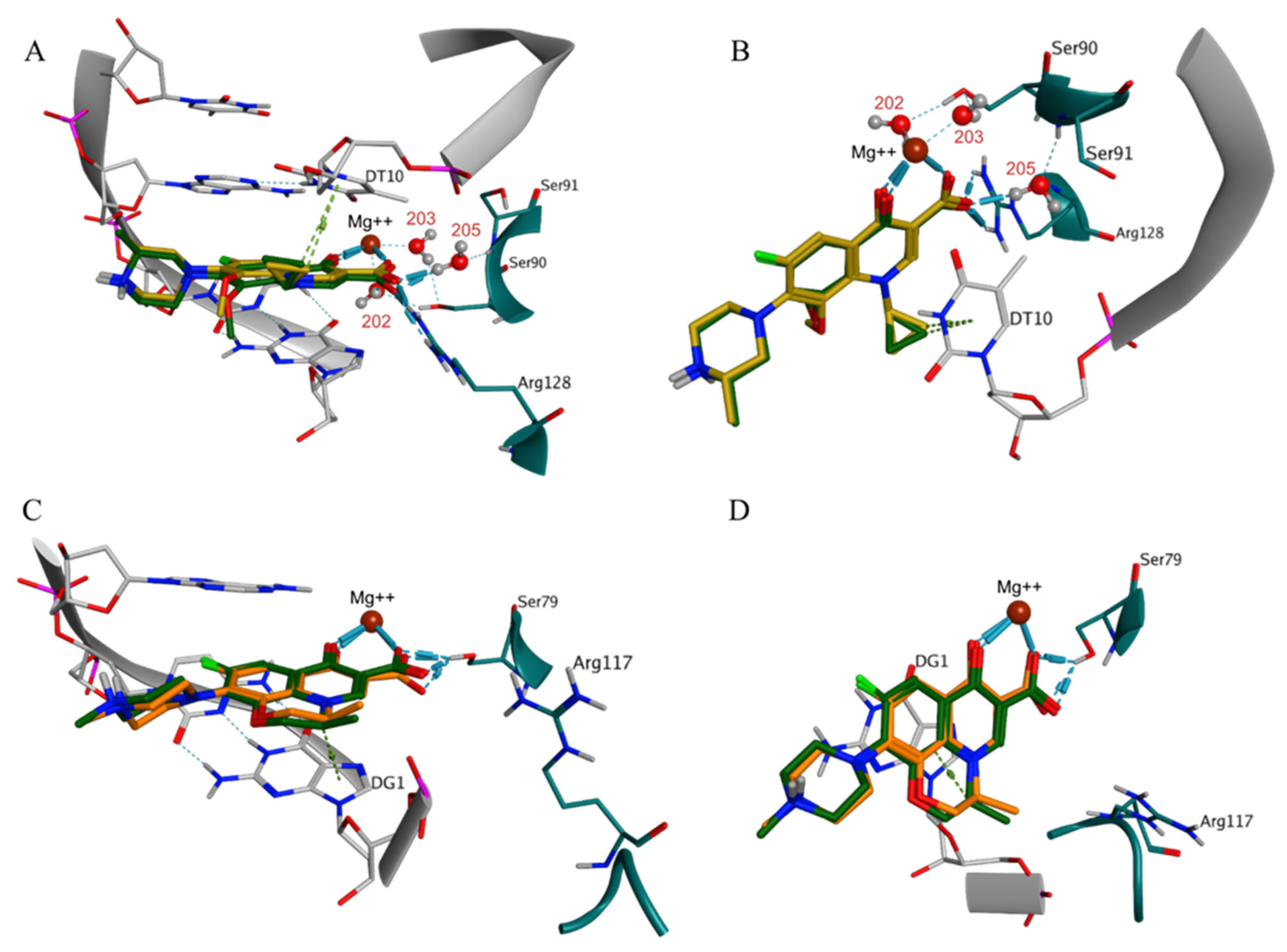
The detailed comparative analysis of the ligand-receptor interaction fingerprints within the subset of poses in highly probable ionization states revealed important trends regarding the structural variability of the studied hydroxyantraquinones. The ranking by the docking scores clearly distinguished affinity-related interaction patterns correlating the top scoring poses to frequently occurring interactions between the Mg++ ion and the 1,2-OH substituents simultaneously. The bottom-scored pattern in most cases involved simultaneous interactions of the 2-OH and the 3-COOH substituents with the Mg++ ion, together with direct interactions of the 3-COOH group with Arg128 or water-bridged ones with Ser90 and Ser91 (
Table S3).
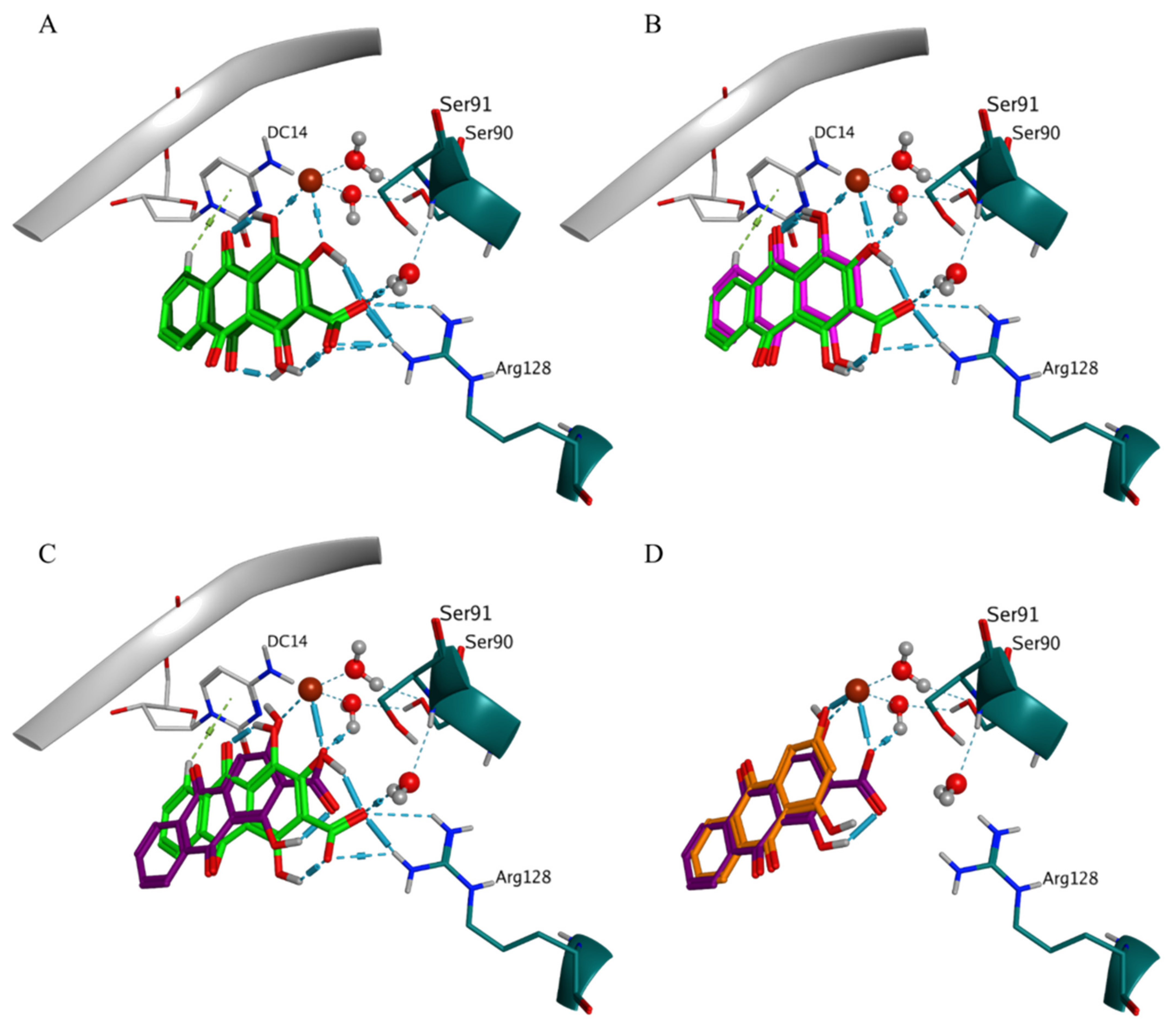
Pseudopurpurin and its non-carboxylated analogue purpurin shared three similar BMs in DNA gyrase, i.e., BM1, BM3, and BM4, and were the compounds with the most negative best and average scores (
Figure 5). Possessing the higher number of substituents, pseudopurpurin also displayed the most diverse ligand-receptor interaction patterns characterized by the highest number of key partnering residues (an amino acid residue, a nucleotide, a Mg++ ion or a water molecule) (
Table S3). Compared to its analogue lacking 1-OH substituent (munjistin), pseudopurpurin performed a higher number of interactions with Arg128, a significantly higher number of water-mediated (HOH205) H-bond interactions with Ser91, and interactions with nucleotide and with a phosphorylated tyrosine 129 (Ptr129) (
Table S3). In one of the BM1 poses pseudopurpurin performed arene-H interaction with DC14 through the distal anthraquinone ring (
Figure 5A). This interaction was predicted neither for purpurin (
Figure 5B), nor for munjistin (
Figure 5C) and xanthopurpurin (
Figure 5D). The lack of interactions with Arg128 in BM1 of munjistin, compared to pseudopurpurin was associated with the engagement of the 3-COOH substituent with the coordination of Mg++, while this was performed by 1-OH and/or 2-OH substituents in the case of pseudopurpurin (
Figure 5C). Due to the lack of 3-COOH substituent, purpurin and xanthopurpurin did not perform interactions with Arg128 (
Figure 5B,D).
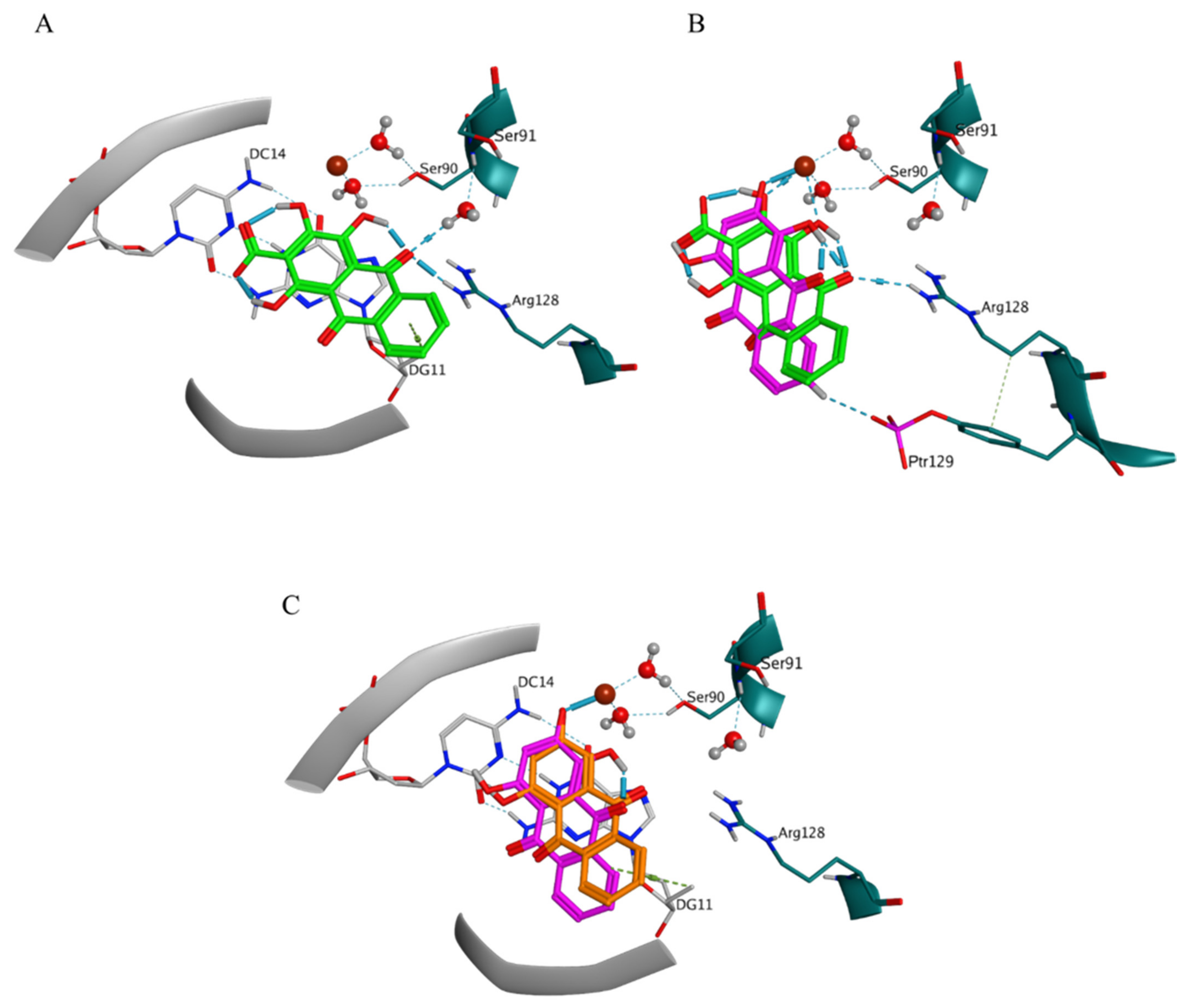
The interactions with DG11 and Ptr129 were rarely predicted (
Table S3). In BM3, DG11 appeared in the ligand-receptor interaction patterns of pseudopurpurin (
Figure 6A) and xanthopurpurin (
Figure 6C), and purpurin displayed H-bond interaction with Ptr129 (
Figure 6B). The ligand-receptor interaction pattern of the two pseudopurpurin’s poses grouped in BM3 involved an obligatory interaction between the carbonyl substituent at position 9 and Arg128 (
Figure 6A,B). The two poses, however, displayed either interaction of the carbonyl group at position 9 with the HOH205-Ser91, combined with interaction of the distal anthraquinone ring and guanine 11 (DG11) (
Figure 6A), or interactions between the Mg++ ion and the 1,2-OH substituents (
Figure 6B).
The binding patterns of purpurin and xanthopurpurin were lacking the interactions with Arg128 and HOH205-Ser91, predicted for their carboxylated analogues (
Table S3).
The deeper analysis of the participation of the different substituents in the performed ligand-receptor interactions explained some of the differences in their binding patterns. The lack of a 3-COOH substituent in purpurin resulted in a higher frequency of simultaneous involvement of the 1-OH and 2-OH substituents in the coordination of the Mg++ ion compared to pseudopurpurin. The metal/ion interaction pattern of the latter engaged with an equal frequency the simultaneous participation of the 2-OH and 3-COOH substituents on the one hand, and of the 1-OH and 2-OH substituents, on the other. Logically, xanthopurpurin relied only on the 2-OH substituent for this interaction due to lack of both 1-OH and 3-COOH substituents. The lack of a 3-COOH substituent affected the binding behavior of purpurin in BM1 and BM4 regarding the water-bridged interaction with Ser90. Purpurin performed H-bond interaction through its 2-OH substituent, instead of involving a 3-COOH substituent as predicted for munjistin and pseudopurpurin in these BMs (
Table S3).
The ligand-receptor interaction patterns in DNA topoisomerase IV were characterized by metal/ion interactions for all hydroxyanthraquinones similarly to the patterns in DNA gyrase (
Table S4). Due to the intrinsic features of the complex in this simulation, the predicted interactions with Ser79 were direct (
Figure 7). The poses differed mostly in the type of the substituents involved in the metal/ion interaction with the Mg++ ion and the H-bond interaction with Ser79. Pseudopurpurin and munjistin interacted with the Mg++ ion exclusively by the 3-COOH substituent alone or in combination with the 2-OH substituent (
Figure 7). In particular, pseudopurpurin performed this interaction also in combination with the 4-OH substituent. Compared to the DNA gyrase docking output in this complex the total number of poses and the variety of binding modes was significantly lower (
Table 2). Nevertheless, pseudopurpurin was still with the highest number of poses. Munjistin, however, was presented only by two poses, both in the shared for the two targets BM1 (
Figure 7A). All hydroxyanthraquinones, except pseudopurpurin, were shown to display this BM. In DNA topoisomerase IV, the interactions of purpurin and xanthopurpurin with Ser79 in BM1 were performed through 1-OH and 2-OH for purpurin (
Figure 7A) and 2-OH for xanthopurpurin (
Figure 7B), respectively, while munjistin involved a combination of its 2-OH and 3-COOH substituents instead (
Figure 7A,B). Pseudopurpurin was the only hydroxyanthraquinone with poses classified in the BM2 group. This binding mode represents a variation of BM1 (
Figure 5A), which was vertically flipped around the imaginary horizontal axis of the three-ring hydroxyanthraquinone scaffold (
Table S1). The two pseudopurpurin’s poses included in BM2 had almost similar ligand-receptor interaction patterns. The only difference between them was the appearance of one more metal/ion interaction between the 4-OH substituent and the Mg++ ion (
Table S4,
Figure 7C). Similarly, to the scoring in the DNA gyrase-based simulation, the best docking scores of the two 1-OH substituted hydroxyanthraquinones were the most negative ones among the best scores of all analogues. This was also valid for their average scores (
Table 2).
The analysis of the frequencies of participation of each individual substituent in ligand-receptor interactions (
Table S5) outlined the central place of the 3-COOH substituent in the receptor-binding patterns of the top scored poses of pseudopurpurin. In addition, the compensatory role of the 4-OH substituent in the interactions of the analogue which lacks the 1-OH substituent (munjistin) was evident. This is related to BMs, which less frequently involve the 3-COOH compared to the interaction pattern of the 1-OH containing analogue (pseudopurpurin). Such a compensatory role in the case of pseudopurpurin’s non-carboxylated analogue purpurin was predicted for the 2-OH substituent. It could be concluded that the absence of a 1-OH substituent results in a spatial shift of the poses to favor a less frequent involvement of the 3-COOH substituent; the absence of the 3-COOH substituent favors the more frequent involvement of the 2-OH substituent. The median values of the hydroxyanthraquinones’ docking scores for DNA topoisomerase IV were comparable. However, in the DNA gyrase-based simulations the presence of the 1-OH substituent is related with better docking scores as evident from the median values of the poses, while the presence of the 3-COOH substituent increases the total number of the predicted poses and the number of ligand-receptor interactions. All these observations underline the role of the 1-OH and 3-COOH substituents for the ligand interactions and point to pseudopurpurin as the most promising molecular scaffold in the series.
Based on these results, further studies could be designed to explore more comprehensively the mechanistically relevant binding behavior of this class of naturally-occurring compounds in the two target complexes. Given the fact that these hydroxyantraquinones have been known as constituents of orally applied traditional medicinal preparations [
14], and even commonly consumed beverages in some regions [
15,
16], an in silico investigation of the potential metabolites of these compounds might be performed as a next step in this modelling workflow. Another intriguing aspect that deserves further attention is the influence of the protonation states of the ligands and their targets on the ligand-receptor interactions upon different pH-related settings. This is especially interesting in relation to the pathological action of
M. tuberculosis and is in line with the therapeutic paradigm exploiting pH-dependent phenomena of relevance to this pathogen [
17,
18].
5. Conclusions
In this study, we combined several in silico methods in a multistep sequential manner—shape and chemical features’ similarity evaluation, structure-based molecular docking, and analysis of the protein-ligand interactions—to comprehensively estimate the potential of selected polyphenols from the chemical class of hydroxyanthraquinones to interact with the bacterial enzymes DNA gyrase and DNA topoisomerase IV. At each of these steps the results were encouraging and supportive for the correctness of our approach proven to be successful in a recent study of structurally more complex polyphenols [
22].
Four hydroxyanthraquinones—pseudopurpurin, purpurin, munjistin, and xanthopurpurin—naturally occurring in Rubia spp. and varying by the substituents at positions 1, 2, and 3 were thoroughly investigated to reveal the relevance of their structural features for the interactions with the enzymes.
Pseudopurpurin and munjistin were found to be more structurally similar to the reference fluoroquinolones (S)-gatifloxacin and (S)-levofloxacin than their non-carboxylated analogues. Docking poses of pseudopurpurin and munjistin in the active sites of the protein-DNA complexes of DNA gyrase and DNA topoisomerase IV revealed their plausible molecular modes of action against Gram-positive and Gram-negative bacteria. The most populated binding modes reproduced the ligand-receptor interaction patterns of the X-ray poses of the referent fluoroquinolones and displayed additional specific interactions with key residues in the binding sites of the complexes. The anthraquinones’ docking scores were better than those of the referent fluoroquinolones, suggesting the possibility for stronger binding of the studied polyphenols to the DNA gyrase and DNA topoisomerase IV protein-DNA complexes compared to their co-crystalized ligands. The binding behavior of pseudopurpurin and munjistin was characterized by a wider range of diverse binding modes, which could be attributed mainly to the contribution of the 1-OH and 3-COOH substituents. This was also evident from the detailed ligand-receptor interaction analysis, which revealed that the presence of both substituents reflects on the ability of the ligands to interact with more key residues in the pocket. A similar trend was observed in the context of predicted ligand’s affinity to the binding sites with emphasis on the 1-OH substituent, present in purpurin and pseudopurpurin.
The results outline the studied hydroxyanthraquinones, and in particular pseudopurpurin, as promising lead structures for further drug development of antibacterial agents with potential to interact with the DNA gyrase and DNA topoisomerase IV. Design of appropriate experimental in vitro studies is ongoing to confirm the relevance of the in silico predictions.
The combined in silico approach used in this study could be applied to any other bioactive compounds of natural origin which putative molecular targets and mechanisms of action are still unknown or need to be elucidated at a molecular level.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}