
Coadsorption Interfered CO Oxidation over Atomically Dispersed Au on h-BN



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theoretical Methods
3. Results and Discussions
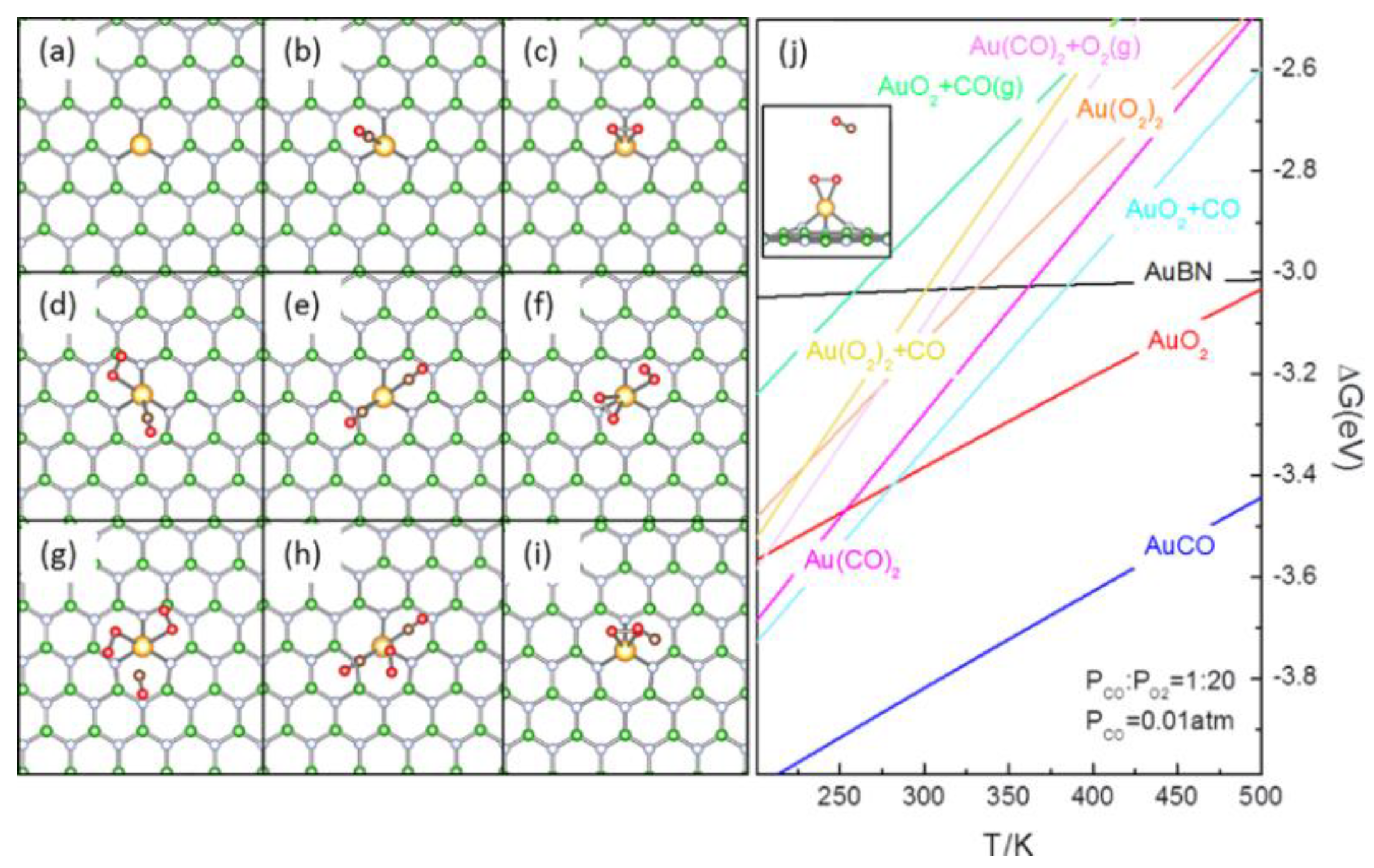
3.1. Thermodynamics Analysis of Potential Reaction Species
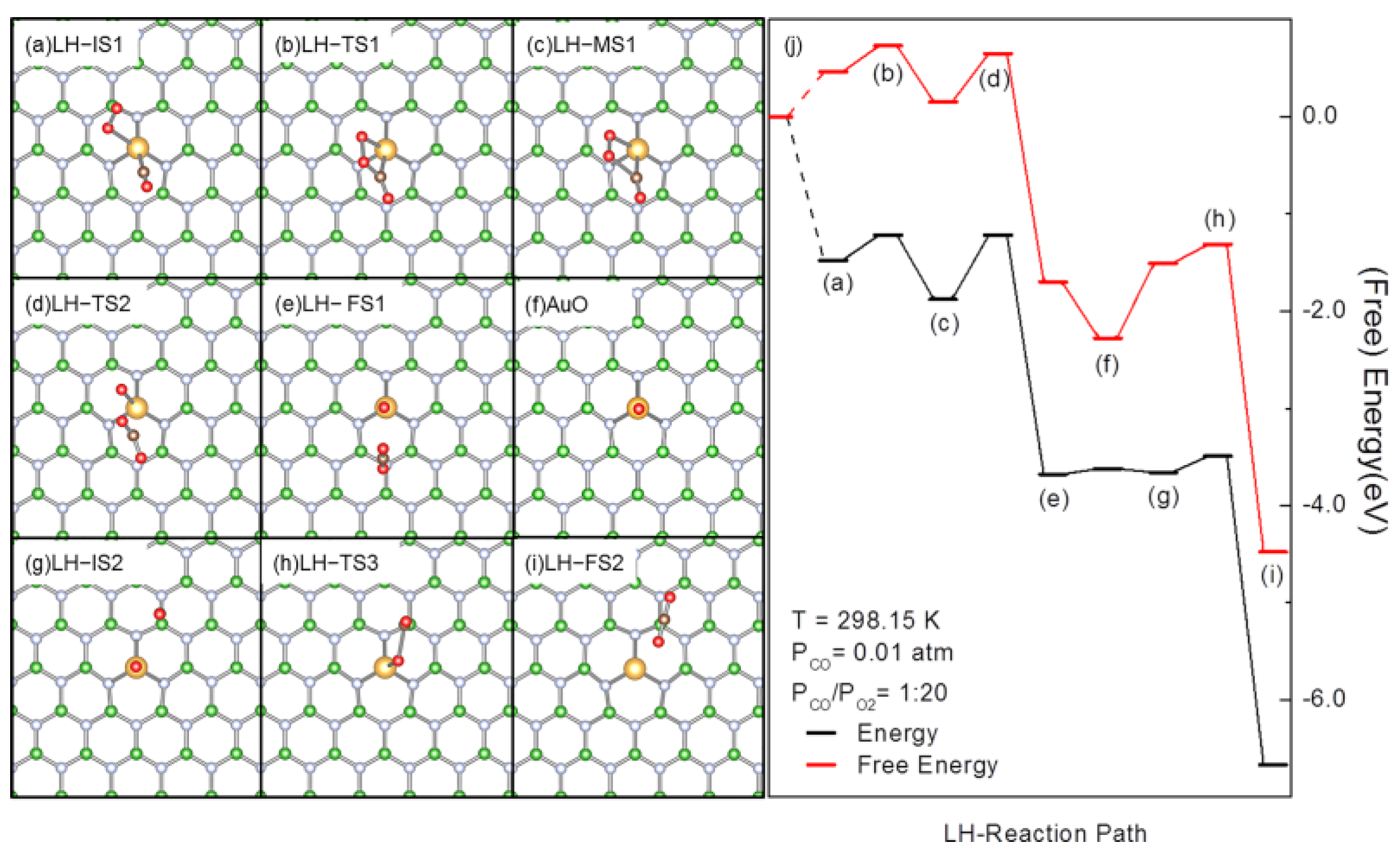
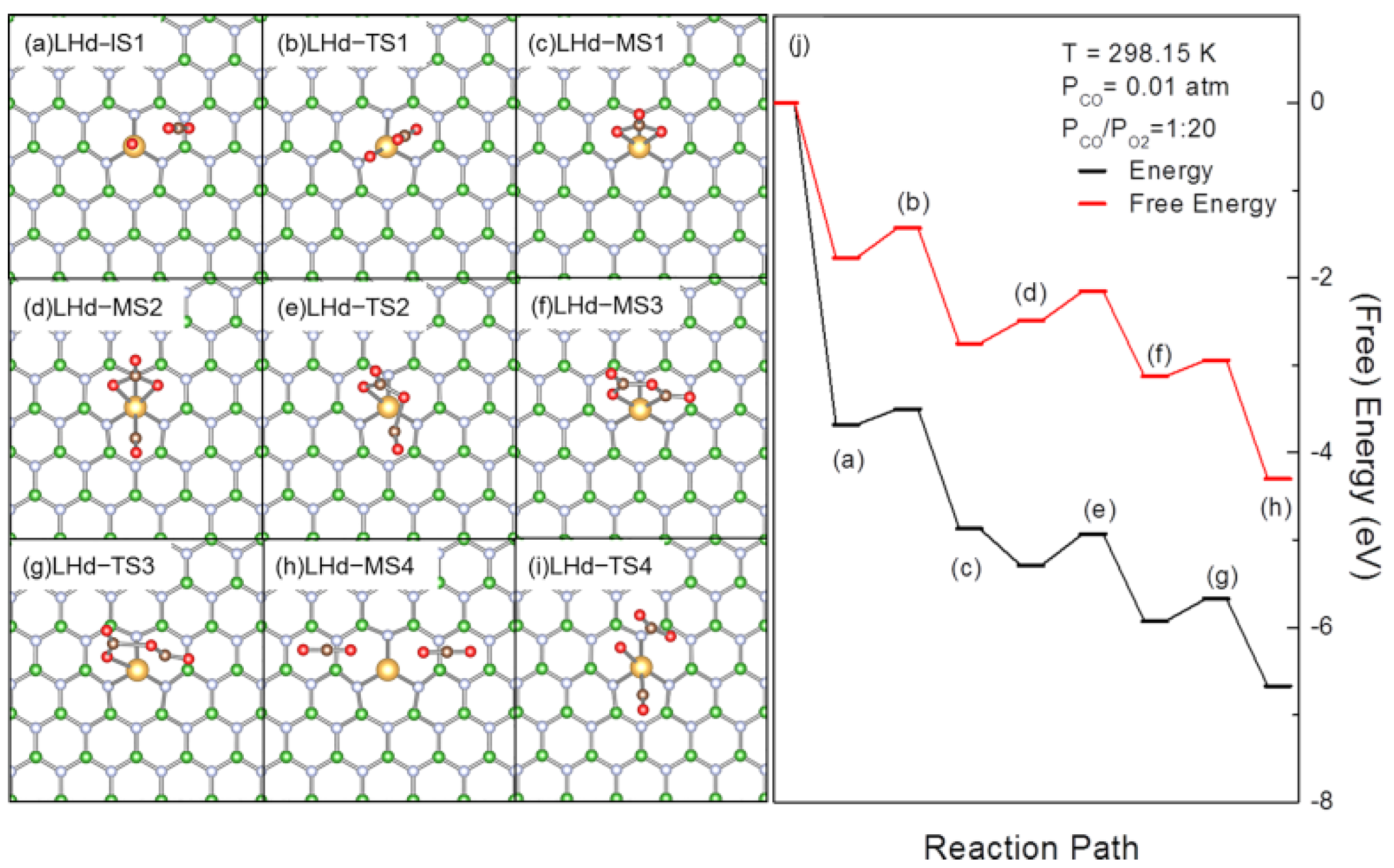
3.2. Revised LH Pathway for CO Oxidation over AuBN
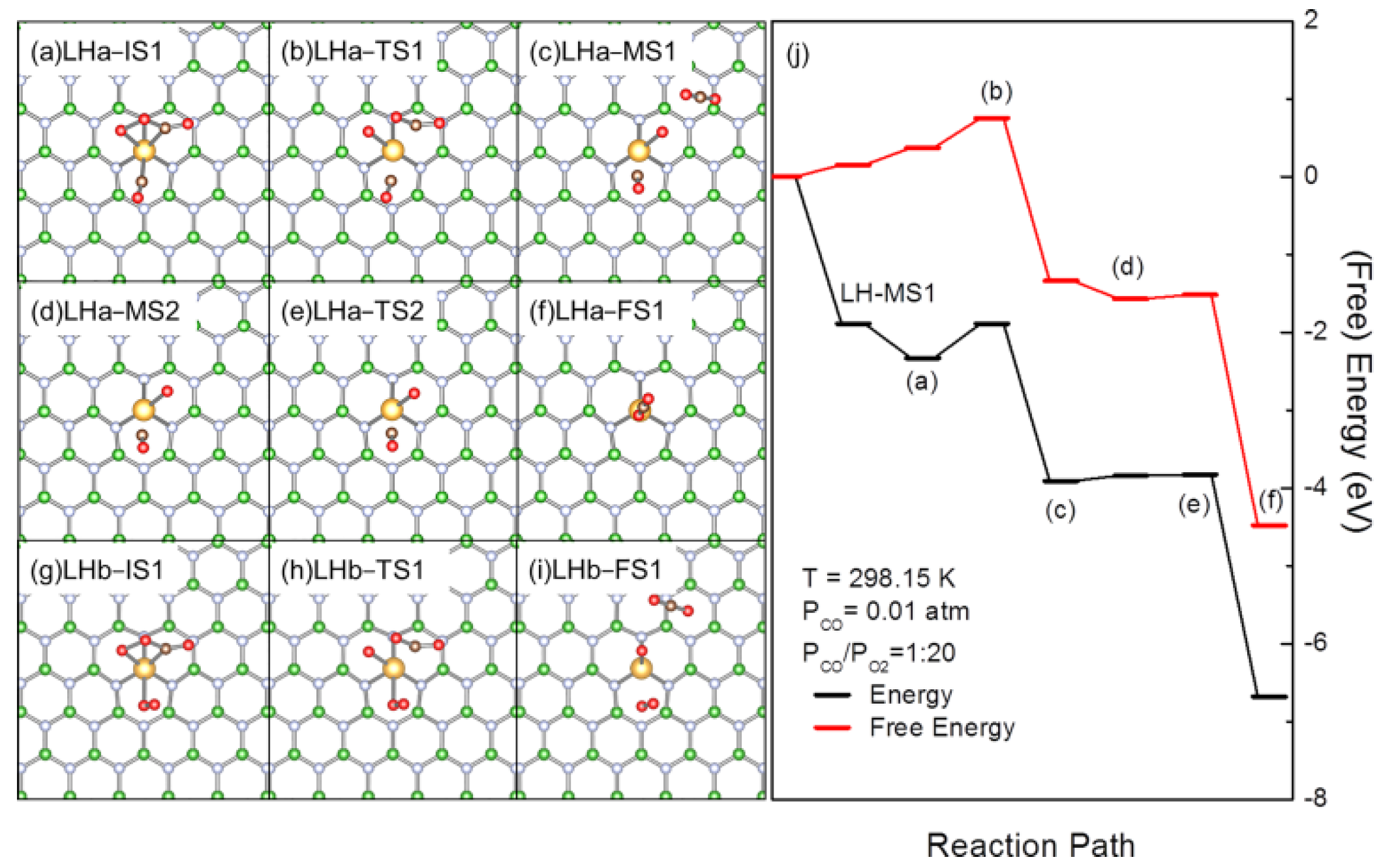
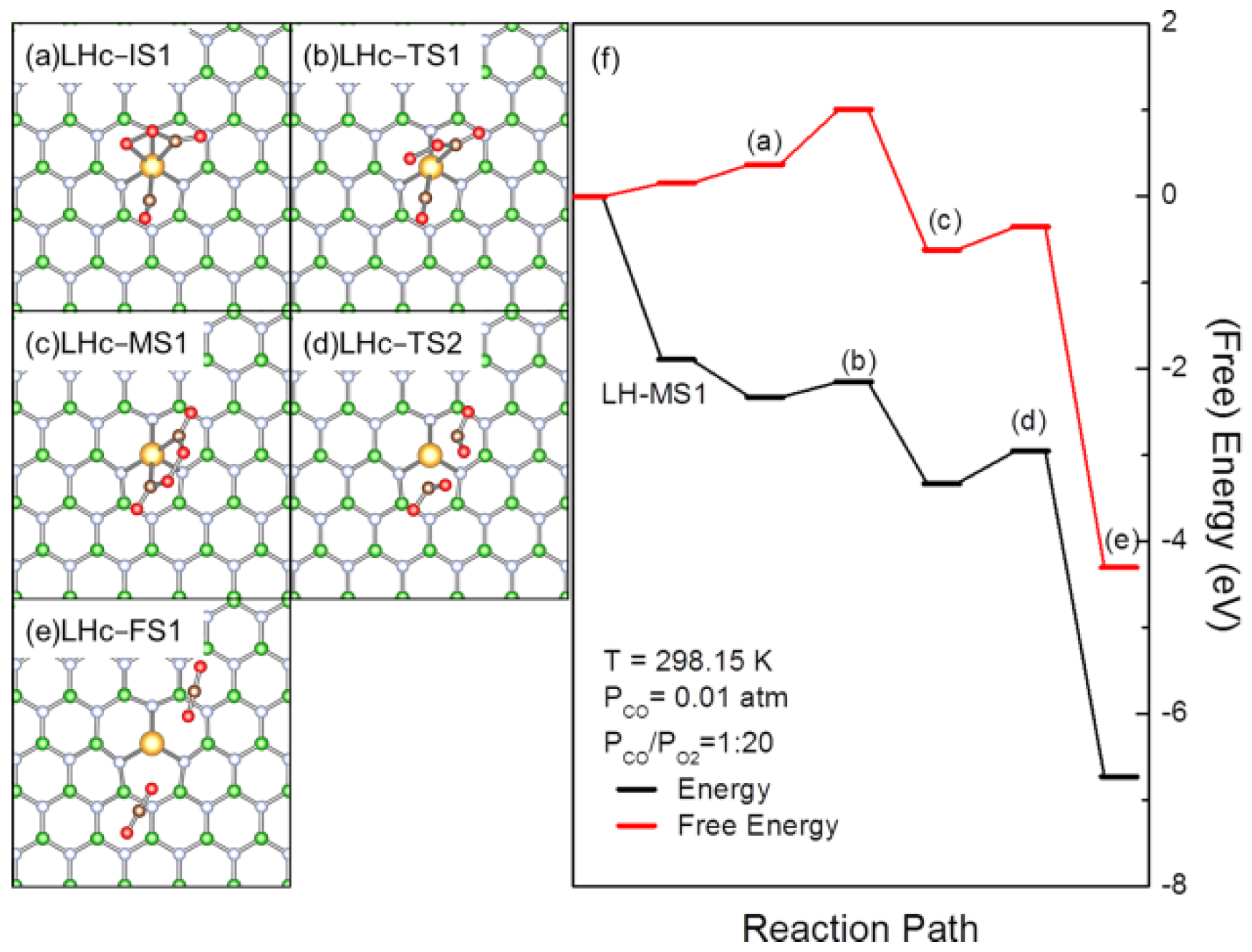
3.3. Interference of the Coadsorption of CO or O2 to CO Oxidation
3.4. Comparison with Other Alternative Pathways
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Liu, J.Y. Catalysis by Supported Single Metal Atoms. ACS Catal. 2017, 7, 34–59. [Google Scholar] [CrossRef]
- Cui, X.J.; Li, W.; Ryabchuk, P.; Junge, K.; Beller, M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nat. Catal. 2018, 1, 385–397. [Google Scholar] [CrossRef]
- Samantaray, M.K.; D’Eia, V.; Pump, E.; Falivene, L.; Harb, M.; Chikh, S.O.; Cavallo, L.; Basset, J.M. The Comparison between Single Atom Catalysis and Surface Organometallic Catalysis. Chem. Rev. 2020, 120, 734–813. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Shen, Q.; Yuan, K.; Yang, W.; Chen, Q.; Geng, Z.; Zhang, J.; Shao, X.; Chen, W.; Xu, G.; et al. Unraveling Charge State of Supported Au Single-Atoms during CO Oxidation. J. Am. Chem. Soc. 2018, 140, 554–557. [Google Scholar] [CrossRef]
- Kaatz, F.H.; Murzin, D.Y.; Bultheel, A. Coordination-Dependent Kinetics in the Catalysis of Gold Nanoclusters. ACS Catal. 2021, 11, 9073–9085. [Google Scholar] [CrossRef]
- Piccolo, L. Restructuring effects of the chemical environment in metal nanocatalysis and single-atom catalysis. Catal. Today 2020, 373, 80–97. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.K.; Baumer, M.; Moskaleva, L.V. Transient Au-CO Complexes Promote the Activity of an Inverse Ceria/Gold Catalyst: An Insight from Ab Initio Molecular Dynamics. J. Phys. Chem. C 2021, 125, 26406–26417. [Google Scholar] [CrossRef]
- Wang, Y.G.; Yoon, Y.; Glezakou, V.A.; Li, J.; Rousseau, R. The Role of Reducible Oxide-Metal Cluster Charge Transfer in Catalytic Processes: New Insights on the Catalytic Mechanism of CO Oxidation on Au/TiO2 from ab Initio Molecular Dynamics. J. Am. Chem. Soc. 2013, 135, 10673–10683. [Google Scholar] [CrossRef]
- Wang, Y.G.; Mei, D.H.; Glezakou, V.A.; Li, J.; Rousseau, R. Dynamic formation of single-atom catalytic active sites on ceria-supported gold nanoparticles. Nat. Commun. 2015, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.C.; Wang, Y.G.; Li, J. Toward Rational Design of Oxide-Supported Single-Atom Catalysts: Atomic Dispersion of Gold on Ceria. J. Am. Chem. Soc. 2017, 139, 6190–6199. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Liu, J.C.; Luo, L.L.; Wang, Y.G.; Zhu, J.F.; Du, Y.G.; Li, J.; Mao, S.X.; Wang, C.M. Size-dependent dynamic structures of supported gold nanoparticles in CO oxidation reaction condition. Proc. Natl. Acad. Sci. USA 2018, 115, 7700–7705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, K.; Gu, Y.T.; Westendorff, K.S.; Li, S.C.; Pihl, J.A.; Grabow, L.C.; Epling, W.S.; Paolucci, C. Condition-Dependent Pd Speciation and NO Adsorption in Pd/Zeolites. ACS Catal. 2020, 10, 12801–12818. [Google Scholar] [CrossRef]
- Ouyang, R.H.; Liu, J.X.; Li, W.X. Atomistic Theory of Ostwald Ripening and Disintegration of Supported Metal Particles under Reaction Conditions. J. Am. Chem. Soc. 2013, 135, 1760–1771. [Google Scholar] [CrossRef]
- Stamatakis, M.; Christiansen, M.A.; Vlachos, D.G.; Mpourmpakis, G. Multiscale Modeling Reveals Poisoning Mechanisms of MgO-Supported Au Clusters in CO Oxidation. Nano Lett. 2012, 12, 3621–3626. [Google Scholar] [CrossRef]
- Negreiros, F.R.; Camellone, M.F.; Fabris, S. Effects of Thermal Fluctuations on the Hydroxylation and Reduction of Ceria Surfaces by Molecular H-2. J. Phys. Chem. C 2015, 119, 21567–21573. [Google Scholar] [CrossRef]
- Ghosh, P.; Camellone, M.F.; Fabris, S. Fluxionality of Au Clusters at Ceria Surfaces during CO Oxidation: Relationships among Reactivity, Size, Cohesion, and Surface Defects from DFT Simulations. J. Phys. Chem. Lett. 2013, 4, 2256–2263. [Google Scholar] [CrossRef]
- Liu, J.C.; Tang, Y.; Chang, C.R.; Wang, Y.G.; Li, J. Mechanistic Insights into Propene Epoxidation with O2-H2O Mixture on Au-7/α-Al2O3: A Hydroproxyl Pathway from ab Initio Molecular Dynamics Simulations. ACS Catal. 2016, 6, 2525–2535. [Google Scholar] [CrossRef]
- Wang, Y.G.; Cantu, D.C.; Lee, M.S.; Li, J.; Glezakou, V.A.; Rousseau, R. CO Oxidation on Au/TiO2: Condition-Dependent Active Sites and Mechanistic Pathways. J. Am. Chem. Soc. 2016, 138, 10467–10476. [Google Scholar] [CrossRef]
- Ding, K.; Gulec, A.; Johnson, A.M.; Schweitzer, N.M.; Stucky, G.D.; Marks, L.D.; Stair, P.C. Identification of active sites in CO oxidation and water-gas shift over supported Pt catalysts. Science 2015, 350, 189–192. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Wang, A.Q.; Zhang, T. Recent Advances in Preferential Oxidation of CO Reaction over Platinum Group Metal Catalysts. ACS Catal. 2012, 2, 1165–1178. [Google Scholar] [CrossRef]
- Beniya, A.; Higashi, S. Towards dense single-atom catalysts for future automotive applications. Nat. Catal. 2019, 2, 590–602. [Google Scholar] [CrossRef]
- Freund, H.J.; Meijer, G.; Scheffler, M.; Schlogl, R.; Wolf, M. CO Oxidation as a Prototypical Reaction for Heterogeneous Processes. Angew. Chem.-Int. Edit. 2011, 50, 10064–10094. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-Atom catalysis of CO oxidation using Pt-1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel Gold Catalysts for the Oxidation of Carbon-Monoxide at a Temperature Far Below 0-Degrees-C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Hutchings, G. A golden future. Nat. Chem. 2009, 1, 584. [Google Scholar] [CrossRef]
- Flytzani-Stephanopoulos, M. Gold Atoms Stabilized on Various Supports Catalyze the Water-Gas Shift Reaction. Acc. Chem. Res. 2014, 47, 783–792. [Google Scholar] [CrossRef]
- Yao, K.X.; Liu, X.; Zhao, L.; Zeng, H.C.; Han, Y. Site-specific growth of Au particles on ZnO nanopyramids under ultraviolet illumination. Nanoscale 2011, 3, 4195–4200. [Google Scholar] [CrossRef]
- Wang, C.Y.; Yang, M.; Flytzani-Stephanopoulos, M. Single gold atoms stabilized on nanoscale metal oxide supports are catalytic active centers for various reactions. AlChe J. 2016, 62, 429–439. [Google Scholar] [CrossRef]
- Gu, X.K.; Qiao, B.T.; Huang, C.Q.; Ding, W.C.; Sun, K.J.; Zhan, E.S.; Zhang, T.; Liu, J.Y.; Li, W.X. Supported Single Pt-1/Au-1 Atoms for Methanol Steam Reforming. ACS Catal. 2014, 4, 3886–3890. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Y.; Chu, M.; Duan, T.; Meng, C.; Han, Y. Defect Stabilized Gold Atoms on Graphene as Potential Catalysts for Ethylene Epoxidation: A First-principles Investigation. Catal. Sci. Technol. 2016, 6, 1632–1641. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Chen, Y.J.; Chao, S.L.; Dong, X.B.; Chen, W.X.; Luo, J.; Liu, C.G.; Wang, D.S.; Chen, C.; Li, W.; et al. Single-Atom Au-I-N-3 Site for Acetylene Hydrochlorination Reaction. ACS Catal. 2020, 10, 1865–1870. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Xu, B.Q. Catalysis by gold: Isolated surface Au3+ ions are active sites for selective hydrogenation of 1,3-butadiene over Au/ZrO2 catalysts. Angew. Chem.-Int. Edit. 2005, 44, 7132–7135. [Google Scholar] [CrossRef]
- Wang, C.Y.; Garbarino, G.; Allard, L.F.; Wilson, F.; Busca, G.; Flytzani-Stephanopoulos, M. Low-Temperature Dehydrogenation of Ethanol on Atomically Dispersed Gold Supported on ZnZrOx. ACS Catal. 2016, 6, 210–218. [Google Scholar] [CrossRef]
- Mochizuki, C.; Inomata, Y.; Yasumura, S.; Lin, M.; Taketoshi, A.; Honma, T.; Sakaguchi, N.; Haruta, M.; Shimizu, K.-I.; Ishida, T.; et al. Defective NiO as a Stabilizer for Au Single-Atom Catalysts. ACS Catal. 2022, 12, 6149–6158. [Google Scholar] [CrossRef]
- Kropp, T.; Lu, Z.; Li, Z.; Chin, Y.-H.C.; Mavrikakis, M. Anionic Single-Atom Catalysts for CO Oxidation: Support-Independent Activity at Low Temperatures. ACS Catal. 2019, 9, 1595–1604. [Google Scholar] [CrossRef]
- Gao, M.; Lyalin, A.; Taketsugu, T. CO oxidation on h-BN supported Au atom. J. Chem. Phys. 2013, 138, 034701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, B.T.; Liu, J.X.; Wang, Y.G.; Lin, Q.Q.; Liu, X.Y.; Wang, A.Q.; Li, J.; Zhang, T.; Liu, J.Y. Highly Efficient Catalysis of Preferential Oxidation of CO in H-2-Rich Stream by Gold Single-Atom Catalysts. ACS Catal. 2015, 5, 6249–6254. [Google Scholar] [CrossRef]
- Lu, Y.H.; Zhou, M.; Zhang, C.; Feng, Y.P. Metal-Embedded Graphene: A Possible Catalyst with High Activity. J. Phys. Chem. C 2009, 113, 20156–20160. [Google Scholar] [CrossRef]
- Weng, Q.H.; Wang, X.B.; Wang, X.; Bando, Y.; Golberg, D. Functionalized hexagonal boron nitride nanomaterials: Emerging properties and applications. Chem. Soc. Rev. 2016, 45, 3989–4012. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.S.; Wu, Z.L.; Foo, G.S.; Gao, X.; Zhou, M.X.; Liu, B.; Veith, G.M.; Wu, P.W.; Browning, K.L.; Lee, H.N.; et al. Taming interfacial electronic properties of platinum nanoparticles on vacancy-abundant boron nitride nanosheets for enhanced catalysis. Nat. Commun. 2017, 8, 15291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.Y.; Cox, M.; Li, B. Surface modification of hexagonal boron nitride nanomaterials: A review. J. Mater. Sci. 2018, 53, 66–99. [Google Scholar] [CrossRef]
- Fang, C.; An, W. Single-Metal-Atom site with high-spin state embedded in defective BN nanosheet promotes electrocatalytic nitrogen reduction. Nano Res. 2021, 14, 4211–4219. [Google Scholar] [CrossRef]
- Fan, M.M.; Jimenez, J.D.; Shirodkar, S.N.; Wu, J.J.; Chen, S.M.; Song, L.; Royko, M.M.; Zhang, J.J.; Guo, H.; Cui, J.W.; et al. Atomic Ru Immobilized on Porous h-BN through Simple Vacuum Filtration for Highly Active and Selective CO2 Methanation. ACS Catal. 2019, 9, 10077–10086. [Google Scholar] [CrossRef]
- Zhang, W.; Fu, Q.; Luo, Q.; Sheng, L.; Yang, J. Understanding Single-Atom Catalysis in View of Theory. JACS Au 2021, 1, 2130–2145. [Google Scholar] [CrossRef]
- Dong, J.H.; Gao, L.J.; Fu, Q. Hexagonal Boron Nitride Meeting Metal: A New Opportunity and Territory in Heterogeneous Catalysis. J. Phys. Chem. Lett. 2021, 12, 9608–9619. [Google Scholar] [CrossRef]
- Zhao, J.X.; Chen, Z.F. Single Mo Atom Supported on Defective Boron Nitride Monolayer as an Efficient Electrocatalyst for Nitrogen Fixation: A Computational Study. J. Am. Chem. Soc. 2017, 139, 12480–12487. [Google Scholar] [CrossRef]
- Gao, S.S.; Ma, Z.J.; Xiao, C.W.; Cui, Z.T.; Du, W.; Sun, X.Q.; Li, Q.H.; Sa, R.J.; Sun, C.H. TM3 (TM = V, Fe, Mo, W) single-cluster catalyst confined on porous BN for electrocatalytic nitrogen reduction. J. Mater. Sci. Technol. 2022, 108, 46–53. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Zeng, Z.Y.; Li, H. Design of 3d transition metal anchored B5N3 catalysts for electrochemical CO2 reduction to methane. J. Mater. Chem. A 2022, 10, 9737–9745. [Google Scholar] [CrossRef]
- Hu, G.X.; Wu, Z.L.; Dai, S.; Jiang, D.E. Interface Engineering of Earth-Abundant Transition Metals Using Boron Nitride for Selective Electroreduction of CO2. ACS Appl. Mater. Interfaces 2018, 10, 6694–6700. [Google Scholar] [CrossRef]
- Li, Z.; Wei, W.; Li, H.; Li, S.; Leng, L.; Zhang, M.; Horton, J.H.; Wang, D.; Sun, W.; Guo, C.; et al. Low-Temperature Synthesis of Single Palladium Atoms Supported on Defective Hexagonal Boron Nitride Nanosheet for Chemoselective Hydrogenation of Cinnamaldehyde. ACS Nano 2021, 15, 10175–10184. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Dai, S.; Wu, Z.; Jiang, D.-e. Single Atoms Anchored in Hexagonal Boron Nitride for Propane Dehydrogenation from First Principles. ChemCatChem 2022, 14, e202200133. [Google Scholar] [CrossRef]
- Mao, K.; Li, L.; Zhang, W.; Pei, Y.; Zeng, X.C.; Wu, X.; Yang, J. A Theoretical Study of Single-Atom Catalysis of CO Oxidation Using Au Embedded 2D h-BN Monolayer: A CO-Promoted O-2 Activation. Sci. Rep. 2014, 4, 5441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.H.; Du, J.B.; Chen, Y.F. Single Cu atom supported on modified h-BN monolayer as n-p codoped catalyst for CO oxidation: A computational study. Catal. Today 2021, 368, 148–160. [Google Scholar] [CrossRef]
- Liu, X.; Xu, M.; Wan, L.; Zhu, H.; Yao, K.; Linguerri, R.; Chambaud, G.; Han, Y.; Meng, C. Superior Catalytic Performance of Atomically Dispersed Palladium on Graphene in CO Oxidation. ACS Catal. 2020, 10, 3084–3093. [Google Scholar] [CrossRef]
- Zhu, J.; Feng, X.; Liu, X.; Zhang, X.; Wu, Y.; Zhu, H.; Yang, Y.; Duan, T.; Sui, Y.; Han, Y.; et al. The formation and evolution of carbonate species in CO oxidation over mono-dispersed Fe on graphene. Phys. Chem. Chem. Phys. 2021, 23, 10509–10517. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Inada, Y.; Orita, H. Efficiency of numerical basis sets for predicting the binding energies of hydrogen bonded complexes: Evidence of small basis set superposition error compared to Gaussian basis sets. J. Comput. Chem. 2008, 29, 225–232. [Google Scholar] [CrossRef]
- Delley, B. An All-Electron Numerical-Method for Solving the Local Density Functional for Polyatomic-Molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol(3) approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comput. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge-Densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Gajdos, M.; Eichler, A.; Hafner, J. CO adsorption on close-packed transition and noble metal surfaces: Trends from ab initio calculations. J. Phys.-Condes. Matter 2004, 16, 1141–1164. [Google Scholar] [CrossRef] [Green Version]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.L.; Gu, X.K.; Yan, H.; Lin, Y.; Li, J.J.; Liu, D.D.; Li, W.X.; Lu, J.L. Water-Mediated Mars-Van Krevelen Mechanism for CO Oxidation on Ceria-Supported Single-Atom Pt-1 Catalyst. ACS Catal. 2017, 7, 887–891. [Google Scholar] [CrossRef]
- Bertoldi, D.S.; Ramos, S.B.; Guillermet, A.F. Interrelations between EOS parameters and cohesive energy of transition metals: Thermostatistical approach, ab initio calculations and analysis of “universality” features. J. Phys. Chem. Solids 2017, 107, 93–99. [Google Scholar] [CrossRef]
- Chan, B.; Yim, W.L. Accurate Computation of Cohesive Energies for Small to Medium-Sized Gold Clusters. J. Chem. Theory Comput. 2013, 9, 1964–1970. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Liu, J.X.; Qian, K.; Jia, A.P.; Li, D.; Shi, L.; Hu, J.; Zhu, J.F.; Huang, W.X. Structure Sensitivity of Au-TiO2 Strong Metal-Support Interactions. Angew. Chem.-Int. Edit. 2021, 60, 12074–12081. [Google Scholar] [CrossRef]
- Kovalskii, A.M.; Volkov, I.N.; Evdokimenko, N.D.; Tkachenko, O.P.; Leybo, D.V.; Chepkasov, I.V.; Popov, Z.I.; Matveev, A.T.; Manakhov, A.; Permyakova, E.S.; et al. Hexagonal BN- and BNO-supported Au and Pt nanocatalysts in carbon monoxide oxidation and carbon dioxide hydrogenation reactions. Appl. Catal. B-Environ. 2022, 303, 16. [Google Scholar] [CrossRef]
- Narula, C.K.; Stocks, G.M. Ab Initio Density Functional Calculations of Adsorption of Transition Metal Atoms on θ-Al2O3(010) Surface. J. Phys. Chem. C 2012, 116, 5628–5636. [Google Scholar] [CrossRef]
- Moses-DeBusk, M.; Yoon, M.; Allard, L.F.; Mullins, D.R.; Wu, Z.; Yang, X.; Veith, G.; Stocks, G.M.; Narula, C.K. CO Oxidation on Supported Single Pt Atoms: Experimental and ab Initio Density Functional Studies of CO Interaction with Pt Atom on θ-Al2O3(010) Surface. J. Am. Chem. Soc. 2013, 135, 12634–12645. [Google Scholar] [CrossRef] [PubMed]
- Narula, C.K.; Allard, L.F.; Wu, Z.L. Ab Initio Density Functional Calculations and Infra-Red Study of CO Interaction with Pd Atoms on theta-Al2O3 (010) Surface. Sci. Rep. 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, M.A.; Ferri, D.; Smolentsev, G.; Marchionni, V.; Nachtegaal, M. Kinetic Studies of the Pt Carbonate-Mediated, Room-Temperature Oxidation of Carbon Monoxide by Oxygen over Pt/Al2O3 Using Combined, Time-Resolved XAFS, DRIFTS, and Mass Spectrometry. J. Am. Chem. Soc. 2016, 138, 13930–13940. [Google Scholar] [CrossRef] [PubMed]
- Ammal, S.C.; Heyden, A. Water-Gas Shift Activity of Atomically Dispersed Cationic Platinum versus Metallic Platinum Clusters on Titania Supports. ACS Catal. 2017, 7, 301–309. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Zhu, Y.H.; Asakura, H.; Zhang, B.; Zhang, J.G.; Zhou, M.X.; Han, Y.; Tanaka, T.; Wang, A.Q.; Zhang, T.; et al. Thermally stable single atom Pt/m-Al2O3 for selective hydrogenation and CO oxidation. Nat. Commun. 2017, 8, 16100. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.B.; Wang, J.M.; Yu, L.; Kovarik, L.; Zhang, X.W.; Hoffman, A.S.; Gallo, A.; Bare, S.R.; Sokaras, D.; Kroll, T.; et al. Identification of the active complex for CO oxidation over single-atom Ir-on-MgAl2O4 catalysts. Nat. Catal. 2019, 2, 149–156. [Google Scholar] [CrossRef]
- Duan, Z.Y.; Henkelman, G. CO Oxidation on the Pd(111) Surface. ACS Catal. 2014, 4, 3435–3443. [Google Scholar] [CrossRef]
- Piccinin, S.; Stamatakis, M. CO Oxidation on Pd(111): A First-Principles-Based Kinetic Monte Carlo Study. ACS Catal. 2014, 4, 2143–2152. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, H.; Linguerri, R.; Han, Y.; Chambaud, G.; Meng, C. Interfacial-Bonding-Regulated CO Oxidation over Pt Atoms Immobilized on Gas-Exfoliated Hexagonal Boron Nitride. ChemistrySelect 2017, 2, 9412–9419. [Google Scholar] [CrossRef]
- Allian, A.D.; Takanabe, K.; Fujdala, K.L.; Hao, X.; Truex, T.J.; Cai, J.; Buda, C.; Neurock, M.; Iglesia, E. Chemisorption of CO and Mechanism of CO Oxidation on Supported Platinum Nanoclusters. J. Am. Chem. Soc. 2011, 133, 4498–4517. [Google Scholar] [CrossRef] [PubMed]
- Kopecky, K.R.; Gomez, R.R. O-O Stretching Frequencies of Cyclic Peroxides—Stabilization of Peroxides by Alkoxy Substituents. Can. J. Chem.-Rev. Can. Chim. 1984, 62, 277–279. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, X.; Yao, K.X.; Zhao, L.; Han, Y. Superior Capture of CO2 Achieved by Introducing Extra-framework Cations into N-doped Microporous Carbon. Chem. Mat. 2012, 24, 4725–4734. [Google Scholar] [CrossRef]
- Newton, M.A.; Ferri, D.; Smolentsev, G.; Marchionni, V.; Nachtegaal, M. Room-Temperature carbon monoxide oxidation by oxygen over Pt/Al2O3 mediated by reactive platinum carbonates. Nat. Commun. 2015, 6, 9675. [Google Scholar] [CrossRef]
- Liu, B.; Li, W.P.; Song, W.Y.; Liu, J. Carbonate-Mediated Mars-van Krevelen mechanism for CO oxidation on cobalt-doped ceria catalysts: Facet-Dependence and coordination-dependence. Phys. Chem. Chem. Phys. 2018, 20, 16045–16059. [Google Scholar] [CrossRef]
- Lin, S.; Ye, X.; Johnson, R.S.; Guo, H. First-Principles Investigations of Metal (Cu, Ag, Au, Pt, Rh, Pd, Fe, Co, and Ir) Doped Hexagonal Boron Nitride Nanosheets: Stability and Catalysis of CO Oxidation. J. Phys. Chem. C 2013, 117, 17319–17326. [Google Scholar] [CrossRef]
- Li, Y.F.; Zhou, Z.; Yu, G.T.; Chen, W.; Chen, Z.F. CO Catalytic Oxidation on Iron-Embedded Graphene: Computational Quest for Low-Cost Nanocatalysts. J. Phys. Chem. C 2010, 114, 6250–6254. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Zhang, X.; Meng, C. Coadsorption Interfered CO Oxidation over Atomically Dispersed Au on h-BN. Molecules 2022, 27, 3627. https://doi.org/10.3390/molecules27113627
Liu X, Zhang X, Meng C. Coadsorption Interfered CO Oxidation over Atomically Dispersed Au on h-BN. Molecules. 2022; 27(11):3627. https://doi.org/10.3390/molecules27113627
Chicago/Turabian StyleLiu, Xin, Xin Zhang, and Changgong Meng. 2022. "Coadsorption Interfered CO Oxidation over Atomically Dispersed Au on h-BN" Molecules 27, no. 11: 3627. https://doi.org/10.3390/molecules27113627
APA StyleLiu, X., Zhang, X., & Meng, C. (2022). Coadsorption Interfered CO Oxidation over Atomically Dispersed Au on h-BN. Molecules, 27(11), 3627. https://doi.org/10.3390/molecules27113627