
Shotgun Proteomics Revealed Preferential Degradation of Misfolded In Vivo Obligate GroE Substrates by Lon Protease in Escherichia coli


Abstract
:1. Introduction
2. Results
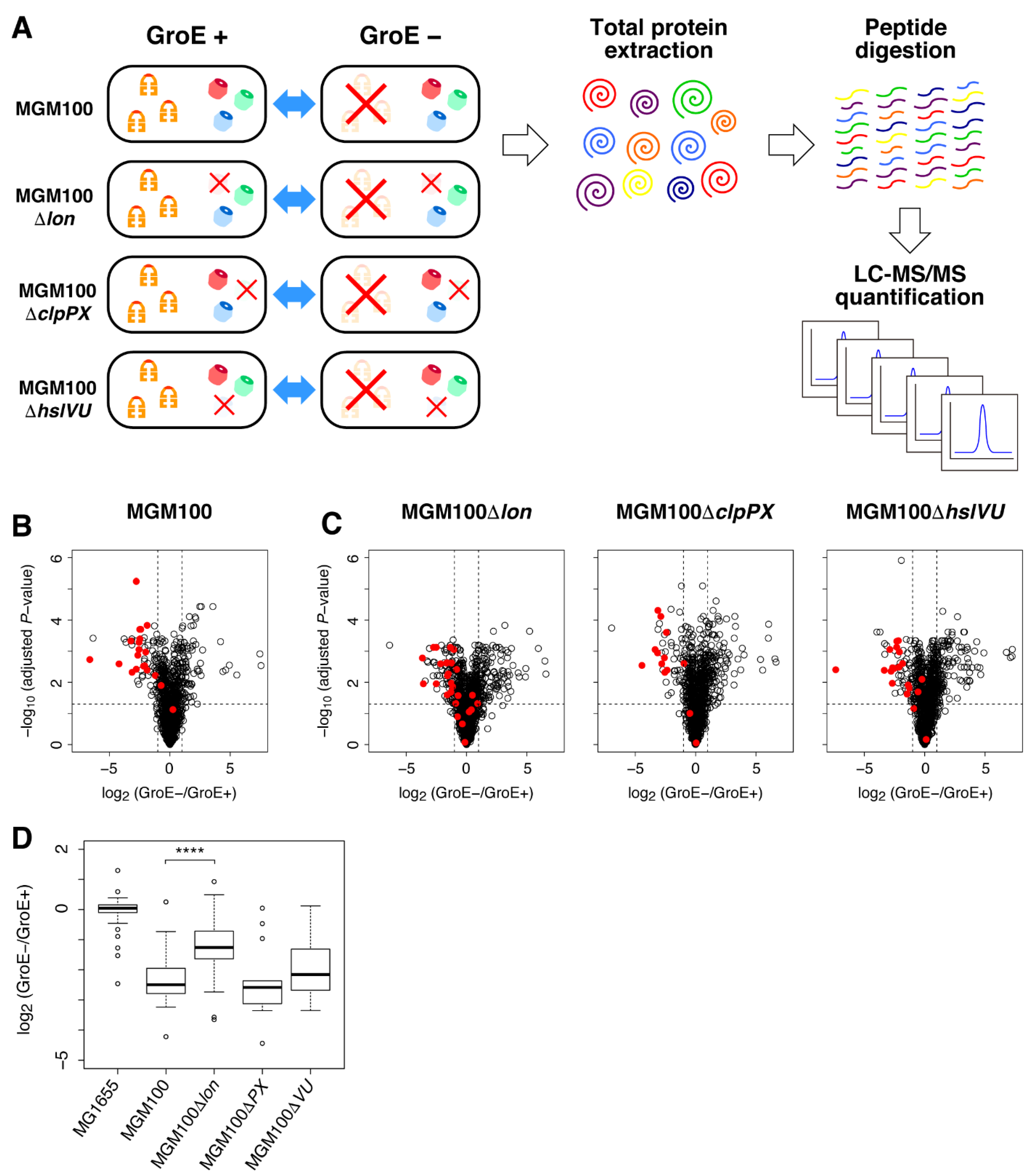
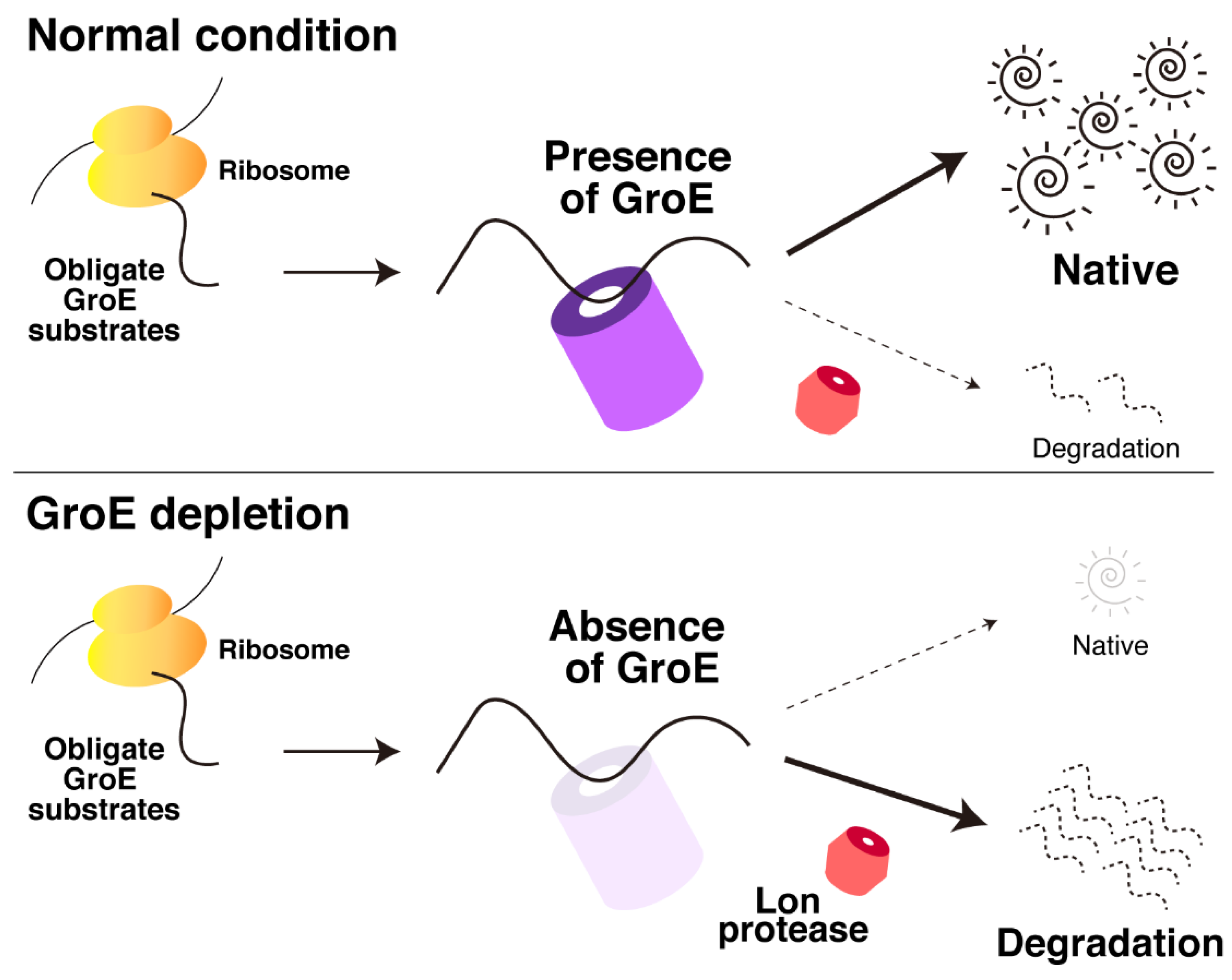
2.1. Obligate GroE Substrates Tend to Be Degraded by Lon under GroE-Depleted Conditions
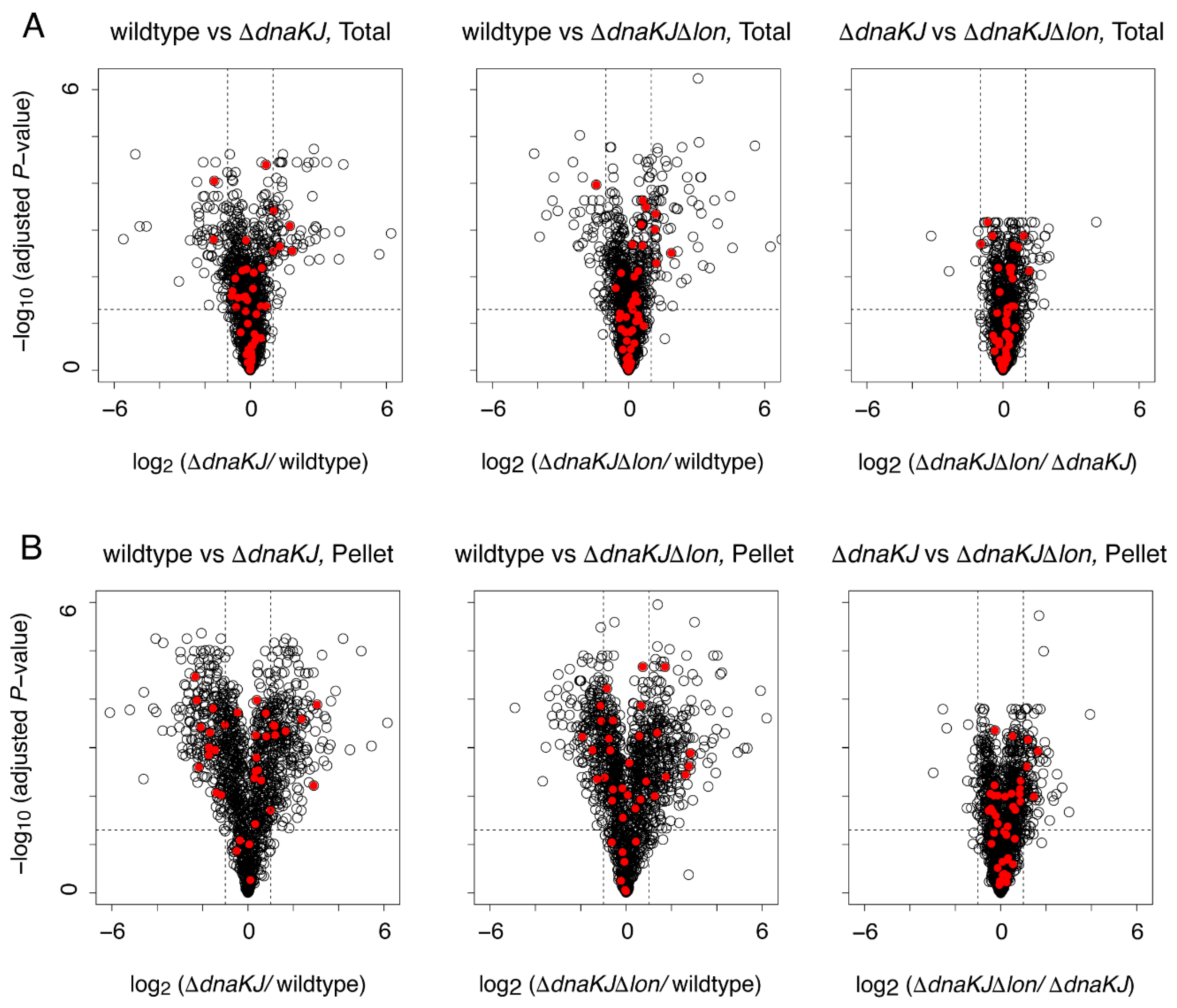
2.2. Deletion of DnaKJ Barely Affects the Folding of Most In Vivo Obligate GroE Substrates
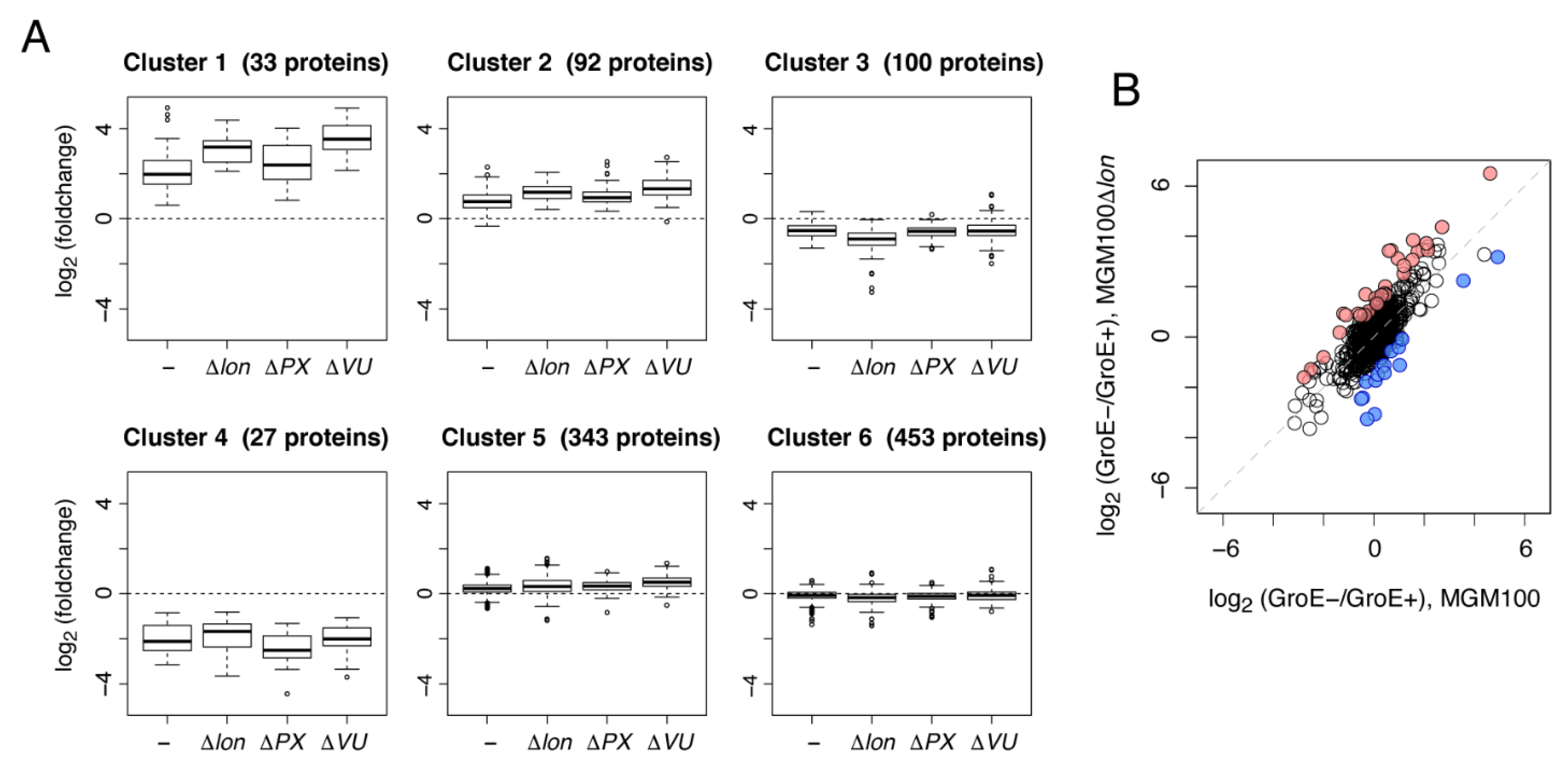
2.3. Metabolic Perturbations by Protease Deletions under GroE-Depleted Conditions Revealed by Clustering Analysis
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Cell Culture and Sample Preparation for the LC-MS/MS Analysis
4.3. LC-MS/MS Measurement and Data Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
References
- Anfinsen, B.C. Principles That Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular Strategies for Controlling Protein Aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In Vivo Aspects of Protein Folding and Quality Control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Fayet, O.; Ziegelhofer, T.; Georgopoulos, C. The GroES and GroEL Heat Shock Gene Products of Escherichia coli Are Essential for Bacterial Growth at All Temperatures. J. Bacteriol. 1989, 171, 1379–1385. [Google Scholar] [CrossRef]
- Kerner, M.J.; Naylor, D.J.; Ishihama, Y.; Maier, T.; Chang, H.C.; Stines, A.P.; Georgopoulos, C.; Frishman, D.; Hayer-Hartl, M.; Mann, M.; et al. Proteome-Wide Analysis of Chaperonin-Dependent Protein Folding in Escherichia coli. Cell 2005, 122, 209–220. [Google Scholar] [CrossRef]
- Fujiwara, K.; Ishihama, Y.; Nakahigashi, K.; Soga, T.; Taguchi, H. A Systematic Survey of In Vivo Obligate Chaperonin-Dependent Substrates. EMBO J. 2010, 29, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- McLennan, N.; Masters, M. GroE Is Vital for Cell-Wall Synthesis. Nature 1998, 392, 139. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Fujiwara, K.; Taguchi, H. Identification of Novel in Vivo Obligate GroEL/ES Substrates Based on Data from a Cell-Free Proteomics Approach. FEBS Lett. 2016, 590, 251–257. [Google Scholar] [CrossRef]
- Chandu, D.; Nandi, D. Comparative Genomics and Functional Roles of the ATP-Dependent Proteases Lon and Clp during Cytosolic Protein Degradation. Res. Microbiol. 2004, 155, 710–719. [Google Scholar] [CrossRef]
- Mahmoud, S.A.; Chien, P. Regulated Proteolysis in Bacteria. Annu. Rev. Biochem. 2018, 87, 677–696. [Google Scholar] [CrossRef] [PubMed]
- Wettstadt, S.; Llamas, M.A. Role of Regulated Proteolysis in the Communication of Bacteria with the Environment. Front. Mol. Biosci. 2020, 7, 294. [Google Scholar] [CrossRef]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Rozanova, S.; Barkovits, K.; Nikolov, M.; Schmidt, C.; Urlaub, H.; Marcus, K. Quantitative Mass Spectrometry-Based Proteomics: An Overview. Methods Mol. Biol. 2021, 2228, 85–116. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-Independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Ying, B.W.; Saito, K.; Jin, W.; Takada, S.; Ueda, T.; Taguchi, H. Bimodal Protein Solubility Distribution Revealed by an Aggregation Analysis of the Entire Ensemble of Escherichia coli Proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 4201–4206. [Google Scholar] [CrossRef]
- Niwa, T.; Kanamori, T.; Ueda, T.; Taguchi, H. Global Analysis of Chaperone Effects Using a Reconstituted Cell-Free Translation System. Proc. Natl. Acad. Sci. USA 2012, 109, 8937–8942. [Google Scholar] [CrossRef]
- Calloni, G.; Chen, T.; Schermann, S.M.; Chang, H.C.; Genevaux, P.; Agostini, F.; Tartaglia, G.G.; Hayer-Hartl, M.; Hartl, F.U. DnaK Functions as a Central Hub in the E. Coli Chaperone Network. Cell Rep. 2012, 1, 251–264. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Weber, H.; Polen, T.; Heuveling, J.; Wendisch, V.F.; Hengge, R. Genome-Wide Analysis of the General Stress Response Network in Escherichia coli: SigmaS-Dependent Genes, Promoters, and Sigma Factor Selectivity. J. Bacteriol. 2005, 187, 1591–1603. [Google Scholar] [CrossRef]
- Hengge-Aronis, R. Signal Transduction and Regulatory Mechanisms Involved in Control of the Sigma(S) (RpoS) Subunit of RNA Polymerase. Microbiol. Mol. Biol. Rev. 2002, 66, 373–395. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-Frame, Single-Gene Knockout Mutants: The Keio Collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-Step Inactivation of Chromosomal Genes in Escherichia coli K-12 Using PCR Products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Masuda, T.; Tomita, M.; Ishihama, Y. Phase Transfer Surfactant-Aided Trypsin Digestion for Membrane Proteome Analysis. J. Proteome Res. 2008, 7, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop And Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Midha, M.K.; Kusebauch, U.; Shteynberg, D.; Kapil, C.; Bader, S.L.; Reddy, P.J.; Campbell, D.S.; Baliga, N.S.; Moritz, R.L. A Comprehensive Spectral Assay Library to Quantify the Escherichia coli Proteome by DIA/SWATH-MS. Sci. Data 2020, 7, 389. [Google Scholar] [CrossRef]
- Mizusawa, S.; Gottesman, S. Protein Degradation in Escherichia coli: The Lon Gene Controls the Stability of SulA Protein. Proc. Natl. Acad. Sci. USA 1983, 80, 358–362. [Google Scholar] [CrossRef]
- Stout, V.; Torres-Cabassa, A.; Maurizi, M.R.; Gutnick, D.; Gottesman, S. RcsA, an Unstable Positive Regulator of Capsular Polysaccharide Synthesis. J. Bacteriol. 1991, 173, 1738–1747. [Google Scholar] [CrossRef]
- Leffers, J.; Gottesman, S. Lambda Xis Degradation In Vivo by Lon and FtsH. J. Bacteriol. 1998, 180, 1573–1577. [Google Scholar] [CrossRef]
- Biran, D.; Gur, E.; Gollan, L.; Ron, E.Z. Control of Methionine Biosynthesis in Escherichia coli by Proteolysis. Mol. Microbiol. 2000, 37, 1436–1443. [Google Scholar] [CrossRef]
- Griffith, K.L.; Shah, I.M.; Wolf, R.E. Proteolytic Degradation of Escherichia coli Transcription Activators SoxS and MarA as the Mechanism for Reversing the Induction of the Superoxide (SoxRS) and Multiple Antibiotic Resistance (Mar) Regulons. Mol. Microbiol. 2004, 51, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Neher, S.B.; Villen, J.; Oakes, E.C.; Bakalarski, C.E.; Sauer, R.T.; Gygi, S.P.; Baker, T.A. Proteomic Profiling of ClpXP Substrates after DNA Damage Reveals Extensive Instability within SOS Regulon. Mol. Cell 2006, 22, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, S.A.; Rivera-Rivera, I.; Sauer, R.T.; Baker, T.A. The IbpA and IbpB Small Heat-Shock Proteins Are Substrates of the AAA+ Lon Protease. Mol. Microbiol. 2010, 75, 1539–1549. [Google Scholar] [CrossRef]
- Langklotz, S.; Narberhaus, F. The Escherichia coli Replication Inhibitor CspD Is Subject to Growth-Regulated Degradation by the Lon Protease. Mol. Microbiol. 2011, 80, 1313–1325. [Google Scholar] [CrossRef] [PubMed]
- Westphal, K.; Langklotz, S.; Thomanek, N.; Narberhaus, F. A Trapping Approach Reveals Novel Substrates and Physiological Functions of the Essential Protease FtsH in Escherichia coli. J. Biol. Chem. 2012, 287, 42962–42971. [Google Scholar] [CrossRef]
- Arends, J.; Griego, M.; Thomanek, N.; Lindemann, C.; Kutscher, B.; Meyer, H.E.; Narberhaus, F. An Integrated Proteomic Approach Uncovers Novel Substrates and Functions of the Lon Protease in Escherichia coli. Proteomics 2018, 18, e1800080. [Google Scholar] [CrossRef]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. JPOSTrepo: An International Standard Data Repository for Proteomes. Nucleic Acids Res. 2017, 45, D1107–D1111. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Kerner 2005 | Fujiwara 2010 | Niwa 2016 | MGM100 | MGM100Δlon | MGM100ΔclpPX | MGM100ΔhslVU | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of Detection | FC | Number of Detection | FC | Number of Detection | FC | Number of Detection | FC | ||||||||
| Ara | Glc | Ara | Glc | Ara | Glc | Ara | Glc | ||||||||
| argP | 3 | 4 | 3 | 3 | 1.191 | 3 | 3 | 1.407 | 3 | 3 | 1.033 | 3 | 3 | 1.089 | |
| ltaE | 3 | 4 | 3 | 3 | 0.604 | 3 | 3 | 1.170 | 3 | 3 | 0.723 | 3 | 3 | 0.857 | |
| metK | 3 | 4 | 3 | 3 | 0.433 | 3 | 3 | 1.902 | 3 | 3 | 0.513 | 3 | 3 | 0.683 | |
| add | 3 | 4 | 3 | 3 | 0.271 | 3 | 3 | 0.316 | 3 | 3 | 0.194 | 3 | 3 | 0.280 | |
| dapA | 3 | 4 | 3 | 3 | 0.269 | 3 | 3 | 0.483 | 3 | 3 | 0.187 | 3 | 3 | 0.234 | |
| asd | 3 | 4 | 3 | 3 | 0.249 | 3 | 3 | 0.569 | 3 | 3 | 0.171 | 3 | 3 | 0.419 | |
| rfbC | 3 | 4 | 3 | 3 | 0.235 | 3 | 3 | 0.450 | 3 | 1 | 3 | 3 | 0.386 | ||
| serC | 2 | 4 | 3 | 3 | 0.221 | 3 | 3 | 0.428 | 3 | 3 | 0.167 | 3 | 3 | 0.363 | |
| hemB | 3 | 4 | 3 | 3 | 0.188 | 3 | 3 | 0.395 | 3 | 3 | 0.112 | 3 | 3 | 0.202 | |
| pmbA | 3 | 4 | 3 | 3 | 0.178 | 3 | 3 | 0.225 | 3 | 0 | 3 | 3 | 0.176 | ||
| lipA | 3 | 4 | 3 | 3 | 0.177 | 3 | 3 | 0.410 | 3 | 3 | 0.136 | 3 | 3 | 0.201 | |
| nuoC | 2 | 4 | 3 | 3 | 0.170 | 3 | 3 | 0.080 | 3 | 3 | 0.115 | 3 | 3 | 0.221 | |
| pepQ | 3 | 4 | 3 | 3 | 0.169 | 3 | 3 | 0.177 | 3 | 3 | 0.143 | 3 | 3 | 0.155 | |
| fabF | 3 | 4 | 3 | 3 | 0.158 | 3 | 3 | 0.336 | 3 | 2 | 3 | 3 | 0.228 | ||
| kdsA | 2 | 4 | 3 | 3 | 0.145 | 3 | 3 | 0.328 | 3 | 3 | 0.098 | 3 | 3 | 0.157 | |
| pyrD | 4 | 3 | 3 | 0.145 | 3 | 3 | 0.176 | 3 | 2 | 3 | 2 | ||||
| gatY | 3 | 4 | 3 | 3 | 0.114 | 3 | 3 | 0.150 | 3 | 3 | 0.046 | 3 | 3 | 0.098 | |
| deoA | 3 | 4 | 3 | 3 | 0.106 | 3 | 1 | 3 | 0 | 3 | 3 | 0.145 | |||
| sdhA | 3 | 4 | 3 | 3 | 0.054 | 3 | 3 | 0.084 | 3 | 0 | 3 | 3 | 0.135 | ||
| araA | 3 | 4 | 3 | 3 | 0.010 | 3 | 1 | 3 | 2 | 3 | 3 | 0.006 | |||
| fbaB | 3 | 4 | 3 | 0 | 3 | 3 | 1.323 | 3 | 1 | 3 | 0 | ||||
| ftsE | 3 | 4 | 3 | 0 | 3 | 3 | 0.620 | 3 | 0 | 3 | 2 | ||||
| nagZ | 3 | 4 | 3 | 0 | 3 | 3 | 0.539 | 3 | 0 | 3 | 0 | ||||
| ybhA | ° | 3 | 0 | 3 | 3 | 0.332 | 3 | 1 | 3 | 0 | |||||
| tas | 2 | ° | 3 | 0 | 3 | 3 | 0.788 | 3 | 0 | 3 | 0 | ||||
| nagD | ° | 3 | 2 | 3 | 3 | 0.598 | 3 | 0 | 3 | 2 | |||||
| alaA | 3 | 4 | 3 | 1 | 3 | 3 | 0.418 | 3 | 0 | 3 | 3 | 0.535 | |||
| dadA | 3 | 4 | 3 | 1 | 3 | 3 | 0.350 | 3 | 0 | 3 | 0 | ||||
| Gene Name | Kerner 2005 | Fujiwara 2010 | Niwa 2016 | Number of Detection | Fold Change | Number of Detection | Fold Change | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wt Total | ΔKJ Total | ΔKJ Δlon Total | ΔKJ/wt Total | ΔKJlon/wt Total | ΔKJlon/ΔKJ Total | wt ppt | ΔKJ ppt | ΔKJ Δlon ppt | ΔKJ/wt ppt | ΔKJlon/wt ppt | ΔKJlon/ΔKJ ppt | ||||
| dadX | 3 | 4 | 3 | 3 | 3 | 3.611 | 3.691 | 1.022 | 3 | 3 | 3 | 5.136 | 7.144 | 1.391 | |
| nanA | 3 | 4 | 3 | 3 | 3 | 3.351 | 1.711 | 0.511 | 3 | 3 | 3 | 1.047 | 0.904 | 0.864 | |
| sdhA | 3 | 4 | 3 | 3 | 3 | 2.466 | 2.230 | 0.905 | 3 | 3 | 3 | 1.725 | 1.578 | 0.915 | |
| lldD | 3 | 4 | 3 | 3 | 3 | 2.035 | 1.477 | 0.726 | 3 | 3 | 3 | 1.218 | 0.890 | 0.731 | |
| dadA | 3 | 4 | 3 | 3 | 3 | 2.022 | 2.320 | 1.147 | 3 | 3 | 3 | 1.754 | 1.847 | 1.053 | |
| gdhA | 4 | 3 | 3 | 3 | 1.632 | 2.291 | 1.404 | 3 | 3 | 3 | 2.147 | 3.361 | 1.566 | ||
| metK | 3 | 4 | 3 | 3 | 3 | 1.612 | 1.008 | 0.625 | 3 | 3 | 3 | 1.264 | 0.996 | 0.788 | |
| nuoC | 2 | 4 | 3 | 3 | 3 | 1.423 | 1.232 | 0.866 | 3 | 3 | 3 | 1.311 | 1.102 | 0.840 | |
| ybjS | 3 | 4 | 3 | 3 | 3 | 1.395 | 1.592 | 1.141 | 3 | 3 | 3 | 2.299 | 2.393 | 1.041 | |
| rfbC | 3 | 4 | 3 | 3 | 3 | 1.378 | 1.079 | 0.783 | 3 | 3 | 3 | 8.265 | 6.135 | 0.742 | |
| yqaB | 3 | 4 | 3 | 3 | 3 | 1.209 | 1.323 | 1.094 | 2 | 3 | 3 | 0.820 | |||
| ycfH | 3 | 4 | 3 | 3 | 3 | 1.206 | 0.935 | 0.775 | 3 | 0 | 2 | ||||
| lsrF | 3 | 4 | 3 | 3 | 3 | 1.154 | 1.381 | 1.196 | 3 | 3 | 3 | 0.713 | 0.855 | 1.200 | |
| ybhA | ° | 3 | 3 | 3 | 1.135 | 1.197 | 1.055 | 3 | 3 | 3 | 7.462 | 6.802 | 0.912 | ||
| gatY | 3 | 4 | 3 | 3 | 3 | 1.114 | 1.541 | 1.384 | 3 | 3 | 3 | 2.262 | 3.266 | 1.443 | |
| fabF | 3 | 4 | 3 | 3 | 3 | 1.088 | 0.910 | 0.836 | 3 | 3 | 3 | 0.496 | 0.592 | 1.194 | |
| nagD | ° | 3 | 3 | 3 | 1.077 | 1.073 | 0.996 | 3 | 3 | 3 | 1.293 | 0.892 | 0.690 | ||
| deoA | 3 | 4 | 3 | 3 | 3 | 1.047 | 0.931 | 0.888 | 3 | 3 | 3 | 0.443 | 0.644 | 1.454 | |
| nagZ | 3 | 4 | 3 | 3 | 3 | 1.045 | 0.954 | 0.914 | 3 | 0 | 2 | ||||
| add | 3 | 4 | 3 | 3 | 3 | 1.039 | 1.135 | 1.092 | 3 | 3 | 3 | 3.164 | 2.571 | 0.813 | |
| ftsE | 3 | 4 | 3 | 3 | 3 | 1.038 | 1.127 | 1.086 | 3 | 3 | 3 | 1.270 | 1.051 | 0.827 | |
| lipA | 3 | 4 | 3 | 3 | 3 | 1.032 | 1.343 | 1.302 | 3 | 3 | 3 | 1.360 | 1.327 | 0.976 | |
| kdsA | 2 | 4 | 3 | 3 | 3 | 1.004 | 1.006 | 1.002 | 3 | 3 | 3 | 0.303 | 0.555 | 1.829 | |
| alaA | 3 | 4 | 3 | 3 | 3 | 0.989 | 1.054 | 1.066 | 3 | 3 | 3 | 1.987 | 1.499 | 0.754 | |
| asd | 3 | 4 | 3 | 3 | 3 | 0.962 | 1.086 | 1.130 | 3 | 3 | 3 | 0.368 | 0.666 | 1.809 | |
| pmbA | 3 | 4 | 3 | 3 | 3 | 0.943 | 1.051 | 1.114 | 3 | 3 | 3 | 1.500 | 1.662 | 1.109 | |
| serC | 2 | 4 | 3 | 3 | 3 | 0.927 | 1.160 | 1.251 | 3 | 3 | 3 | 0.223 | 0.409 | 1.837 | |
| argP | 3 | 4 | 3 | 3 | 3 | 0.920 | 1.339 | 1.456 | 3 | 3 | 3 | 1.252 | 1.548 | 1.236 | |
| pepQ | 3 | 4 | 3 | 3 | 3 | 0.916 | 1.127 | 1.230 | 3 | 3 | 3 | 0.199 | 0.458 | 2.297 | |
| ltaE | 3 | 4 | 3 | 3 | 3 | 0.890 | 1.192 | 1.339 | 3 | 3 | 3 | 0.344 | 0.615 | 1.787 | |
| pyrD | 4 | 3 | 3 | 3 | 0.886 | 0.838 | 0.947 | 3 | 3 | 3 | 0.728 | 0.667 | 0.916 | ||
| dapA | 3 | 4 | 3 | 3 | 3 | 0.879 | 0.949 | 1.080 | 3 | 3 | 3 | 0.210 | 0.466 | 2.217 | |
| uxaC | 3 | 4 | 3 | 3 | 3 | 0.870 | 0.772 | 0.887 | 3 | 2 | 3 | 0.263 | |||
| tldD | 3 | 4 | 3 | 3 | 3 | 0.859 | 0.944 | 1.100 | 3 | 3 | 3 | 0.784 | 0.966 | 1.232 | |
| hemB | 3 | 4 | 3 | 3 | 3 | 0.812 | 1.531 | 1.887 | 3 | 3 | 3 | 0.303 | 0.949 | 3.135 | |
| pyrC | 2 | 4 | 3 | 3 | 3 | 0.785 | 0.797 | 1.015 | 3 | 2 | 3 | 0.520 | |||
| argE | 3 | 4 | 3 | 3 | 2 | 0.745 | 3 | 3 | 3 | 1.076 | 1.346 | 1.251 | |||
| nfo | 3 | 4 | 3 | 3 | 3 | 0.686 | 0.676 | 0.984 | 3 | 0 | 2 | ||||
| yajO | 3 | 4 | 3 | 3 | 3 | 0.648 | 0.802 | 1.239 | 3 | 0 | 0 | ||||
| tas | 2 | ° | 3 | 3 | 3 | 0.633 | 1.002 | 1.583 | 3 | 1 | 0 | ||||
| gpr | 2 | ° | 3 | 3 | 3 | 0.581 | 0.756 | 1.300 | 3 | 3 | 3 | 0.236 | 0.651 | 2.762 | |
| cysH | ° | 3 | 3 | 3 | 0.566 | 1.257 | 2.221 | 3 | 3 | 2 | 0.380 | ||||
| fbaB | 3 | 4 | 3 | 3 | 3 | 0.332 | 0.372 | 1.120 | 3 | 0 | 0 | ||||
| frdA | 3 | 4 | 3 | 3 | 3 | 0.326 | 0.373 | 1.147 | 3 | 3 | 3 | 0.315 | 0.359 | 1.141 | |
| yafD | 3 | 4 | 0 | 3 | 3 | 1.206 | 1 | 3 | 3 | 1.461 | |||||
| fadA | 4 | 2 | 3 | 3 | 0.740 | 1 | 3 | 3 | 1.068 | ||||||
| yigB | ° | 2 | 2 | 2 | 2 | 3 | 3 | 1.542 | |||||||
| yjhH | 4 | 0 | 3 | 3 | 1.219 | ||||||||||
| dusB | 3 | 4 | 1 | 3 | 3 | 1.119 | |||||||||
| eutB | 3 | 4 | 1 | 0 | 1 | 1 | 3 | 3 | 0.962 | ||||||
| Cluster | Annotation (KEGG BRITE Hierarchy3) | Odds Ratio | p-Value * | Number of Proteins in Population | Number of Proteins in Subgroup |
|---|---|---|---|---|---|
| Cluster 1 | Selenocompound metabolism | 24.95 | 0.0009 | 7 | 3 |
| Monobactam biosynthesis | 16.15 | 0.0133 | 6 | 2 | |
| Folate biosynthesis | 9.23 | 0.0302 | 9 | 2 | |
| Cysteine and methionine metabolism | 8.83 | 0.0008 | 25 | 5 | |
| Lysine biosynthesis | 8.08 | 0.0370 | 10 | 2 | |
| Glycine, serine and threonine metabolism | 5.93 | 0.0086 | 27 | 4 | |
| Cluster 2 | Histidine metabolism | 31.93 | 0.0025 | 4 | 3 |
| Nitrogen metabolism | 10.55 | 0.0407 | 4 | 2 | |
| Chaperones and folding catalysts | 5.41 | 0.0002 | 31 | 10 | |
| Starch and sucrose metabolism | 4.77 | 0.0215 | 13 | 4 | |
| Glutathione metabolism | 3.90 | 0.0357 | 15 | 4 | |
| Cluster 3 | C5-Branched dibasic acid metabolism | Inf | 0.0090 | 2 | 2 |
| Lysine degradation | Inf | 0.0090 | 2 | 2 | |
| Photosynthesis proteins | 16.48 | 0.0003 | 8 | 5 | |
| Oxidative phosphorylation | 11.54 | 5.5 × 10−7 | 21 | 11 | |
| Tetracycline biosynthesis | 9.61 | 0.0476 | 4 | 2 | |
| Citrate cycle (TCA cycle) | 6.64 | 0.0016 | 15 | 6 | |
| Purine metabolism | 5.33 | 1.2 × 10−5 | 42 | 14 | |
| Pyrimidine metabolism | 4.89 | 0.0003 | 31 | 10 | |
| One carbon pool by folate | 4.84 | 0.0463 | 9 | 3 | |
| Propanoate metabolism | 3.90 | 0.0369 | 14 | 4 | |
| Enzymes | 1.96 | 0.0015 | 561 | 68 | |
| Cluster 4 | Bacterial chemotaxis | Inf | 0.0006 | 2 | 2 |
| Galactose metabolism | 43.52 | 1.8 × 10−8 | 15 | 7 | |
| Monobactam biosynthesis | 20.12 | 0.0090 | 6 | 2 | |
| Lysine biosynthesis | 10.07 | 0.0253 | 10 | 2 | |
| Glycine, serine and threonine metabolism | 5.18 | 0.0295 | 27 | 3 | |
| Enzymes | 3.12 | 0.0076 | 561 | 21 |
| Group | Annotation (KEGG BRITE Hierarchy3) | Odds Ratio | p-Value * | Number of Proteins in Population | Number of Proteins in Subgroup |
|---|---|---|---|---|---|
| Up-regulated in MGM100Δlon | Bacterial motility proteins | Inf | 0.0344 | 1 | 1 |
| Monobactam biosynthesis | 14.69 | 0.0158 | 6 | 2 | |
| Selenocompound metabolism | 11.76 | 0.0216 | 7 | 2 | |
| Cysteine and methionine metabolism | 7.96 | 0.0012 | 25 | 5 | |
| Lysine biosynthesis | 7.35 | 0.0434 | 10 | 2 | |
| Glyoxylate and dicarboxylate metabolism | 6.02 | 0.0213 | 18 | 3 | |
| Glycine, serine and threonine metabolism | 5.36 | 0.0117 | 27 | 4 | |
| Down-regulated in MGM100Δlon | Glycosylphosphatidylinositol (GPI)-anchored proteins | Inf | 0.0162 | 1 | 1 |
| Aminobenzoate degradation | Inf | 0.0162 | 1 | 1 | |
| Glyoxylate and dicarboxylate metabolism | 14.38 | 0.0025 | 18 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niwa, T.; Chadani, Y.; Taguchi, H. Shotgun Proteomics Revealed Preferential Degradation of Misfolded In Vivo Obligate GroE Substrates by Lon Protease in Escherichia coli. Molecules 2022, 27, 3772. https://doi.org/10.3390/molecules27123772
Niwa T, Chadani Y, Taguchi H. Shotgun Proteomics Revealed Preferential Degradation of Misfolded In Vivo Obligate GroE Substrates by Lon Protease in Escherichia coli. Molecules. 2022; 27(12):3772. https://doi.org/10.3390/molecules27123772
Chicago/Turabian StyleNiwa, Tatsuya, Yuhei Chadani, and Hideki Taguchi. 2022. "Shotgun Proteomics Revealed Preferential Degradation of Misfolded In Vivo Obligate GroE Substrates by Lon Protease in Escherichia coli" Molecules 27, no. 12: 3772. https://doi.org/10.3390/molecules27123772
APA StyleNiwa, T., Chadani, Y., & Taguchi, H. (2022). Shotgun Proteomics Revealed Preferential Degradation of Misfolded In Vivo Obligate GroE Substrates by Lon Protease in Escherichia coli. Molecules, 27(12), 3772. https://doi.org/10.3390/molecules27123772