
In-Silico Screening and Molecular Dynamics Simulation of Drug Bank Experimental Compounds against SARS-CoV-2

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
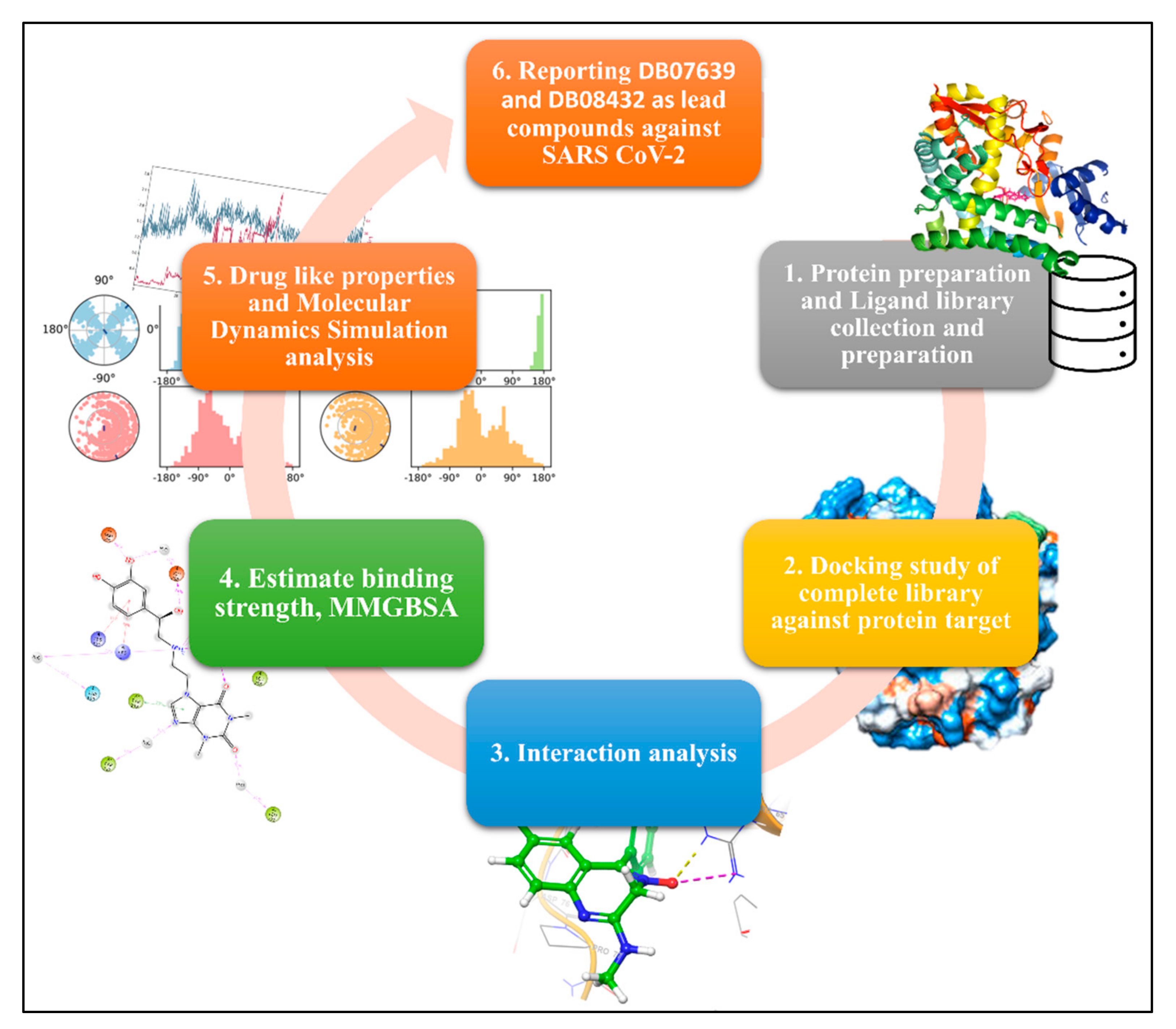
2. Methodology
2.1. Protein Preparation
2.2. Ligand Library Collection and Preparation
2.3. Active Site Calculation and Glide Grid Generation
2.4. Molecular Docking and ADMET Analysis
2.5. Molecular Dynamics Simulation
3. Results
3.1. Molecular Docking
3.2. Molecular Dynamics Simulation
3.2.1. RMSD and RMSF
3.2.2. Intermolecular Interaction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ahmad, S.; Bhanu, P.; Kumar, J.; Pathak, R.K.; Mallick, D.; Uttarkar, A.; Niranjan, V.; Mishra, V. Molecular dynamics simulation and docking studies reveal NF-κB as a promising therapeutic drug target for COVID-19. Bioinformation 2021, 18, 170–179. [Google Scholar]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): An update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [Green Version]
- Raza, K. (Ed.) Computational Intelligence Methods in COVID-19: Surveillance, Prevention, Prediction and Diagnosis; Springer: Berlin/Heidelberg, Germany, 2021; pp. 79–108. [Google Scholar]
- Struyf, T.; Deeks, J.J.; Dinnes, J.; Takwoingi, Y.; Davenport, C.; Leeflang, M.M.; Spijker, R.; Hooft, L.; Emperador, D.; Dittrich, S.; et al. Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19. Cochrane Database Syst. Rev. 2021, 2, CD013665. [Google Scholar] [PubMed]
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76. [Google Scholar] [CrossRef]
- Tarique, M.; Ahmad, S.; Malik, A.; Ahmad, I.; Saeed, M.; Almatroudi, A.; Qatah, T.; Murad, M.A.; Mashraqi, M.; Alam, Q.; et al. Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2) and Other Coronaviruses: A Genome-wide Comparative Annotation and Analysis. Mol. Cell. Biochem. 2021, 476, 2203–2217. [Google Scholar] [CrossRef] [PubMed]
- Rizou, M.; Galanakis, I.M.; Aldawoud, T.M.; Galanakis, C.M. Safety of foods, food supply chain and environment within the COVID-19 pandemic. Trends Food Sci. Technol. 2020, 102, 293–299. [Google Scholar] [CrossRef]
- Tang, S.; Mao, Y.; Jones, R.M.; Tan, Q.; Ji, J.S.; Li, N.; Shen, J.; Lv, Y.; Pan, L.; Ding, P.; et al. Aerosol transmission of SARS-CoV-2? Evidence, prevention and control. Environ. Int. 2020, 144, 106039. [Google Scholar] [CrossRef]
- Yang, Q.; Zhou, Y.; Ai, J.; Ma, J.; Cao, F.; Cao, W.; Zhang, W.; Wang, S.; Chen, W.; Ma, X.; et al. Collaborated effort against SARS-CoV-2 outbreak in China. Clin. Transl. Med. 2020, 10, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, M.; Ren, R.; Li, L.; Chen, E.Q.; Li, W.; Ying, B. International expansion of a novel SARS-CoV-2 mutant. J. Virol. 2020, 94, e00567-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. -Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated modeling program, applied chemical theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef] [PubMed]
- 2: LigPrep, Schrödinger LLC: New York, NY, USA, 2017.
- Halgren, T.A. Identifying and characterising binding sites and assessing druggability. J. Chem. Inf. Modeling 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2019-3: Glide, Schrödinger LLC: New York, NY, USA, 2019.
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins: Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [Green Version]
- Kadioglu, O.; Saeed, M.; Greten, H.J.; Efferth, T. Identification of novel compounds against three targets of SARS CoV-2 coronavirus by combined virtual screening and supervised machine learning. Comput. Biol. Med. 2021, 133, 104359. [Google Scholar] [CrossRef]
- Walters, W.P. Going further than Lipinski’s rule in drug design. Expert Opin. Drug Discov. 2012, 7, 99–107. [Google Scholar] [CrossRef]
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the reliability of QikProp. Correlation between experimental and predicted values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- QikProp, Schrödinger LLC: New York, NY, USA, 2017.
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, SC’06, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Huang, C.; Li, C.; Choi, P.Y.; Nandakumar, K.; Kostiuk, L.W. A novel method for molecular dynamics simulation in the isothermal–isobaric ensemble. Mol. Phys. 2011, 109, 191–202. [Google Scholar] [CrossRef]
- Katz, B.A.; Luong, C. Recruiting Zn2+ to mediate potent, specific inhibition of serine proteases. J. Mol. Biol. 1999, 292, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, G.; Möllmann, H. Pharmacokinetic/pharmacodynamic characteristics of the beta-2-agonists terbutaline, salbutamol and fenoterol. Int. J. Clin. Pharmacol. Ther. Toxicol. 1992, 30, 342–362. [Google Scholar] [PubMed]
- Amanat, F.; Krammer, F. SARS-CoV-2 vaccines: Status report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Drug Bank ID | Protein Name | Docking Score | MMGBSA dG Bind | Rotatable Bonds | Ligand Efficiency Sa | Ligand Efficiency Ln | Evdw | Ecoul |
|---|---|---|---|---|---|---|---|---|---|
| 1 | DB07639 | RNA dependent-polymerase | −8.781 | 45.78 | 9 | −1.955 | −9.34 | −33 | −35.6 |
| 2 | DB08432 | Replication transcription | −8.582 | 55.492 | 5 | −1.042 | −3.609 | −27.6 | −43.4 |
| Title | Normal Values | DB07639 | DB08432 | Title | Normal Values | DB07639 | DB08432 |
|---|---|---|---|---|---|---|---|
| #acid | 0–1 | 0 | 2 | IP(eV) | 7.9–10.5 | 0 | 0 |
| #amide | 0–1 | 0 | 0 | Jm | N/A | 0 | 0.003 |
| #amidine | 0 | 0 | 0 | mol MW | 130.0–725.0 | 433.549 | 338.271 |
| #amine | 0–1 | 3 | 0 | % HumanOralAbsorption | >80% is high, <25% is poor | 50.112 | 42.465 |
| #in34 | N/A | 0 | 0 | PISA | 0.0–450.0 | 260.165 | 29.219 |
| #in56 | N/A | 21 | 11 | PSA | 7.0–200.0 | 108.278 | 153.962 |
| #metab | 1–8 | 7 | 3 | QPlogBB | −3.0–1.2 | −1.188 | −1.776 |
| #NandO | 2–15 | 6 | 9 | QPlogHERG | concern below −5 | −8.495 | −0.274 |
| #noncon | N/A | 4 | 4 | QPlogKhsa | −1.5–1.5 | 0.55 | −0.909 |
| #nonHatm | N/A | 32 | 21 | QPlogKp | −8.0–−1.0 | −7.049 | −5.278 |
| #ringatoms | N/A | 21 | 11 | QPlogPC16 | 4.0–18.0 | 15.966 | 10.053 |
| #rotor | 0–15 | 10 | 6 | QPlogPo/w | −2.0–6.5 | 2.434 | 1.062 |
| #rtvFG | 0–2 | 1 | 1 | QPlogPoct | 8.0–35.0 | 26.265 | 20.346 |
| #stars | 0–5 | 0 | 0 | QPlogPw | 4.0–45.0 | 15.875 | 15.661 |
| accptHB | 2.0–20.0 | 6.25 | 8.4 | QPlogS | −6.5–0.5 | −2.394 | −2.741 |
| ACxDN^.5/SA | 0.0–0.05 | 0.0176843 | 0.0317999 | QPPCaco | <25 poor, >500 great | 3.15 | 3.31 |
| CIQPlogS | −6.5–0.5 | −2.997 | −3.155 | QPPMDCK | <25 poor, >500 great | 1.324 | 4.15 |
| CNS | −2 (inactive), +2 (active) | −2 | −2 | QPpolrz | 13.0–70.0 | 47.608 | 26.903 |
| dip^2/V | 0.0–0.13 | 0 | 0 | RuleOfFive | maximum is 4 | 0 | 0 |
| dipole | 1.0–12.5 | 0 | 0 | RuleOfThree | maximum is 3 | 2 | 1 |
| donorHB | 0.0–6.0 | 5 | 4 | SAamideO | 0.0–35.0 | 0 | 0 |
| EA(eV) | −0.9–1.7 | 0 | 0 | SAfluorine | 0.0–100.0 | 0 | 0 |
| FISA | 7.0–330.0 | 178.038 | 240.775 | SASA | 300.0–1000.0 | 790.275 | 528.304 |
| FOSA | 0.0–750.0 | 352.072 | 186.095 | volume | 500.0–2000.0 | 1441.735 | 915.318 |
| glob | 0.75–0.95 | 0.7809869 | 0.8629708 | WPSA | 0.0–175.0 | 0 | 72.214 |
| HumanOralAbsorption | N/A | 2 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alturki, N.A.; Mashraqi, M.M.; Alzamami, A.; Alghamdi, Y.S.; Alharthi, A.A.; Asiri, S.A.; Ahmad, S.; Alshamrani, S. In-Silico Screening and Molecular Dynamics Simulation of Drug Bank Experimental Compounds against SARS-CoV-2. Molecules 2022, 27, 4391. https://doi.org/10.3390/molecules27144391
Alturki NA, Mashraqi MM, Alzamami A, Alghamdi YS, Alharthi AA, Asiri SA, Ahmad S, Alshamrani S. In-Silico Screening and Molecular Dynamics Simulation of Drug Bank Experimental Compounds against SARS-CoV-2. Molecules. 2022; 27(14):4391. https://doi.org/10.3390/molecules27144391
Chicago/Turabian StyleAlturki, Norah A., Mutaib M. Mashraqi, Ahmad Alzamami, Youssef S. Alghamdi, Afaf A. Alharthi, Saeed A. Asiri, Shaban Ahmad, and Saleh Alshamrani. 2022. "In-Silico Screening and Molecular Dynamics Simulation of Drug Bank Experimental Compounds against SARS-CoV-2" Molecules 27, no. 14: 4391. https://doi.org/10.3390/molecules27144391
APA StyleAlturki, N. A., Mashraqi, M. M., Alzamami, A., Alghamdi, Y. S., Alharthi, A. A., Asiri, S. A., Ahmad, S., & Alshamrani, S. (2022). In-Silico Screening and Molecular Dynamics Simulation of Drug Bank Experimental Compounds against SARS-CoV-2. Molecules, 27(14), 4391. https://doi.org/10.3390/molecules27144391