
Electrochemical Site-Selective Alkylation of Azobenzenes with (Thio)Xanthenes


Abstract
:1. Introduction
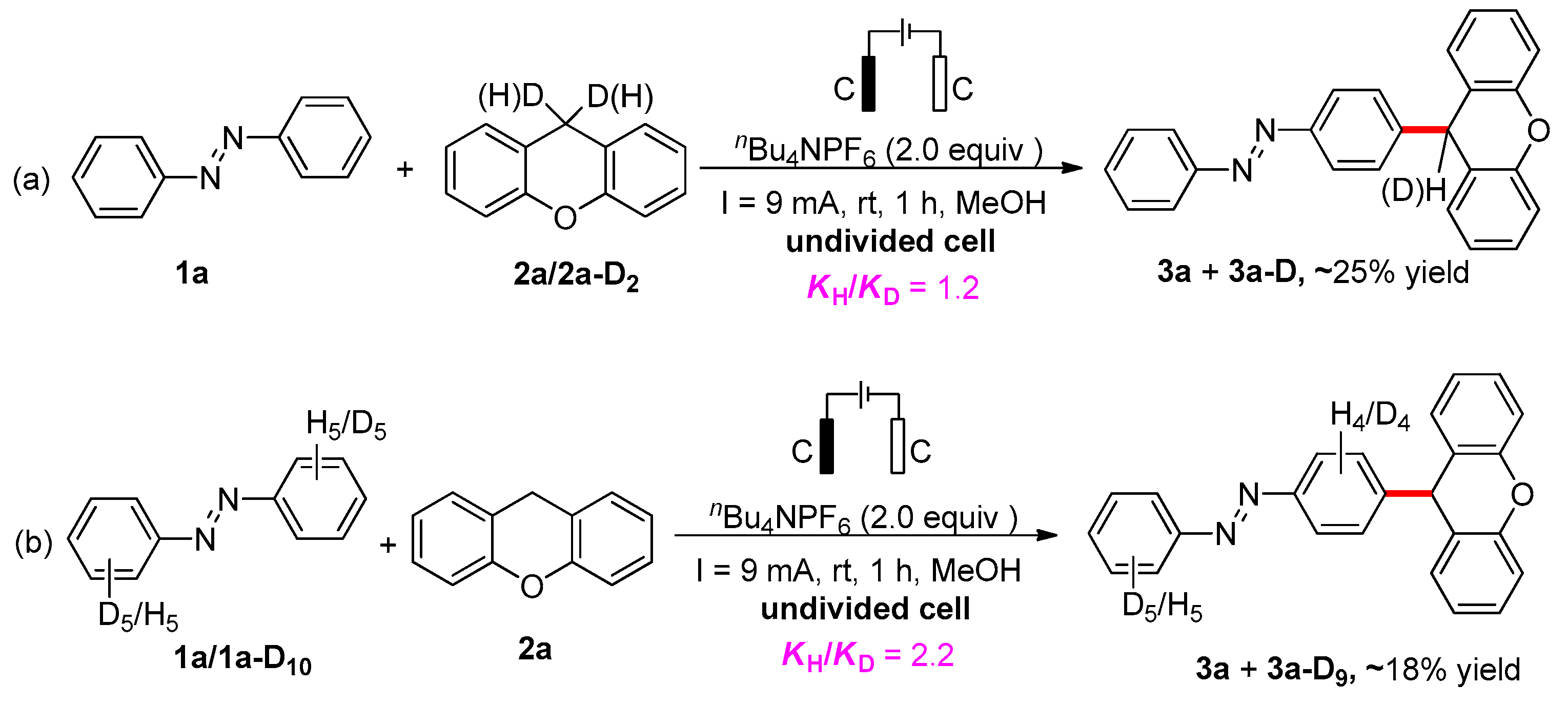
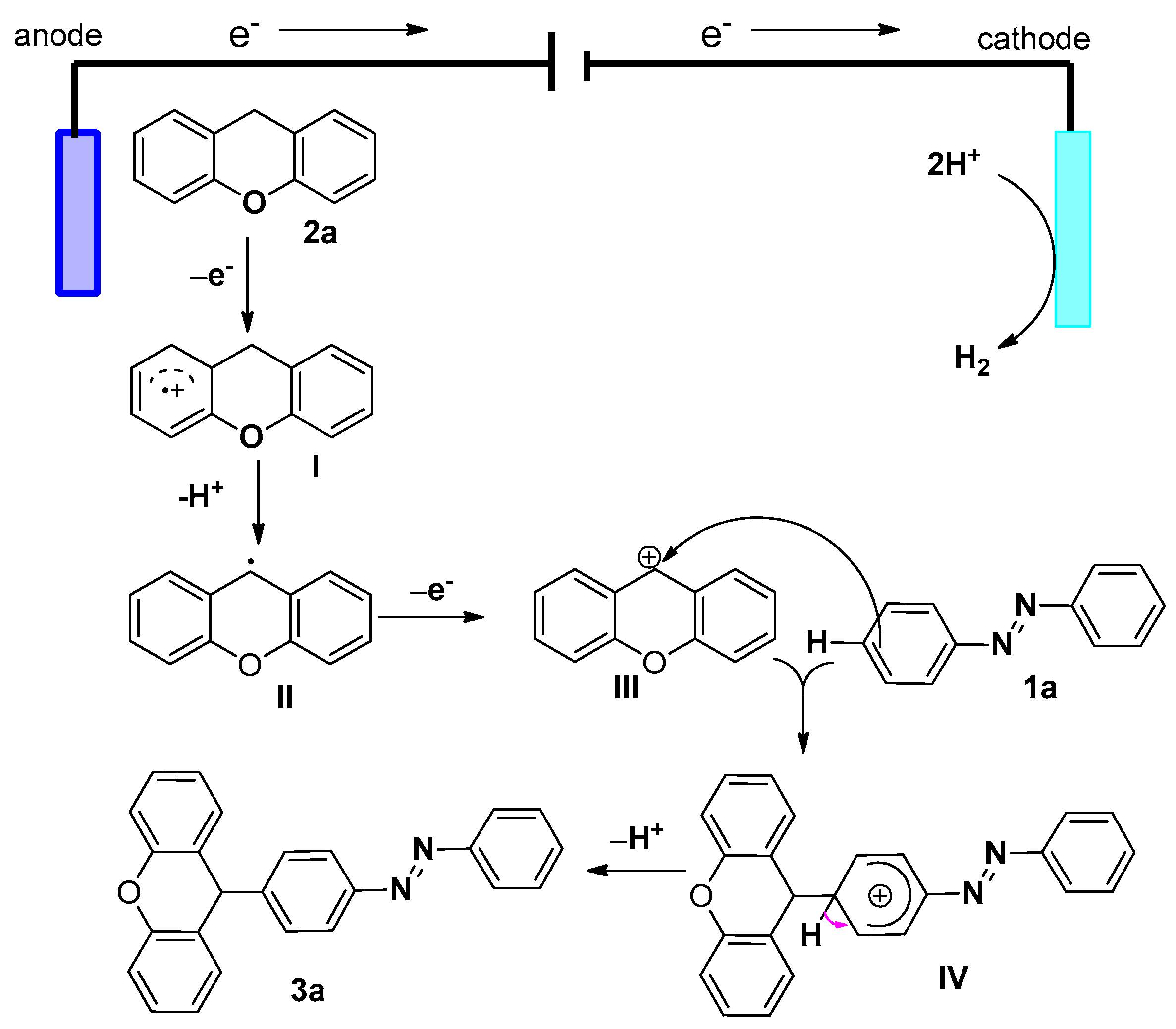
2. Results and Discussion
3. Materials and Methods
3.1. General Considerations
3.2. Typical Procedure for the Synthesis of 3a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Léonard, E.; Mangin, F.; Villette, C.; Billamboz, M.; Len, C. Azobenzenes and catalysis. Catal. Sci. Technol. 2016, 6, 379–398. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, P.; Wu, B.; Xing, Y.; Shi, K.; Fang, W.; Yu, H.; Wang, G. Photochromic dendrimers for photoswitched solid-to-liquid transitions and solar thermal fuels. ACS Appl. Mater. Interfaces 2020, 12, 50135–50142. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-B.; Zhang, S.; Qi, J.; Liang, X.-J.; Yoon, J. Advances in application of azobenzene as a trigger in biomedicine: Molecular design and spontaneous assembly. Adv. Mater. 2021, 33, 2007290. [Google Scholar] [CrossRef] [PubMed]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Yanai, N.; Uemura, T.; Inoue, M.; Matsuda, R.; Fukushima, T.; Tsujimoto, M.; Isoda, S.; Kitagawa, S. Guest-to-host transmission of structural changes for stimuli-responsive adsorption property. J. Am. Chem. Soc. 2012, 134, 4501–4504. [Google Scholar] [CrossRef]
- Donovan, B.R.; Matavulj, V.M.; Ahn, S.; Guin, T.; White, T.J. All-optical control of shape. Adv. Mater. 2019, 31, 1805750. [Google Scholar] [CrossRef]
- Jiang, Y.; Tan, P.; Qi, S.-C.; Liu, X.-Q.; Yan, J.-H.; Fan, F.; Sun, L.-B. Metal–organic frameworks with target-specific active sites switched by photoresponsive motifs: Efficient adsorbents for tailorable CO2 capture. Angew. Chem. Int. Ed. 2019, 58, 6600–6604. [Google Scholar] [CrossRef]
- Kondo, M.; Nakamura, K.; Krishnan, C.G.; Takizawa, S.; Abe, T.; Sasai, H. Photoswitchable chiral phase transfer catalyst. ACS Catal. 2021, 11, 1863–1867. [Google Scholar] [CrossRef]
- Hansen, M.J.; Lerch, M.M.; Szymanski, W.; Feringa, B.L. Direct and versatile synthesis of red-shifted azobenzenes. Angew. Chem. Int. Ed. 2016, 55, 13514–13518. [Google Scholar] [CrossRef]
- Trads, J.B.; Hüll, K.; Matsuura, B.S.; Laprell, L.; Fehrentz, T.; Görldt, N.; Kozek, K.A.; Weaver, C.D.; Klöcker, N.; Barber, D.M.; et al. Sign inversion in photopharmacology: Incorporation of cyclic azobenzenes in photoswitchable potassium channel blockers and openers. Angew. Chem. Int. Ed. 2019, 58, 15421–15428. [Google Scholar] [CrossRef]
- Jerca, F.A.; Jerca, V.V.; Hoogenboom, R. Advances and opportunities in the exciting world of azobenzenes. Nat. Rev. Chem. 2021, 6, 51–69. [Google Scholar] [CrossRef]
- Wencel-Delord, J.; Dröge, T.; Liu, F.; Glorius, F. Towards mild metal-catalyzed C-H bond activation. Chem. Soc. Rev. 2011, 40, 4740–4761. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lan, J.; You, J. Oxidative C-H/C-H coupling reactions between two (hetero)arenes. Chem. Rev. 2017, 117, 8787–8863. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Meng, G.; Nolan, S.P.; Szostak, M. N-heterocyclic carbene complexes in C-H activation reactions. Chem. Rev. 2020, 120, 1981–2048. [Google Scholar] [CrossRef]
- Nguyen, T.H.L.; Gigant, N.; Joseph, D. Advances in direct metal-catalyzed functionalization of azobenzenes. ACS Catal. 2018, 8, 1546–1579. [Google Scholar] [CrossRef]
- Li, H.; Li, P.; Wang, L. Direct access to acylated azobenzenes via Pd-catalyzed C–H functionalization and further transformation into an indazole backbone. Org. Lett. 2013, 15, 620–623. [Google Scholar] [CrossRef]
- Lian, Y.; Bergman, R.G.; Lavis, L.D.; Ellman, J.A. Rhodium(III)-catalyzed indazole synthesis by C–H bond functionalization and cyclative capture. J. Am. Chem. Soc. 2013, 135, 7122–7125. [Google Scholar] [CrossRef]
- Deng, H.; Li, H.; Wang, L. ortho-Heteroarylation of azobenzenes by Rh-catalyzed cross-dehydrogenative coupling: An approach to conjugated biaryls. Org. Lett. 2016, 18, 3110–3113. [Google Scholar] [CrossRef]
- Li, H.; Li, P.; Tan, H.; Wang, L. A highly efficient palladium-catalyzed decarboxylative ortho-acylation of azobenzenes with α-oxocarboxylic acids: Direct access to acylated azo compounds. Chem. Eur. J. 2013, 19, 14432–14436. [Google Scholar] [CrossRef]
- Deng, H.; Li, H.; Wang, L. A unique alkylation of azobenzenes with allyl acetates by RhIII-catalyzed C–H functionalization. Org. Lett. 2015, 17, 2450–2453. [Google Scholar] [CrossRef]
- Li, H.; Xie, X.; Wang, L. Ruthenium-catalyzed alkenylation of azoxybenzenes with alkenes through ortho-selective C–H activation. Chem. Commun. 2014, 50, 4218–4221. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Y.; Li, D.-D.; Wang, G.-W. Palladium-catalyzed decarboxylative ortho acylation of azobenzenes with α-oxocarboxylic acids. J. Org. Chem. 2013, 78, 10414–10420. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Jiang, X.; Sun, P. Palladium-catalyzed direct ortho alkoxylation of aromatic azo compounds with alcohols. J. Org. Chem. 2013, 78, 10002–10007. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xian, Y.; Lan, J.; Luo, Y.; Ma, W.; You, J. Fusion of aromatic ring to azoarenes: One-pot access to 5,6-phenanthroliniums for mitochondria-targeted far-red/nir fluorescent probes. Org. Lett. 2019, 21, 1037–1041. [Google Scholar] [CrossRef]
- Hu, W.; Pi, C.; Hu, D.; Han, X.; Wu, Y.; Cui, X. Rh(III)-catalyzed synthesis of indazolo [2,3-α]quinolines: Vinylene carbonate as C1 and C2 building blocks. Org. Lett. 2022, 24, 2613–2618. [Google Scholar] [CrossRef]
- Zhang, W.; Bu, J.; Wang, L.; Li, P.; Li, H. Sunlight-mediated [3 + 2] cycloaddition of azobenzenes with arynes: An approach toward the carbazole skeleton. Org. Chem. Front. 2021, 8, 5045–5051. [Google Scholar] [CrossRef]
- Li, G.; Ma, X.; Jia, C.; Han, Q.; Wang, Y.; Wang, J.; Yu, L.; Yang, S. Ruthenium-catalyzed meta/ortho-selective C–H alkylation of azoarenes using alkyl bromides. Chem. Commun. 2017, 53, 1261–1264. [Google Scholar] [CrossRef]
- Tao, Y.; Hu, R.; Xie, Z.; Lin, P.; Su, W. Cobalt-catalyzed regioselective para-amination of azobenzenes via nucleophilic aromatic substitution of hydrogen. J. Org. Chem. 2022, 87, 4724–4731. [Google Scholar] [CrossRef]
- Lu, Q.; Cembellín, S.; Greßies, S.; Singha, S.; Daniliuc, C.G.; Glorius, F. Manganese(I)-catalyzed C–H (2-indolyl)methylation: Expedient access to diheteroarylmethanes. Angew. Chem. Int. Ed. 2018, 57, 1399–1403. [Google Scholar] [CrossRef]
- Xiong, P.; Xu, H.-C. Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res. 2019, 52, 3339–3350. [Google Scholar] [CrossRef]
- Xiang, J.; Shang, M.; Kawamata, Y.; Lundberg, H.; Reisberg, S.H.; Chen, M.; Mykhailiuk, P.; Beutner, G.; Collins, M.R.; Davies, A.; et al. Hindered dialkyl ether synthesis with electrogenerated carbocations. Nature 2019, 573, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution reactions. Acc. Chem. Res. 2019, 52, 3309–3324. [Google Scholar] [CrossRef]
- Shi, S.-H.; Liang, Y.; Jiao, N. Electrochemical oxidation induced selective C–C bond cleavage. Chem. Rev. 2020, 121, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.-J.; Xing, Y.-K.; Yang, Q.-L.; Qiu, H.; Mei, T.-S. Site-selective C–H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res. 2020, 53, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Ang, N.W.J.; Meyer, T.H.; Qiu, Y.; Ackermann, L. Organic electrochemistry: Molecular syntheses with potential. ACS Cent. Sci. 2021, 7, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.R.D.; Cox, J.H.; Chiappini, N.D.; Roos, C.B.; McLoughlin, E.A.; Hejna, B.G.; Nguyen, S.T.; Ripberger, H.H.; Ganley, J.M.; Tsui, E.; et al. Photochemical and electrochemical applications of proton-coupled electron transfer in organic synthesis. Chem. Rev. 2021, 122, 2017–2291. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Gao, P.-S.; Wang, X.; Gao, J.-Q.; Xu, X.-T.; He, Z.; Ma, C.; Mei, T.-S. TEMPO-enabled electrochemical enantioselective oxidative coupling of secondary acyclic amines with ketones. J. Am. Chem. Soc. 2021, 143, 15599–15605. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, L.; Li, H.; Cao, Y.; Hu, J.; Xu, M.; Shi, R.; Yi, H.; Lei, A. Electrochemical palladium-catalyzed oxidative sonogashira carbonylation of arylhydrazines and alkynes to ynones. J. Am. Chem. Soc. 2021, 143, 12460–12466. [Google Scholar] [CrossRef]
- Huang, C.; Ma, W.; Zheng, X.; Xu, M.; Qi, X.; Lu, Q. Epoxide electroreduction. J. Am. Chem. Soc. 2021, 144, 1389–1395. [Google Scholar] [CrossRef]
- Jie, L.-H.; Guo, B.; Song, J.; Xu, H.-C. Organoelectrocatalysis enables direct cyclopropanation of methylene compounds. J. Am. Chem. Soc. 2022, 144, 2343–2350. [Google Scholar] [CrossRef]
- Li, C.; Zhong, Q.; Tang, S.; Wang, L.; Li, P.; Li, H. Electrochemical formal [3 + 2] cycloaddition of azobenzenes with hexahydro-1,3,5-triazines. Org. Chem. Front. 2022, 9, 3769–3774. [Google Scholar] [CrossRef]
- Chen, X.; Liu, H.; Gao, H.; Li, P.; Miao, T.; Li, H. Electrochemical regioselective cross-dehydrogenative coupling of indoles with xanthenes. J. Org. Chem. 2022, 87, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chen, X.; Wang, P.-L.; Shi, M.-M.; Shang, L.-L.; Guo, H.-Y.; Li, H.; Li, P. Electrochemical benzylic C–H arylation of xanthenes and thioxanthenes without a catalyst and oxidant. Org. Chem. Front. 2022, 9, 1911–1916. [Google Scholar] [CrossRef]
- CCDC Number: 2178297 for 3d; the Regarding Crystallographic Data Can Be Obtained from the Cambridge Crystallographic Data Centre. Available online: www.ccdc.cam.ac.uk (accessed on 10 June 2022).
- Pintér, Á.; Klussmann, M. Sulfonic acid-catalyzed autoxidative carbon-carbon coupling reaction under elevated partial pressure of oxygen. Adv. Synth. Catal. 2012, 354, 701–711. [Google Scholar] [CrossRef]
- Wu, H.; Su, C.; Tandiana, R.; Liu, C.; Qiu, C.; Bao, Y.; Wu, J.; Xu, Y.; Lu, J.; Fan, D.; et al. Graphene-oxide-catalyzed direct CH–CH-type cross-coupling: The intrinsic catalytic activities of zigzag edges. Angew. Chem. Int. Ed. 2018, 57, 10848–10853. [Google Scholar] [CrossRef]
- Liang, Y.; Niu, L.; Liang, X.-A.; Wang, S.; Wang, P.; Lei, A. Electrooxidation-induced C(sp3)–H/C(sp2)–H radical-radical cross-coupling between xanthanes and electron-rich arenes. Chin. J. Chem. 2022, 40, 1422–1428. [Google Scholar] [CrossRef]
- Wei, B.; Qin, J.-H.; Yang, Y.-Z.; Xie, Y.-X.; Ouyang, X.-H.; Song, R.-J. Electrochemical radical C(sp3)–H arylation of xanthenes with electron-rich arenes. Org. Chem. Front. 2022, 9, 816–821. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, S.; She, M.; Nie, J.; Huo, Y.; Chen, Q.; Li, X.; Hu, X.-Q. Practical synthesis of 3-aryl anthranils via an electrophilic aromatic substitution strategy. Chem. Sci. 2022, 13, 2105–2114. [Google Scholar] [CrossRef]
- Zubar, V.; Dewanji, A.; Rueping, M. Chemoselective hydrogenation of nitroarenes using an air-stable base-metal catalyst. Org. Lett. 2021, 23, 2742–2747. [Google Scholar] [CrossRef]
- Zhang, C.; Jiao, N. Copper-catalyzed aerobic oxidative dehydrogenative coupling of anilines leading to aromatic azo compounds using dioxygen as an oxidant. Angew. Chem. Int. Ed. 2010, 49, 6174–6177. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry | Variation from the “Standard Conditions” | Yield (%) b |
| 1 | none | 76 |
| 2 | nBu4NBF4 instead of nBu4NPF6 | 62 |
| 3 | nBu4NI instead of nBu4NPF6 | 35 |
| 4 | LiClO4 instead of nBu4NPF6 | n.d. |
| 5 | Pt(+)|Pt(−) instead of C(+)|C(−) | n.d. |
| 6 | Pt(+)|C(−) instead of C(+)|C(−) | 68 |
| 7 | C(+)|Pt (−) instead of C(+)|C(−) | 64 |
| 8 | C(+)|GF(−) instead of C(+)|C(−) | 19 |
| 9 | GF(+)|C(−) instead of C(+)|C(−) | 65 |
| 10 | C(+)|Ni(−) instead of C(+)|C(−) | 11 |
| 11 | DCE instead of MeOH | 55 |
| 12 | CH3CN instead of MeOH | 43 |
| 13 | THF instead of MeOH | 40 |
| 14 | DMF instead of MeOH | 29 |
| 15 | Acetone instead of MeOH | n.d. |
| 16 | 3 h instead of 4 h | 55 |
| 17 | 5 h instead of 4 h | 68 |
| 18 | 8 mA instead of 9 mA | 46 |
| 19 | 10 mA instead of 9 mA | 67 |
| 20 | no electric current | n.d. |
| 21 | N2 | 74 c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, Q.; Gao, H.; Wang, P.-L.; Zhou, C.; Miao, T.; Li, H. Electrochemical Site-Selective Alkylation of Azobenzenes with (Thio)Xanthenes. Molecules 2022, 27, 4967. https://doi.org/10.3390/molecules27154967
Zhong Q, Gao H, Wang P-L, Zhou C, Miao T, Li H. Electrochemical Site-Selective Alkylation of Azobenzenes with (Thio)Xanthenes. Molecules. 2022; 27(15):4967. https://doi.org/10.3390/molecules27154967
Chicago/Turabian StyleZhong, Qiang, Hui Gao, Pei-Long Wang, Chao Zhou, Tao Miao, and Hongji Li. 2022. "Electrochemical Site-Selective Alkylation of Azobenzenes with (Thio)Xanthenes" Molecules 27, no. 15: 4967. https://doi.org/10.3390/molecules27154967
APA StyleZhong, Q., Gao, H., Wang, P. -L., Zhou, C., Miao, T., & Li, H. (2022). Electrochemical Site-Selective Alkylation of Azobenzenes with (Thio)Xanthenes. Molecules, 27(15), 4967. https://doi.org/10.3390/molecules27154967