
Aβ and Tau Interact with Metal Ions, Lipid Membranes and Peptide-Based Amyloid Inhibitors: Are These Common Features Relevant in Alzheimer’s Disease?

 , ,
, ,  and
and 
Abstract
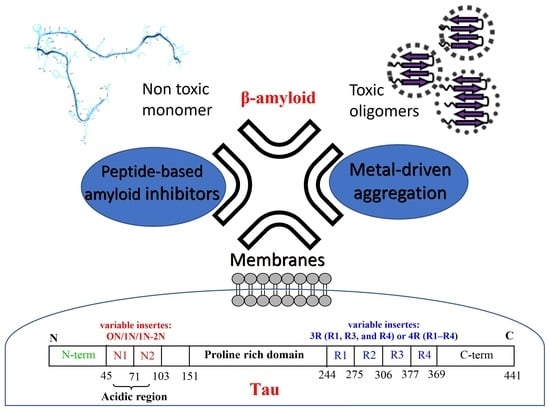
:
1. Introduction
2. Aβ and Tau Proteins: Molecular Structure and Physiological Functions
3. Could Aβ and Tau Be Colocalized in Lipid Membranes?
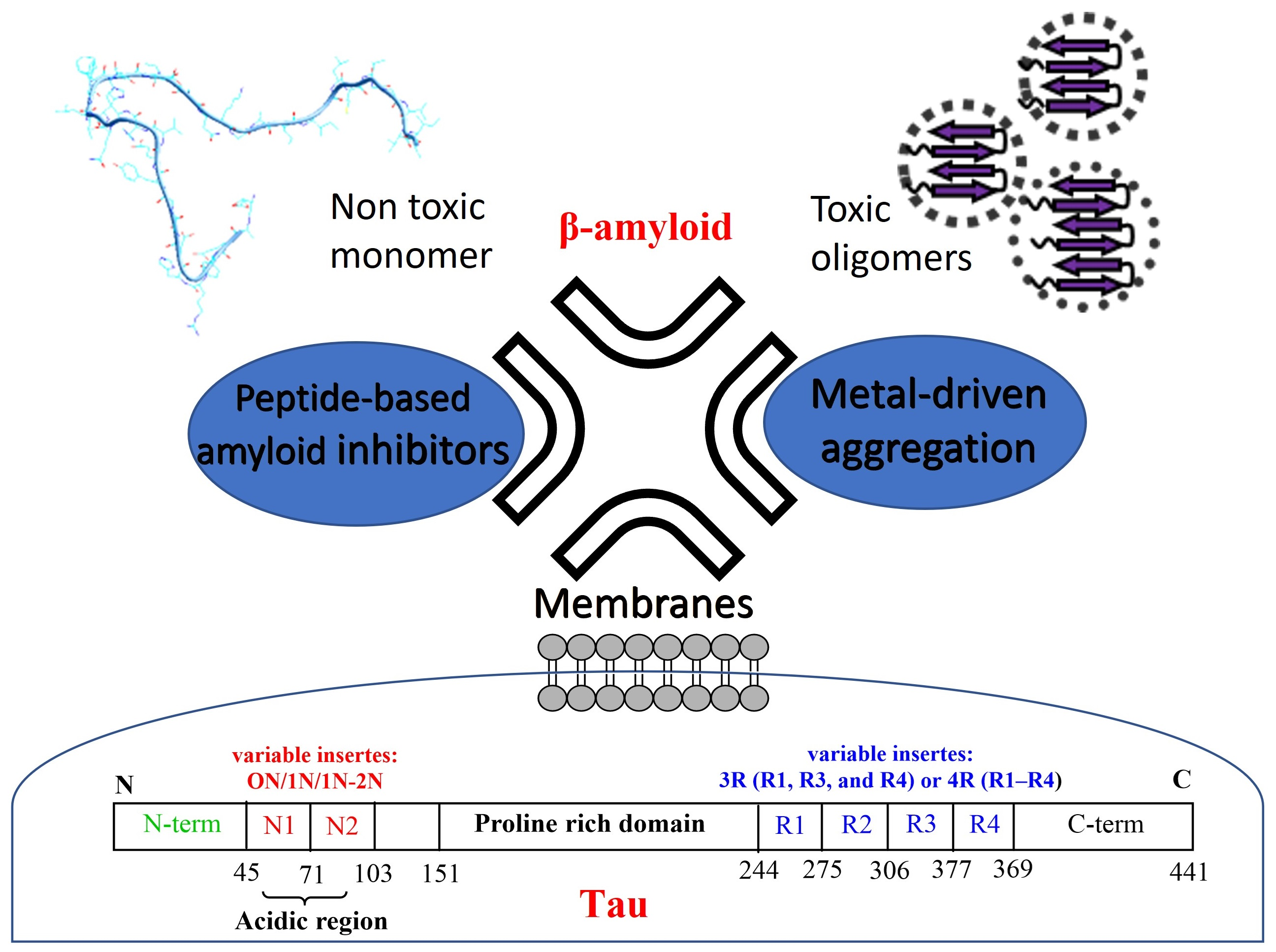
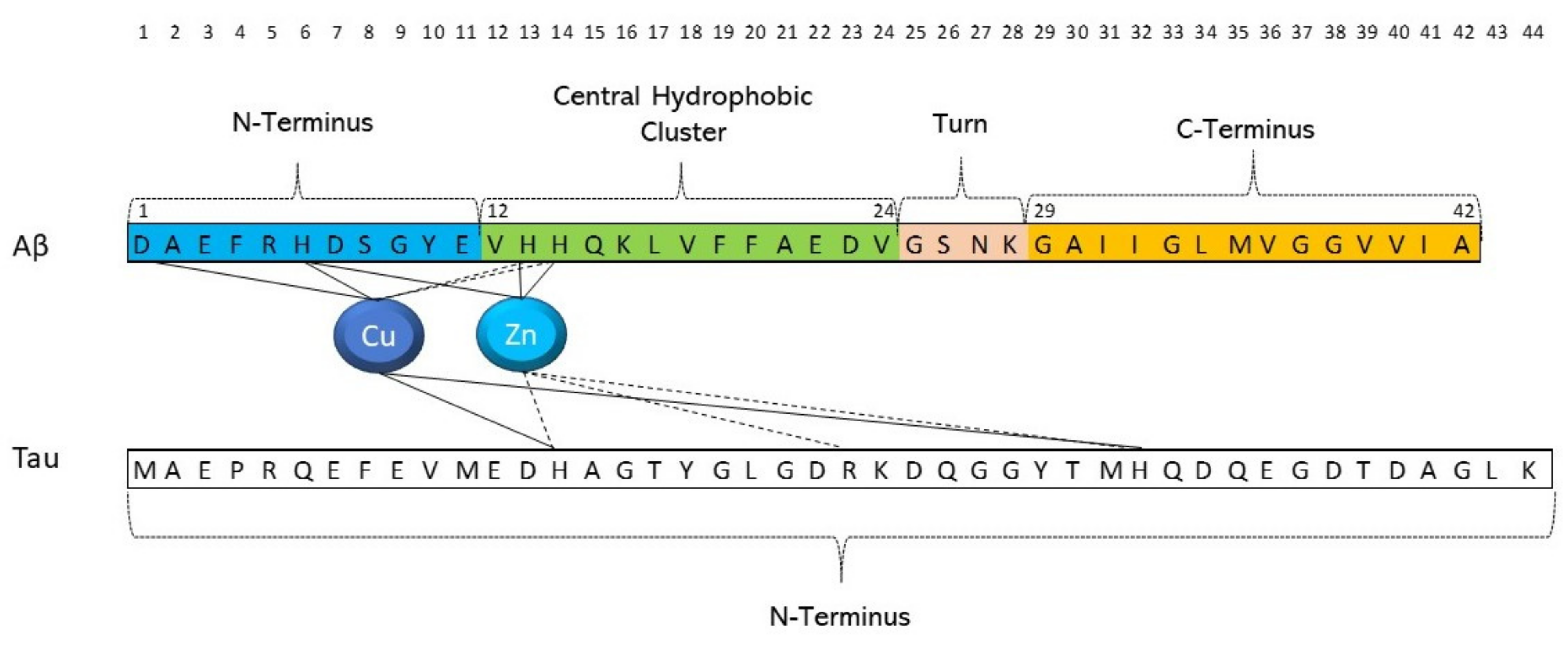
4. Aβ and Tau Can Interact with Metal Ions
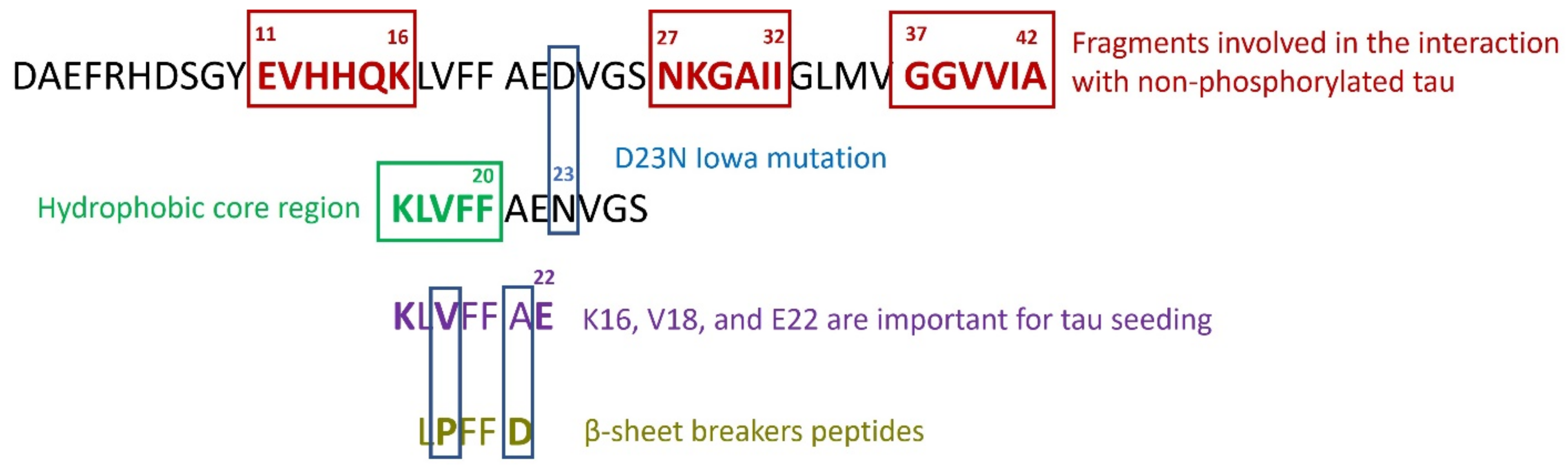
5. Can Targeted Peptide-Based Inhibitors Prevent Aβ/Tau Cross-Seeding AD?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | Description | References |
|---|---|---|
| Ac-EVMEDHAKLVFF-NH2 (τ9-16-KLVFF) Ac-QGGYTMHQKLVFF-NH2 (τ26-33-KLVFF) | Chimera Tau/Aβ tau 9–16/Aβ 16–20 tau 26–33/Aβ 16–20 | [90,141] |
| (D)-LYIWIWRT (D)-LYWIQKT | Peptide-based inhibitors effectiveness for both Aβ and tau designed against Aβ 16–26 D23N | [169] |
| Thymine-Sr-L-Sr-F-Sr-A Thymine-K-Sr-V-Sr-F-A | Introduction of thymine and sarcosine (Sr) inhibiting aggregates of tau and β-amyloid. | [168] |
| cyclic D,L-α-peptides [lJwHsK] 1 | Designed tau aggregation peptide inhibitors that may cross-react with Aβ | [170,171] |
6. Conclusions
Funding
Conflicts of Interest
References
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English Translation of Alzheimer’s 1907 Paper, “Über Eine Eigenartige Erkankung Der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Towards a Dementia Plan: A WHO Guide; World Health Organization: Geneva, Switzerland, 2018.
- 2015 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. J. Alzheimers Assoc. 2015, 11, 332–384. [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Lee, V.M.; Balin, B.J.; Otvos, L.; Trojanowski, J.Q. A68: A Major Subunit of Paired Helical Filaments and Derivatized Forms of Normal Tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alzheimer’s and Parkinson’s Diseases: The Prion Concept in Relation to Assembled Aβ, Tau, and α-Synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A. Straight and Paired Helical Filaments in Alzheimer Disease Have a Common Structural Unit. Proc. Natl. Acad. Sci. USA 1991, 88, 2288–2292. [Google Scholar] [CrossRef] [Green Version]
- Meraz-Ríos, M.A.; Lira-De León, K.I.; Campos-Peña, V.; De Anda-Hernández, M.A.; Mena-López, R. Tau Oligomers and Aggregation in Alzheimer’s Disease. J. Neurochem. 2010, 112, 1353–1367. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Tan, L.; Liu, J.; Yu, J.-T. The Essential Role of Soluble Aβ Oligomers in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 1905–1924. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. The Past and the Future of Alzheimer’s Disease CSF Biomarkers—A Journey toward Validated Biochemical Tests Covering the Whole Spectrum of Molecular Events. Front. Neurosci. 2015, 9, 345. [Google Scholar] [CrossRef] [Green Version]
- Sandwall, E.; O’Callaghan, P.; Zhang, X.; Lindahl, U.; Lannfelt, L.; Li, J.-P. Heparan Sulfate Mediates Amyloid-Beta Internalization and Cytotoxicity. Glycobiology 2010, 20, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iribarren, P.; Zhou, Y.; Hu, J.; Le, Y.; Wang, J.M. Role of Formyl Peptide Receptor-like 1 (FPRL1/FPR2) in Mononuclear Phagocyte Responses in Alzheimer Disease. Immunol. Res. 2005, 31, 165–176. [Google Scholar] [CrossRef]
- Wang, H.; Chen, F.; Du, Y.-F.; Long, Y.; Reed, M.N.; Hu, M.; Suppiramaniam, V.; Hong, H.; Tang, S.-S. Targeted Inhibition of RAGE Reduces Amyloid-β Influx across the Blood-Brain Barrier and Improves Cognitive Deficits in Db/Db Mice. Neuropharmacology 2018, 131, 143–153. [Google Scholar] [CrossRef]
- Yuyama, K.; Yamamoto, N.; Yanagisawa, K. Accelerated Release of Exosome-Associated GM1 Ganglioside (GM1) by Endocytic Pathway Abnormality: Another Putative Pathway for GM1-Induced Amyloid Fibril Formation. J. Neurochem. 2008, 105, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Sakono, M.; Zako, T.; Ueda, H.; Yohda, M.; Maeda, M. Formation of Highly Toxic Soluble Amyloid Beta Oligomers by the Molecular Chaperone Prefoldin. FEBS J. 2008, 275, 5982–5993. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and Tau—A Toxic Pas de Deux in Alzheimer’s Disease. Nat. Rev. Neurosci. 2011, 12, 67–72. [Google Scholar] [CrossRef]
- Naveh Tassa, S.; Ben Zichri, S.; Lacham-Hartman, S.; Oren, O.; Slobodnik, Z.; Eremenko, E.; Toiber, D.; Jelinek, R.; Papo, N. A Mechanism for the Inhibition of Tau Neurotoxicity: Studies with Artificial Membranes, Isolated Mitochondria, and Intact Cells. ACS Chem. Neurosci. 2021, 12, 1563–1577. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Pickett, E.K.; Herrmann, A.G.; McQueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldstad, M.P. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer’s Disease. Cell Rep. 2019, 29, 3592–3604. [Google Scholar] [CrossRef] [Green Version]
- McInnes, J.; Wierda, K.; Snellinx, A.; Bounti, L.; Wang, Y.-C.; Stancu, I.-C.; Apóstolo, N.; Gevaert, K.; Dewachter, I.; Spires-Jones, T.L.; et al. Synaptogyrin-3 Mediates Presynaptic Dysfunction Induced by Tau. Neuron 2018, 97, 823–835.e8. [Google Scholar] [CrossRef] [Green Version]
- Ovsepian, S.V.; O’Leary, V.B.; Zaborszky, L.; Ntziachristos, V.; Dolly, J.O. Synaptic Vesicle Cycle and Amyloid β: Biting the Hand That Feeds. Alzheimers Dement. 2018, 14, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Zhang, Z.; Zhao, W.; Yang, J. Interactions between Amyloid β Peptide and Lipid Membranes. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, E.R.; Xiao, S.; Borbat, P.P.; Freed, J.H.; Eliezer, D. Tau Binds to Lipid Membrane Surfaces via Short Amphipathic Helices Located in Its Microtubule-Binding Repeats. Biophys. J. 2014, 107, 1441–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallin, C.; Hiruma, Y.; Wärmländer, S.K.T.S.; Huvent, I.; Jarvet, J.; Abrahams, J.P.; Gräslund, A.; Lippens, G.; Luo, J. The Neuronal Tau Protein Blocks in Vitro Fibrillation of the Amyloid-β (Aβ) Peptide at the Oligomeric Stage. J. Am. Chem. Soc. 2018, 140, 8138–8146. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Hyman, B.T. Synergy between Amyloid-β and Tau in Alzheimer’s Disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s Disease: Clinical Trials and Drug Development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A. Hormesis and Amyloid-β Protein: Physiology or Pathology? J. Alzheimers Dis. JAD 2012, 29, 487–492. [Google Scholar] [CrossRef]
- Hillen, H. The Beta Amyloid Dysfunction (BAD) Hypothesis for Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1154. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, M.L.; Caraci, F.; Pignataro, B.; Cataldo, S.; De Bona, P.; Bruno, V.; Molinaro, G.; Pappalardo, G.; Messina, A.; Palmigiano, A.; et al. Beta-Amyloid Monomers Are Neuroprotective. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 10582–10587. [Google Scholar] [CrossRef]
- Cuesto, G.; Enriquez-Barreto, L.; Caramés, C.; Cantarero, M.; Gasull, X.; Sandi, C.; Ferrús, A.; Acebes, Á.; Morales, M. Phosphoinositide-3-Kinase Activation Controls Synaptogenesis and Spinogenesis in Hippocampal Neurons. J. Neurosci. 2011, 31, 2721–2733. [Google Scholar] [CrossRef] [Green Version]
- Ml, G.; Hm, B.; Sr, R. Alzheimer’s Amyloid-β Is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimers Dis. JAD 2018, 62, 1495–1506. [Google Scholar] [CrossRef] [Green Version]
- Freedman, D.M.; Wu, J.; Chen, H.; Kuncl, R.W.; Enewold, L.R.; Engels, E.A.; Freedman, N.D.; Pfeiffer, R.M. Associations between Cancer and Alzheimer’s Disease in a U.S. Medicare Population. Cancer Med. 2016, 5, 2965–2976. [Google Scholar] [CrossRef] [PubMed]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid Plaque Core Protein in Alzheimer Disease and Down Syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Normal and Abnormal Biology of the Beta-Amyloid Precursor Protein. Annu. Rev. Neurosci. 1994, 17, 489–517. [Google Scholar] [CrossRef]
- Thornton, E.; Vink, R.; Blumbergs, P.C.; Van Den Heuvel, C. Soluble Amyloid Precursor Protein Alpha Reduces Neuronal Injury and Improves Functional Outcome Following Diffuse Traumatic Brain Injury in Rats. Brain Res. 2006, 1094, 38–46. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Qu, D.-H.; Wang, K. Therapeutic Approaches to Alzheimer’s Disease through Stimulating of Non-Amyloidogenic Processing of Amyloid Precursor Protein. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2389–2403. [Google Scholar]
- Luna-Viramontes, N.I.; Campa-Córdoba, B.B.; Ontiveros-Torres, M.Á.; Harrington, C.R.; Villanueva-Fierro, I.; Guadarrama-Ortíz, P.; Garcés-Ramírez, L.; de la Cruz, F.; Hernandes-Alejandro, M.; Martínez-Robles, S.; et al. PHF-Core Tau as the Potential Initiating Event for Tau Pathology in Alzheimer’s Disease. Front. Cell. Neurosci. 2020, 14, 247. [Google Scholar] [CrossRef]
- Do, T.D.; Economou, N.J.; Chamas, A.; Buratto, S.K.; Shea, J.-E.; Bowers, M.T. Interactions between Amyloid-β and Tau Fragments Promote Aberrant Aggregates: Implications for Amyloid Toxicity. J. Phys. Chem. B 2014, 118, 11220–11230. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, S.; Cubinkova, V.; Zimova, I.; Brezovakova, V.; Madari, A.; Cigankova, V.; Zilka, N. Tau-Mediated Synaptic Damage in Alzheimer’s Disease. Transl. Neurosci. 2015, 6, 214–226. [Google Scholar] [CrossRef]
- Gonçalves, R.A.; Wijesekara, N.; Fraser, P.E.; De Felice, F.G. Behavioral Abnormalities in Knockout and Humanized Tau Mice. Front. Endocrinol. 2020, 11, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LoPresti, P. Tau in Oligodendrocytes Takes Neurons in Sickness and in Health. Int. J. Mol. Sci. 2018, 19, 2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The Physiological Roles of Tau and Aβ: Implications for Alzheimer’s Disease Pathology and Therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of Tau Protein in Health and Disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Martín, T.; Cuchillo-Ibáñez, I.; Noble, W.; Nyenya, F.; Anderton, B.H.; Hanger, D.P. Tau Phosphorylation Affects Its Axonal Transport and Degradation. Neurobiol. Aging 2013, 34, 2146–2157. [Google Scholar] [CrossRef] [Green Version]
- Pallas-Bazarra, N.; Jurado-Arjona, J.; Navarrete, M.; Esteban, J.A.; Hernández, F.; Ávila, J.; Llorens-Martín, M. Novel Function of Tau in Regulating the Effects of External Stimuli on Adult Hippocampal Neurogenesis. EMBO J. 2016, 35, 1417–1436. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Terro, F. Post-Translational Modifications of Tau Protein: Implications for Alzheimer’s Disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Tempra, C.; Scollo, F.; Milardi, D.; La Rosa, C. Amyloid Growth and Membrane Damage: Current Themes and Emerging Perspectives from Theory and Experiments on Aβ and HIAPP. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1625–1638. [Google Scholar] [CrossRef]
- García-Viñuales, S.; Sciacca, M.F.M.; Lanza, V.; Santoro, A.M.; Grasso, G.; Tundo, G.R.; Sbardella, D.; Coletta, M.; Grasso, G.; La Rosa, C.; et al. The Interplay between Lipid and Aβ Amyloid Homeostasis in Alzheimer’s Disease: Risk Factors and Therapeutic Opportunities. Chem. Phys. Lipids 2021, 236, 105072. [Google Scholar] [CrossRef]
- Drolle, E.; Negoda, A.; Hammond, K.; Pavlov, E.; Leonenko, Z. Changes in Lipid Membranes May Trigger Amyloid Toxicity in Alzheimer’s Disease. PLoS ONE 2017, 12, e0182194. [Google Scholar] [CrossRef]
- Künze, G.; Barré, P.; Scheidt, H.A.; Thomas, L.; Eliezer, D.; Huster, D. Binding of the Three-Repeat Domain of Tau to Phospholipid Membranes Induces an Aggregated-like State of the Protein. Biochim. Biophys. Acta 2012, 1818, 2302–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, T.; Eidenmüller, J.; Brandt, R. Interaction of Tau with the Neural Membrane Cortex Is Regulated by Phosphorylation at Sites That Are Modified in Paired Helical Filaments. J. Biol. Chem. 2000, 275, 15733–15740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, R.; Léger, J.; Lee, G. Interaction of Tau with the Neural Plasma Membrane Mediated by Tau’s Amino-Terminal Projection Domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef]
- Sallaberry, C.A.; Voss, B.J.; Majewski, J.; Biernat, J.; Mandelkow, E.; Chi, E.Y.; Vander Zanden, C.M. Tau and Membranes: Interactions That Promote Folding and Condensation. Front. Cell Dev. Biol. 2021, 2632. [Google Scholar] [CrossRef] [PubMed]
- Ait-Bouziad, N.; Lv, G.; Mahul-Mellier, A.-L.; Xiao, S.; Zorludemir, G.; Eliezer, D.; Walz, T.; Lashuel, H.A. Discovery and Characterization of Stable and Toxic Tau/Phospholipid Oligomeric Complexes. Nat. Commun. 2017, 8, 1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbaum-Garfinkle, S.; Ramlall, T.; Rhoades, E. The Role of the Lipid Bilayer in Tau Aggregation. Biophys. J. 2010, 98, 2722–2730. [Google Scholar] [CrossRef] [Green Version]
- Katsinelos, T.; Zeitler, M.; Dimou, E.; Karakatsani, A.; Müller, H.-M.; Nachman, E.; Steringer, J.P.; Ruiz de Almodovar, C.; Nickel, W.; Jahn, T.R. Unconventional Secretion Mediates the Trans-Cellular Spreading of Tau. Cell Rep. 2018, 23, 2039–2055. [Google Scholar] [CrossRef]
- Fabiani, C.; Antollini, S.S. Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Beta-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts. Front. Cell. Neurosci. 2019, 13, 309. [Google Scholar] [CrossRef] [Green Version]
- Lahdo, R.; De La Fournière-Bessueille, L. Insertion of the Amyloid Precursor Protein into Lipid Monolayers: Effects of Cholesterol and Apolipoprotein E. Biochem. J. 2004, 382, 987–994. [Google Scholar] [CrossRef]
- Matsuzaki, K. Physicochemical Interactions of Amyloid β-Peptide with Lipid Bilayers. Biochim. Biophys. Acta BBA-Biomembr. 2007, 1768, 1935–1942. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.-S.; Chen, L.-Y.; Wang, S.S.S.; Chang, Y.; Chen, W.-Y. Examining the Levels of Ganglioside and Cholesterol in Cell Membrane on Attenuation the Cytotoxicity of Beta-Amyloid Peptide. Colloids Surf. B Biointerfaces 2008, 65, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Fabelo, N.; Martín, V.; Marín, R.; Moreno, D.; Ferrer, I.; Díaz, M. Altered Lipid Composition in Cortical Lipid Rafts Occurs at Early Stages of Sporadic Alzheimer’s Disease and Facilitates APP/BACE1 Interactions. Neurobiol. Aging 2014, 35, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Kosicek, M.; Hecimovic, S. Phospholipids and Alzheimer’s Disease: Alterations, Mechanisms and Potential Biomarkers. Int. J. Mol. Sci. 2013, 14, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Beel, A.J.; Sakakura, M.; Barrett, P.J.; Sanders, C.R. Direct Binding of Cholesterol to the Amyloid Precursor Protein: An Important Interaction in Lipid-Alzheimer’s Disease Relationships? Biochim. Biophys. Acta 2010, 1801, 975–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.R. Cholesterol Binding to Amyloid-Beta Fibrils: A TEM Study. Micron. Oxf. Engl. 2008, 39, 1192–1196. [Google Scholar] [CrossRef]
- Barrett, P.J.; Song, Y.; Van Horn, W.D.; Hustedt, E.J.; Schafer, J.M.; Hadziselimovic, A.; Beel, A.J.; Sanders, C.R. The Amyloid Precursor Protein Has a Flexible Transmembrane Domain and Binds Cholesterol. Science 2012, 336, 1168–1171. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, M.; Matsuzaki, K. Formation of Amyloids by Aβ-(1–42) on NGF-Differentiated PC12 Cells: Roles of Gangliosides and Cholesterol. J. Mol. Biol. 2007, 371, 924–933. [Google Scholar] [CrossRef]
- Merezhko, M.; Brunello, C.A.; Yan, X.; Vihinen, H.; Jokitalo, E.; Uronen, R.-L.; Huttunen, H.J. Secretion of Tau via an Unconventional Non-Vesicular Mechanism. Cell Rep. 2018, 25, 2027–2035. [Google Scholar] [CrossRef] [Green Version]
- Feringa, F.M.; van der Kant, R. Cholesterol and Alzheimer’s Disease; From Risk Genes to Pathological Effects. Front. Aging Neurosci. 2021, 13, 333. [Google Scholar] [CrossRef]
- Matsuzaki, K. How Do Membranes Initiate Alzheimer’s Disease? Formation of Toxic Amyloid Fibrils by the Amyloid β-Protein on Ganglioside Clusters. Acc. Chem. Res. 2014, 47, 2397–2404. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Kato, K.; Yanagisawa, K. Aβ Polymerization through Interaction with Membrane Gangliosides. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2010, 1801, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, M.; Okada, T.; Kozutsumi, Y.; Matsuzaki, K. GM1 Ganglioside-Mediated Accumulation of Amyloid β-Protein on Cell Membranes. Biochem. Biophys. Res. Commun. 2005, 328, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric Amyloid β Protein Rapidly Accumulates in Lipid Rafts Followed by Apolipoprotein E and Phosphorylated Tau Accumulation in the Tg2576 Mouse Model of Alzheimer’s Disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Krämer, E.-M.; Cardine, A.-M.; Schraven, B.; Brandt, R.; Trotter, J. Process Outgrowth of Oligodendrocytes Is Promoted by Interaction of Fyn Kinase with the Cytoskeletal Protein Tau. J. Neurosci. 2002, 22, 698–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, R.; Usardi, A.; Hanger, D.P.; Anderton, B.H. Membrane-Bound β-Amyloid Oligomers Are Recruited into Lipid Rafts by a Fyn-Dependent Mechanism. FASEB J. 2008, 22, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- Campos-Peña, V.; Tapia-Ramírez, J.; Sánchez-Torres, C.; Meraz-Rios, M.A. Pathological-like Assembly of Tau Induced by a Paired Helical Filament Core Expressed at the Plasma Membrane. J. Alzheimers Dis. JAD 2009, 18, 919–933. [Google Scholar] [CrossRef]
- Jones, E.M.; Dubey, M.; Camp, P.J.; Vernon, B.C.; Biernat, J.; Mandelkow, E.; Majewski, J.; Chi, E.Y. Interaction of Tau Protein with Model Lipid Membranes Induces Tau Structural Compaction and Membrane Disruption. Biochemistry 2012, 51, 2539–2550. [Google Scholar] [CrossRef] [Green Version]
- Kawarabayashi, T.; Nakamura, T.; Sato, K.; Seino, Y.; Ichii, S.; Nakahata, N.; Takatama, M.; Westaway, D.; George-Hyslop, P.S.; Shoji, M. Lipid Rafts Act as a Common Platform for Amyloid-β Oligomer-Induced Alzheimer’s Disease Pathology. J. Alzheimers Dis. JAD 2022, 87, 1189–1203. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing Endogenous Tau Ameliorates Amyloid Beta-Induced Deficits in an Alzheimer’s Disease Mouse Model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Ferreira, A. The Generation of a 17 KDa Neurotoxic Fragment: An Alternative Mechanism by Which Tau Mediates β-Amyloid-Induced Neurodegeneration. J. Neurosci. 2005, 25, 5365–5375. [Google Scholar] [CrossRef]
- Nicholson, A.M.; Ferreira, A. Increased Membrane Cholesterol Might Render Mature Hippocampal Neurons More Susceptible to β-Amyloid-Induced Calpain Activation and Tau Toxicity. J. Neurosci. 2009, 29, 4640–4651. [Google Scholar] [CrossRef] [PubMed]
- Wray, S.; Noble, W. Linking Amyloid and Tau Pathology in Alzheimer’s Disease: The Role of Membrane Cholesterol in Aβ-Mediated Tau Toxicity. J. Neurosci. 2009, 29, 9665–9667. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Jenner, P.; Schapira, A.H.; Marsden, C.D. Alterations in Levels of Iron, Ferritin, and Other Trace Metals in Neurodegenerative Diseases Affecting the Basal Ganglia. Ann. Neurol. 1992, 32 (Suppl. 1), S94–S100. [Google Scholar] [CrossRef] [PubMed]
- Cuajungco, M.P.; Goldstein, L.E.; Nunomura, A.; Smith, M.A.; Lim, J.T.; Atwood, C.S.; Huang, X.; Farrag, Y.W.; Perry, G.; Bush, A.I. Evidence That the β-Amyloid Plaques of Alzheimer’s Disease Represent the Redox-Silencing and Entombment of Aβ by Zinc. J. Biol. Chem. 2000, 275, 19439–19442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogh, B.D.; Szakács, B.; Di Natale, G.; Tabbì, G.; Pappalardo, G.; Sóvágó, I.; Várnagy, K. Copper (II) Binding Properties of an Octapeptide Fragment from the R3 Region of Tau Protein: A Combined Potentiometric, Spectroscopic and Mass Spectrometric Study. J. Inorg. Biochem. 2021, 217, 111358. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Wu, B.; Song, R.; Zhu, S.; Simpson, A.; Wilson, D.J.; Kraatz, H.B. Exploring the Interactions of Iron and Zinc with the Microtubule Binding Repeats R1 and R4. J. Inorg. Biochem. 2020, 205, 110987. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Zhu, S.; Sharma, R.; Wilson, D.J.; Kraatz, H.B. Interaction of Metal Ions with Tau Protein. The Case for a Metal-Mediated Tau Aggregation. J. Inorg. Biochem. 2019, 194, 44–51. [Google Scholar] [CrossRef]
- Ma, Q.F.; Li, Y.M.; Du, J.T.; Kanazawa, K.; Nemoto, T.; Nakanishi, H.; Zhao, Y.F. Binding of Copper (II) Ion to an Alzheimer’s Tau Peptide as Revealed by MALDI-TOF MS, CD, and NMR. Biopolymers 2005, 79, 74–85. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Di Natale, G.; Tosto, R.; Milardi, D.; Pappalardo, G. Tau/Aβ Chimera Peptides: Evaluating the Dual Function of Metal Coordination and Membrane Interaction in One Sequence. J. Inorg. Biochem. 2020, 205, 110996. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Paradis, M.D.; Tanzi, R.E. Modulation of A Beta Adhesiveness and Secretase Site Cleavage by Zinc. J. Biol. Chem. 1994, 269, 12152–12158. [Google Scholar] [CrossRef]
- Jakob-Roetne, R.; Jacobsen, H. Alzheimer’s Disease: From Pathology to Therapeutic Approaches. Angew. Chem. Int. Ed. 2009, 48, 3030–3059. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Lee, S.; Liu, Y.; Lim, M.H. Untangling Amyloid-β, Tau, and Metals in Alzheimer’s Disease. ACS Chem. Biol. 2013, 8, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Luo, D.-J.; Wang, X.-L.; Qiu, M.; Yang, Y.; Yan, X.; Wang, J.-Z.; Ye, Q.-F.; Liu, R. Zinc Binds to and Directly Inhibits Protein Phosphatase 2A in Vitro. Neurosci. Bull. 2015, 31, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Boom, A.; Authelet, M.; Dedecker, R.; Frédérick, C.; Van Heurck, R.; Daubie, V.; Leroy, K.; Pochet, R.; Brion, J.-P. Bimodal Modulation of Tau Protein Phosphorylation and Conformation by Extracellular Zn2+ in Human-Tau Transfected Cells. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2009, 1793, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Neve, R.L.; Kosik, K.S. The Microtubule Binding Domain of Tau Protein. Neuron 1989, 2, 1615–1624. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal Dyshomeostasis and Oxidative Stress in Alzheimer’s Disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, Z.; Cao, Y.; Lang, M.; Lu, B.; Zhou, B. Zinc Binding Directly Regulates Tau Toxicity Independent of Tau Hyperphosphorylation. Cell Rep. 2014, 8, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Grasso, G.; Santoro, A.M.; Lanza, V.; Sbardella, D.; Tundo, G.R.; Ciaccio, C.; Marini, S.; Coletta, M.; Milardi, D. The Double Faced Role of Copper in Aβ Homeostasis: A Survey on the Interrelationship between Metal Dyshomeostasis, UPS Functioning and Autophagy in Neurodegeneration. Coord. Chem. Rev. 2017, 347, 1–22. [Google Scholar] [CrossRef]
- Kepp, K.P. Bioinorganic Chemistry of Alzheimer’s Disease. Chem. Rev. 2012, 112, 5193–5239. [Google Scholar] [CrossRef] [Green Version]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in Neurobiology: Probing Their Chemistry and Biology with Molecular Imaging. Chem. Rev. 2008, 108, 1517–1549. [Google Scholar] [CrossRef]
- Duce, J.A.; Bush, A.I. Biological Metals and Alzheimer’s Disease: Implications for Therapeutics and Diagnostics. Prog. Neurobiol. 2010, 92, 1–18. [Google Scholar] [CrossRef]
- Cheignon, C.; Jones, M.; Atrián-Blasco, E.; Kieffer, I.; Faller, P.; Collin, F.; Hureau, C. Identification of Key Structural Features of the Elusive Cu–Aβ Complex That Generates ROS in Alzheimer’s Disease. Chem. Sci. 2017, 8, 5107–5118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskici, G.; Axelsen, P.H. Copper and Oxidative Stress in the Pathogenesis of Alzheimer’s Disease. Biochemistry 2012, 51, 6289–6311. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Vitek, M.P.; Mason, R.P. Cupric–Amyloid β Peptide Complex Stimulates Oxidation of Ascorbate and Generation of Hydroxyl Radical. Free Radic. Biol. Med. 2004, 36, 340–347. [Google Scholar] [CrossRef]
- Ali, F.E.; Separovic, F.; Barrow, C.J.; Cherny, R.A.; Fraser, F.; Bush, A.I.; Masters, C.L.; Barnham, K.J. Methionine Regulates Copper/Hydrogen Peroxide Oxidation Products of Aβ. J. Pept. Sci. 2005, 11, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Cassagnes, L.-E.; Hervé, V.; Nepveu, F.; Hureau, C.; Faller, P.; Collin, F. The Catalytically Active Copper-Amyloid-Beta State: Coordination Site Responsible for Reactive Oxygen Species Production. Angew. Chem. Int. Ed. 2013, 52, 11110–11113. [Google Scholar] [CrossRef]
- Kim, A.C.; Lim, S.; Kim, Y.K. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci. 2018, 19, 128. [Google Scholar] [CrossRef] [Green Version]
- Sarell, C.J.; Wilkinson, S.R.; Viles, J.H. Substoichiometric Levels of Cu2+ Ions Accelerate the Kinetics of Fiber Formation and Promote Cell Toxicity of Amyloid-{beta} from Alzheimer Disease. J. Biol. Chem. 2010, 285, 41533–41540. [Google Scholar] [CrossRef] [Green Version]
- Garzon-Rodriguez, W.; Yatsimirsky, A.K.; Glabe, C.G. Binding of Zn(II), Cu(II), and Fe(II) Ions to Alzheimer’s Aß Peptide Studied by Fluorescence. Bioorg. Med. Chem. Lett. 1999, 9, 2243–2248. [Google Scholar] [CrossRef]
- Atwood, C.S.; Scarpa, R.C.; Huang, X.; Moir, R.D.; Jones, W.D.; Fairlie, D.P.; Tanzi, R.E.; Bush, A.I. Characterization of Copper Interactions with Alzheimer Amyloid Beta Peptides: Identification of an Attomolar-Affinity Copper Binding Site on Amyloid Beta1-42. J. Neurochem. 2000, 75, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Zambelli, B.; Mukrasch, M.D.; Biernat, J.; Jeganathan, S.; Griesinger, C.; Ciurli, S.; Mandelkow, E.; Zweckstetter, M. Structural Characterization of Binding of Cu(II) to Tau Protein. Biochemistry 2008, 47, 10841–10851. [Google Scholar] [CrossRef] [PubMed]
- Bacchella, C.; Gentili, S.; Bellotti, D.; Quartieri, E.; Draghi, S.; Baratto, M.C.; Remelli, M.; Valensin, D.; Monzani, E.; Nicolis, S.; et al. Binding and Reactivity of Copper to R1 and R3 Fragments of Tau Protein. Inorg. Chem. 2020, 59, 274–286. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45. [Google Scholar] [CrossRef]
- Hung, Y.H.; Bush, A.I.; Cherny, R.A. Copper in the Brain and Alzheimer’s Disease. JBIC J. Biol. Inorg. Chem. 2010, 15, 61–76. [Google Scholar] [CrossRef]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s Disease: Synaptic Dysfunction and Aβ. Mol. Neurodegener. 2009, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.-C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau Association with Synaptic Vesicles Causes Presynaptic Dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C. Bioinorganic Chemistry of Copper and Zinc Ions Coordinated to Amyloid-β Peptide. Dalton Trans. 2009, 1080–1094. [Google Scholar] [CrossRef]
- Faller, P. Copper and Zinc Binding to Amyloid-Beta: Coordination, Dynamics, Aggregation, Reactivity and Metal-Ion Transfer. Chembiochem Eur. J. Chem. Biol. 2009, 10, 2837–2845. [Google Scholar] [CrossRef]
- Syme, C.D.; Nadal, R.C.; Rigby, S.E.J.; Viles, J.H. Copper Binding to the Amyloid-Beta (Abeta) Peptide Associated with Alzheimer’s Disease: Folding, Coordination Geometry, PH Dependence, Stoichiometry, and Affinity of Abeta-(1-28): Insights from a Range of Complementary Spectroscopic Techniques. J. Biol. Chem. 2004, 279, 18169–18177. [Google Scholar] [CrossRef] [Green Version]
- Kowalik-Jankowska, T.; Ruta, M.; Wiśniewska, K.; Lankiewicz, L. Coordination Abilities of the 1-16 and 1-28 Fragments of Beta-Amyloid Peptide towards Copper(II) Ions: A Combined Potentiometric and Spectroscopic Study. J. Inorg. Biochem. 2003, 95, 270–282. [Google Scholar] [CrossRef]
- Damante, C.A.; Ösz, K.; Nagy, Z.; Pappalardo, G.; Grasso, G.; Impellizzeri, G.; Rizzarelli, E.; Sóvágó, I. The Metal Loading Ability of β-Amyloid N-Terminus: A Combined Potentiometric and Spectroscopic Study of Copper(II) Complexes with β-Amyloid(1−16), Its Short or Mutated Peptide Fragments, and Its Polyethylene Glycol (PEG)-Ylated Analogue. Inorg. Chem. 2008, 47, 9669–9683. [Google Scholar] [CrossRef]
- Natale, G.D.; Sinopoli, A.; Grenács, Á.; Sanna, D.; Sóvágó, I.; Pappalardo, G. Copper(II) Coordination Properties of the Aβ(1–16)2 Peptidomimetic: Experimental Evidence of Intermolecular Macrochelate Complex Species in the Aβ Dimer. New J. Chem. 2016, 40, 10274–10284. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Long, F.; Miller, Y.; Xiao, Y.; McElheny, D.; Thurber, K.; Ma, B.; Nussinov, R.; Ishii, Y. Molecular-Level Examination of Cu2+ Binding Structure for Amyloid Fibrils of 40-Residue Alzheimer’s β by Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2011, 133, 3390–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damante, C.A.; Osz, K.; Nagy, Z.; Pappalardo, G.; Grasso, G.; Impellizzeri, G.; Rizzarelli, E.; Sóvágó, I. Metal Loading Capacity of Abeta N-Terminus: A Combined Potentiometric and Spectroscopic Study of Zinc(II) Complexes with Abeta(1-16), Its Short or Mutated Peptide Fragments and Its Polyethylene Glycol-Ylated Analogue. Inorg. Chem. 2009, 48, 10405–10415. [Google Scholar] [CrossRef] [PubMed]
- Mithu, V.S.; Sarkar, B.; Bhowmik, D.; Chandrakesan, M.; Maiti, S.; Madhu, P.K. Zn(++) Binding Disrupts the Asp(23)-Lys(28) Salt Bridge without Altering the Hairpin-Shaped Cross-β Structure of Aβ(42) Amyloid Aggregates. Biophys. J. 2011, 101, 2825–2832. [Google Scholar] [CrossRef] [Green Version]
- Damante, C.A.; Ősz, K.; Nagy, Z.; Grasso, G.; Pappalardo, G.; Rizzarelli, E.; Sóvágó, I. Zn2+’s Ability to Alter the Distribution of Cu2+ among the Available Binding Sites of Aβ(1-16)-Polyethylenglycol-Ylated Peptide: Implications in Alzheimer’s Disease. Inorg. Chem. 2011, 50, 5342–5350. [Google Scholar] [CrossRef]
- Moreira, P.; Pereira, C.; Santos, M.S.; Oliveira, C. Effect of Zinc Ions on the Cytotoxicity Induced by the Amyloid β-Peptide. Antioxid. Redox Signal. 2000, 2, 317–325. [Google Scholar] [CrossRef]
- Di Natale, G.; Bellia, F.; Sciacca, M.F.M.; Campagna, T.; Pappalardo, G. Tau-Peptide Fragments and Their Copper(II) Complexes: Effects on Amyloid-β Aggregation. Inorganica Chim. Acta 2018, 472, 82–92. [Google Scholar] [CrossRef]
- Lukács, M.; Szunyog, G.; Grenács, A.; Lihi, N.; Kállay, C.; Di Natale, G.; Campagna, T.; Lanza, V.; Tabbì, G.; Pappalardo, G.; et al. Copper(II) Coordination Abilities of the N-Terminal Peptide Fragments of Tau Protein: A Combined Potentiometric, Spectroscopic and ESI-MS Study. ChemPlusChem 2019, 84, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Magrì, A.; Di Natale, G.; Rizzarelli, E. Copper-Assisted Interaction between Amyloid-β and Prion: Ternary Metal Complexes with Aβ N-Terminus and Octarepeat. Inorganica Chim. Acta 2018, 472, 93–102. [Google Scholar] [CrossRef]
- Di Natale, G.; Pappalardo, G.; Milardi, D.; Sciacca, M.F.M.; Attanasio, F.; La Mendola, D.; Rizzarelli, E. Membrane Interactions and Conformational Preferences of Human and Avian Prion N-Terminal Tandem Repeats: The Role of Copper(II) Ions, PH, and Membrane Mimicking Environments. J. Phys. Chem. B 2010, 114, 13830–13838. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Shi, C.; di Lorenzo, D.; Van Baelen, A.-C.; Tonali, N. The Positive Side of the Alzheimer’s Disease Amyloid Cross-Interactions: The Case of the Aβ 1-42 Peptide with Tau, TTR, CysC, and ApoA1. Molecules 2020, 25, 2439. [Google Scholar] [CrossRef] [PubMed]
- Subedi, S.; Sasidharan, S.; Nag, N.; Saudagar, P.; Tripathi, T. Amyloid Cross-Seeding: Mechanism, Implication, and Inhibition. Molecules 2022, 27, 1776. [Google Scholar] [CrossRef] [PubMed]
- Roda, A.R.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-Beta Peptide and Tau Protein Crosstalk in Alzheimer’s Disease. Neural Regen. Res. 2022, 17, 1666. [Google Scholar]
- Malafaia, D.; Albuquerque, H.M.T.; Silva, A.M.S. Amyloid-β and Tau Aggregation Dual-Inhibitors: A Synthetic and Structure-Activity Relationship Focused Review. Eur. J. Med. Chem. 2021, 214, 113209. [Google Scholar] [CrossRef]
- Szałaj, N.; Godyń, J.; Jończyk, J.; Pasieka, A.; Panek, D.; Wichur, T.; Więckowski, K.; Zaręba, P.; Bajda, M.; Pislar, A.; et al. Multidirectional in Vitro and in Cellulo Studies as a Tool for Identification of Multi-Target-Directed Ligands Aiming at Symptoms and Causes of Alzheimer’s Disease. J. Enzyme Inhib. Med. Chem. 2020, 35, 1944. [Google Scholar] [CrossRef]
- Lv, P.; Xia, C.-L.; Wang, N.; Liu, Z.-Q.; Huang, Z.-S.; Huang, S.-L. Synthesis and Evaluation of 1,2,3,4-Tetrahydro-1-Acridone Analogues as Potential Dual Inhibitors for Amyloid-Beta and Tau Aggregation. Bioorg. Med. Chem. 2018, 26, 4693–4705. [Google Scholar] [CrossRef]
- Okuda, M.; Fujita, Y.; Hijikuro, I.; Wada, M.; Uemura, T.; Kobayashi, Y.; Waku, T.; Tanaka, N.; Nishimoto, T.; Izumi, Y.; et al. PE859, A Novel Curcumin Derivative, Inhibits Amyloid-β and Tau Aggregation, and Ameliorates Cognitive Dysfunction in Senescence-Accelerated Mouse Prone 8. J. Alzheimers Dis. JAD 2017, 59, 313–328. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Di Natale, G.; Milardi, D.; Pappalardo, G. Tau/Aβ Chimera Peptides: A Thioflavin-T and MALDI-TOF Study of Aβ Amyloidosis in the Presence of Cu(II) or Zn(II) Ions and Total Lipid Brain Extract (TLBE) Vesicles. Chem. Phys. Lipids 2021, 237, 105085. [Google Scholar] [CrossRef]
- Guo, J.-P.; Arai, T.; Miklossy, J.; McGeer, P.L. Aβ and Tau Form Soluble Complexes That May Promote Self Aggregation of Both into the Insoluble Forms Observed in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2006, 103, 1953–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadros, R.; Pérez, M.; Ruiz-Gabarre, D.; Hernández, F.; García-Escudero, V.; Avila, J. Specific Peptide from the Novel W-Tau Isoform Inhibits Tau and Amyloid β Peptide Aggregation In Vitro. ACS Chem. Neurosci. 2022, 13, 1974–1978. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.; Ma, B.; Nussinov, R. Synergistic Interactions between Repeats in Tau Protein and Aβ Amyloids May Be Responsible for Accelerated Aggregation via Polymorphic States. Biochemistry 2011, 50, 5172–5181. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, L.O.; Callaway, D.J.E.; Tjernberg, A.; Hahne, S.; Lilliehöök, C.; Terenius, L.; Thyberg, J.; Nordstedt, C. A Molecular Model of Alzheimer Amyloid β-Peptide Fibril Formation. J. Biol. Chem. 1999, 274, 12619–12625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, S.L.; Wyttenbach, T.; Baumketner, A.; Shea, J.-E.; Bitan, G.; Teplow, D.B.; Bowers, M.T. Amyloid Beta-Protein: Monomer Structure and Early Aggregation States of Abeta42 and Its Pro19 Alloform. J. Am. Chem. Soc. 2005, 127, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.E.; Vadukul, D.M.; Dahal, L.; Theisen, A.; Fowler, M.W.; Al-Hilaly, Y.; Ford, L.; Kemenes, G.; Day, I.J.; Staras, K.; et al. A Critical Role for the Self-Assembly of Amyloid-Β1-42 in Neurodegeneration. Sci. Rep. 2016, 6, 30182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjernberg, L.O.; Näslund, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of Beta-Amyloid Fibril Formation by a Pentapeptide Ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [Green Version]
- Soto, C.; Kindy, M.S.; Baumann, M.; Frangione, B. Inhibition of Alzheimer’s Amyloidosis by Peptides That Prevent Beta-Sheet Conformation. Biochem. Biophys. Res. Commun. 1996, 226, 672–680. [Google Scholar] [CrossRef]
- Funke, S.A.; Willbold, D. Peptides for Therapy and Diagnosis of Alzheimer’s Disease. Curr. Pharm. Des. 2012, 18, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.J.; Sciarretta, K.L.; Meredith, S.C. Inhibition of β-Amyloid(40) Fibrillogenesis and Disassembly of β-Amyloid(40) Fibrils by Short β-Amyloid Congeners Containing N-Methyl Amino Acids at Alternate Residues. Biochemistry 2001, 40, 8237–8245. [Google Scholar] [CrossRef]
- Soto, P.; Griffin, M.A.; Shea, J.-E. New Insights into the Mechanism of Alzheimer Amyloid-β Fibrillogenesis Inhibition by N-Methylated Peptides. Biophys. J. 2007, 93, 3015–3025. [Google Scholar] [CrossRef] [Green Version]
- Kokkoni, N.; Stott, K.; Amijee, H.; Mason, J.M.; Doig, A.J. N-Methylated Peptide Inhibitors of β-Amyloid Aggregation and Toxicity. Optimization of the Inhibitor Structure. Biochemistry 2006, 45, 9906–9918. [Google Scholar] [CrossRef] [PubMed]
- Bett, C.K.; Serem, W.K.; Fontenot, K.R.; Hammer, R.P.; Garno, J.C. Effects of Peptides Derived from Terminal Modifications of the Aβ Central Hydrophobic Core on Aβ Fibrillization. ACS Chem. Neurosci. 2010, 1, 661–678. [Google Scholar] [CrossRef] [Green Version]
- Pallitto, M.M.; Ghanta, J.; Heinzelman, P.; Kiessling, L.L.; Murphy, R.M. Recognition Sequence Design for Peptidyl Modulators of β-Amyloid Aggregation and Toxicity. Biochemistry 1999, 38, 3570–3578. [Google Scholar] [CrossRef] [PubMed]
- Austen, B.M.; Paleologou, K.E.; Ali, S.A.E.; Qureshi, M.M.; Allsop, D.; El-Agnaf, O.M.A. Designing Peptide Inhibitors for Oligomerization and Toxicity of Alzheimer’s β-Amyloid Peptide. Biochemistry 2008, 47, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Chafekar, S.M.; Malda, H.; Merkx, M.; Meijer, E.W.; Viertl, D.; Lashuel, H.A.; Baas, F.; Scheper, W. Branched KLVFF Tetramers Strongly Potentiate Inhibition of β-Amyloid Aggregation. ChemBioChem 2007, 8, 1857–1864. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Leibowitz, M.J.; Sinko, P.J.; Stein, S. Multiple-Peptide Conjugates for Binding Beta-Amyloid Plaques of Alzheimer’s Disease. Bioconjug. Chem. 2003, 14, 86–92. [Google Scholar] [CrossRef]
- Kino, R.; Araya, T.; Arai, T.; Sohma, Y.; Kanai, M. Covalent Modifier-Type Aggregation Inhibitor of Amyloid-β Based on a Cyclo-KLVFF Motif. Bioorg. Med. Chem. Lett. 2015, 25, 2972–2975. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Sasaki, D.; Araya, T.; Sato, T.; Sohma, Y.; Kanai, M. A Cyclic KLVFF-Derived Peptide Aggregation Inhibitor Induces the Formation of Less-Toxic off-Pathway Amyloid-β Oligomers. Chembiochem Eur. J. Chem. Biol. 2014, 15, 2577–2583. [Google Scholar] [CrossRef]
- Villari, V.; Tosto, R.; Natale, G.D.; Sinopoli, A.; Tomasello, M.F.; Lazzaro, S.; Micali, N.; Pappalardo, G. A Metalloporphyrin-Peptide Conjugate as an Effective Inhibitor of Amyloid-β Peptide Fibrillation and Cytotoxicity. ChemistrySelect 2017, 2, 9122–9129. [Google Scholar] [CrossRef]
- Rocha, S.; Cardoso, I.; Börner, H.; Pereira, M.C.; Saraiva, M.J.; Coelho, M. Design and Biological Activity of β-Sheet Breaker Peptide Conjugates. Biochem. Biophys. Res. Commun. 2009, 380, 397–401. [Google Scholar] [CrossRef]
- Giordano, C.; Sansone, A.; Masi, A.; Masci, A.; Mosca, L.; Chiaraluce, R.; Pasquo, A.; Consalvi, V. Inhibition of Amyloid Peptide Fragment Aβ25–35 Fibrillogenesis and Toxicity by N-Terminal β-Amino Acid-Containing Esapeptides: Is Taurine Moiety Essential for In Vivo Effects? Chem. Biol. Drug Des. 2012, 79, 30–37. [Google Scholar] [CrossRef]
- Adessi, C.; Frossard, M.-J.; Boissard, C.; Fraga, S.; Bieler, S.; Ruckle, T.; Vilbois, F.; Robinson, S.M.; Mutter, M.; Banks, W.A.; et al. Pharmacological Profiles of Peptide Drug Candidates for the Treatment of Alzheimer’s Disease. J. Biol. Chem. 2003, 278, 13905–13911. [Google Scholar] [CrossRef] [Green Version]
- Bona, P.D.; Giuffrida, M.L.; Caraci, F.; Copani, A.; Pignataro, B.; Attanasio, F.; Cataldo, S.; Pappalardo, G.; Rizzarelli, E. Design and Synthesis of New Trehalose-Conjugated Pentapeptides as Inhibitors of Aβ(1–42) Fibrillogenesis and Toxicity. J. Pept. Sci. 2009, 15, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Sinopoli, A.; Giuffrida, A.; Tomasello, M.F.; Giuffrida, M.L.; Leone, M.; Attanasio, F.; Caraci, F.; De Bona, P.; Naletova, I.; Saviano, M.; et al. Ac-LPFFD-Th: A Trehalose-Conjugated Peptidomimetic as a Strong Suppressor of Amyloid-β Oligomer Formation and Cytotoxicity. ChemBioChem 2016, 17, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, F.; Cascio, C.; Fisichella, S.; Nicoletti, V.G.; Pignataro, B.; Savarino, A.; Rizzarelli, E. Trehalose Effects on α-Crystallin Aggregates. Biochem. Biophys. Res. Commun. 2007, 354, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Gorantla, N.V.; Sunny, L.P.; Rajasekhar, K.; Nagaraju, P.G.; Cg, P.P.; Govindaraju, T.; Chinnathambi, S. Amyloid-β-Derived Peptidomimetics Inhibits Tau Aggregation. ACS Omega 2021, 6, 11131–11138. [Google Scholar] [CrossRef]
- Griner, S.L.; Seidler, P.; Bowler, J.; Murray, K.A.; Yang, T.P.; Sahay, S.; Sawaya, M.R.; Cascio, D.; Rodriguez, J.A.; Philipp, S.; et al. Structure-Based Inhibitors of Amyloid Beta Core Suggest a Common Interface with Tau. eLife 2019, 8, e46924. [Google Scholar] [CrossRef] [PubMed]
- Richman, M.; Wilk, S.; Chemerovski, M.; Wärmländer, S.K.T.S.; Wahlström, A.; Gräslund, A.; Rahimipour, S. In Vitro and Mechanistic Studies of an Antiamyloidogenic Self-Assembled Cyclic d,l-α-Peptide Architecture. J. Am. Chem. Soc. 2013, 135, 3474–3484. [Google Scholar] [CrossRef] [PubMed]
- Belostozky, A.; Richman, M.; Lisniansky, E.; Tovchygrechko, A.; Chill, J.H.; Rahimipour, S. Inhibition of Tau-Derived Hexapeptide Aggregation and Toxicity by a Self-Assembled Cyclic D,L-α-Peptide Conformational Inhibitor. Chem. Commun. 2018, 54, 5980–5983. [Google Scholar] [CrossRef] [Green Version]
- Aggidis, A.; Chatterjee, S.; Townsend, D.; Fullwood, N.J.; Ortega, E.R.; Tarutani, A.; Hasegawa, M.; Lucas, H.; Mudher, A.; Allsop, D. Peptide-Based Inhibitors of Tau Aggregation as a Potential Therapeutic for Alzheimer’s Disease and Other Tauopathies. bioRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Shamloo, A.; Asadbegi, M.; Khandan, V.; Amanzadi, A. Designing a New Multifunctional Peptide for Metal Chelation and Aβ Inhibition. Arch. Biochem. Biophys. 2018, 653, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, T.; Khan, H. Direct Interaction between the β-Amyloid Core and Tau Facilitates Cross-Seeding: A Novel Target for Therapeutic Intervention. Biochemistry 2020, 59, 341–342. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K. Extracellular Tau and Its Potential Role in the Propagation of Tau Pathology. Front. Neurosci. 2017, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Dickson, D.W.; Lin, W.-L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.-H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced Neurofibrillary Degeneration in Transgenic Mice Expressing Mutant Tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [Green Version]
- Seidler, P.M.; Boyer, D.R.; Rodriguez, J.A.; Sawaya, M.R.; Cascio, D.; Murray, K.; Gonen, T.; Eisenberg, D.S. Structure-Based Inhibitors of Tau Aggregation. Nat. Chem. 2018, 10, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Doğanyiğit, Z.; Erbakan, K.; Akyuz, E.; Polat, A.K.; Arulsamy, A.; Shaikh, M.F. The Role of Neuroinflammatory Mediators in the Pathogenesis of Traumatic Brain Injury: A Narrative Review. ACS Chem. Neurosci. 2022, 13, 1835–1848. [Google Scholar] [CrossRef]
- Majewski, J.; Jones, E.M.; Vander Zanden, C.M.; Biernat, J.; Mandelkow, E.; Chi, E.Y. Lipid Membrane Templated Misfolding and Self-Assembly of Intrinsically Disordered Tau Protein. Sci. Rep. 2020, 10, 13324. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Natale, G.; Sabatino, G.; Sciacca, M.F.M.; Tosto, R.; Milardi, D.; Pappalardo, G. Aβ and Tau Interact with Metal Ions, Lipid Membranes and Peptide-Based Amyloid Inhibitors: Are These Common Features Relevant in Alzheimer’s Disease? Molecules 2022, 27, 5066. https://doi.org/10.3390/molecules27165066
Di Natale G, Sabatino G, Sciacca MFM, Tosto R, Milardi D, Pappalardo G. Aβ and Tau Interact with Metal Ions, Lipid Membranes and Peptide-Based Amyloid Inhibitors: Are These Common Features Relevant in Alzheimer’s Disease? Molecules. 2022; 27(16):5066. https://doi.org/10.3390/molecules27165066
Chicago/Turabian StyleDi Natale, Giuseppe, Giuseppina Sabatino, Michele Francesco Maria Sciacca, Rita Tosto, Danilo Milardi, and Giuseppe Pappalardo. 2022. "Aβ and Tau Interact with Metal Ions, Lipid Membranes and Peptide-Based Amyloid Inhibitors: Are These Common Features Relevant in Alzheimer’s Disease?" Molecules 27, no. 16: 5066. https://doi.org/10.3390/molecules27165066
APA StyleDi Natale, G., Sabatino, G., Sciacca, M. F. M., Tosto, R., Milardi, D., & Pappalardo, G. (2022). Aβ and Tau Interact with Metal Ions, Lipid Membranes and Peptide-Based Amyloid Inhibitors: Are These Common Features Relevant in Alzheimer’s Disease? Molecules, 27(16), 5066. https://doi.org/10.3390/molecules27165066