
Singlet Fission, Polaron Generation and Intersystem Crossing in Hexaphenyl Film

Abstract
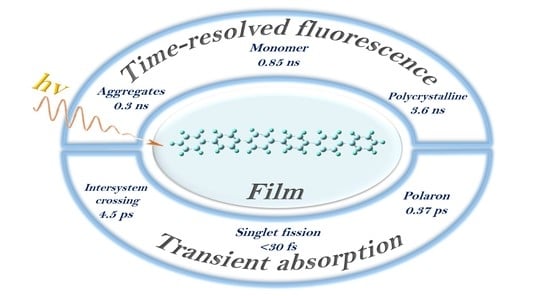
:
1. Introduction
2. Materials and Methods
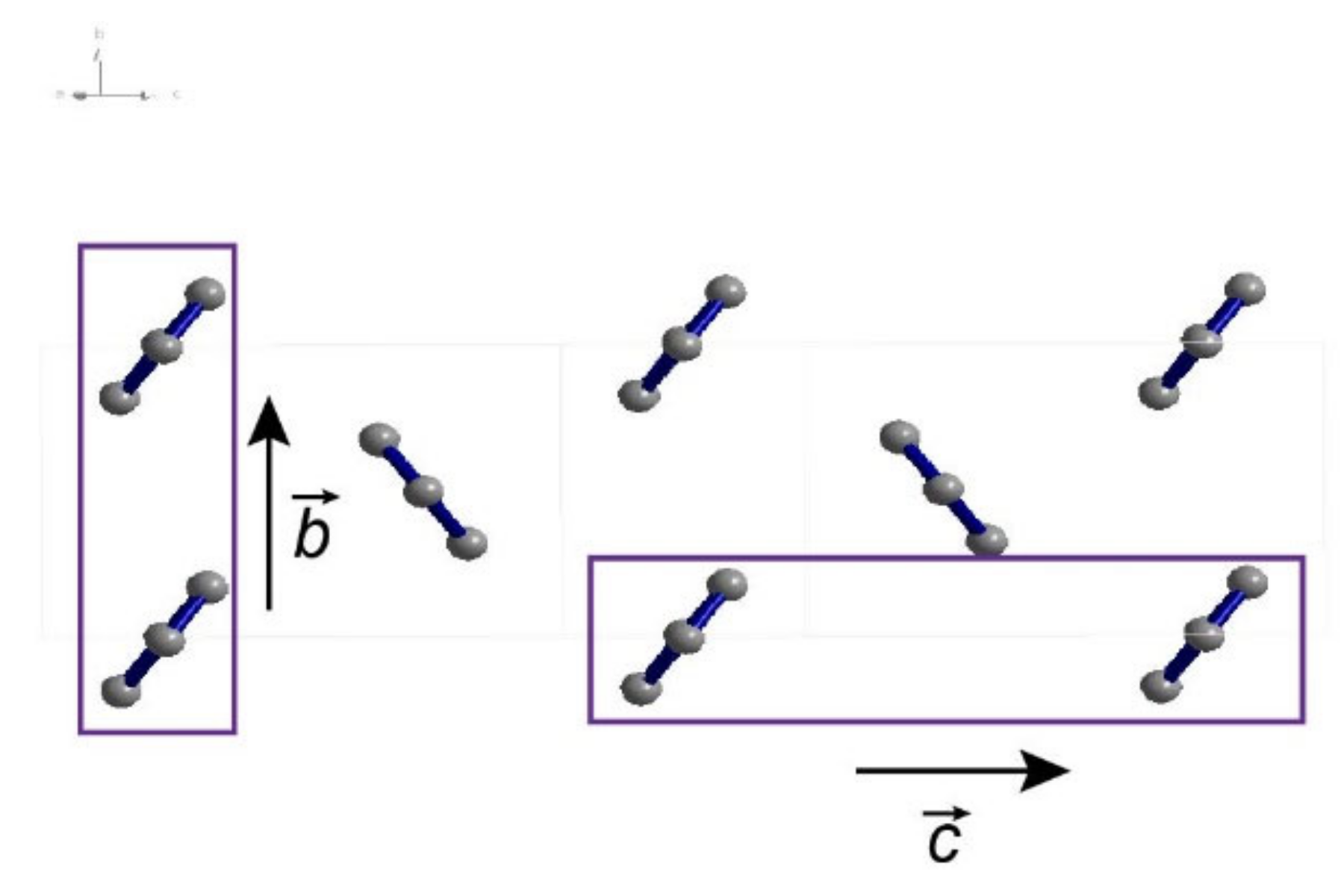
2.1. Preparation of the Hexaphenyl Thin Film
2.2. Characterization of the Hexaphenyl Thin Film
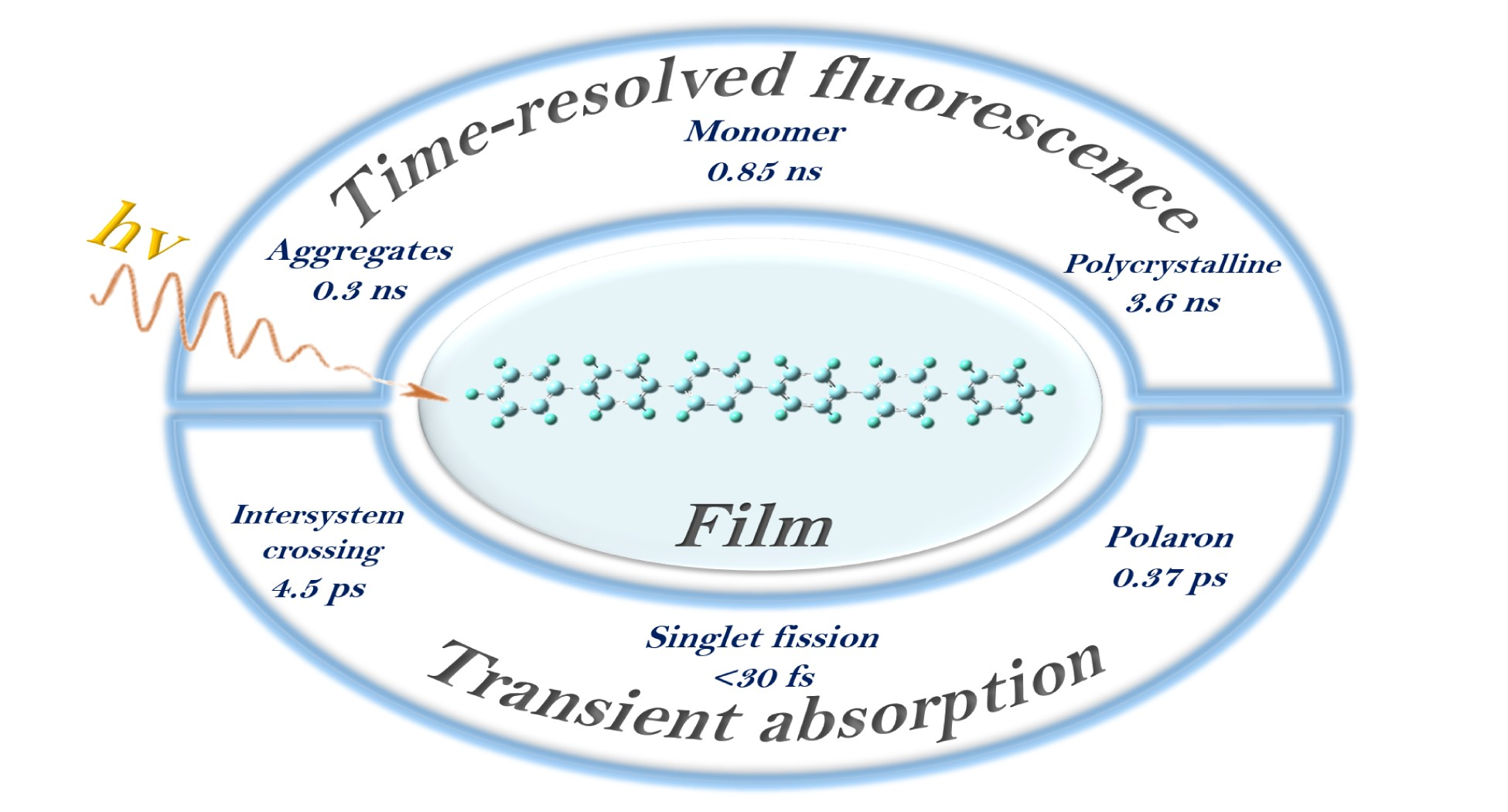
2.3. Steady-State Absorption and Fluorescence Spectroscopy
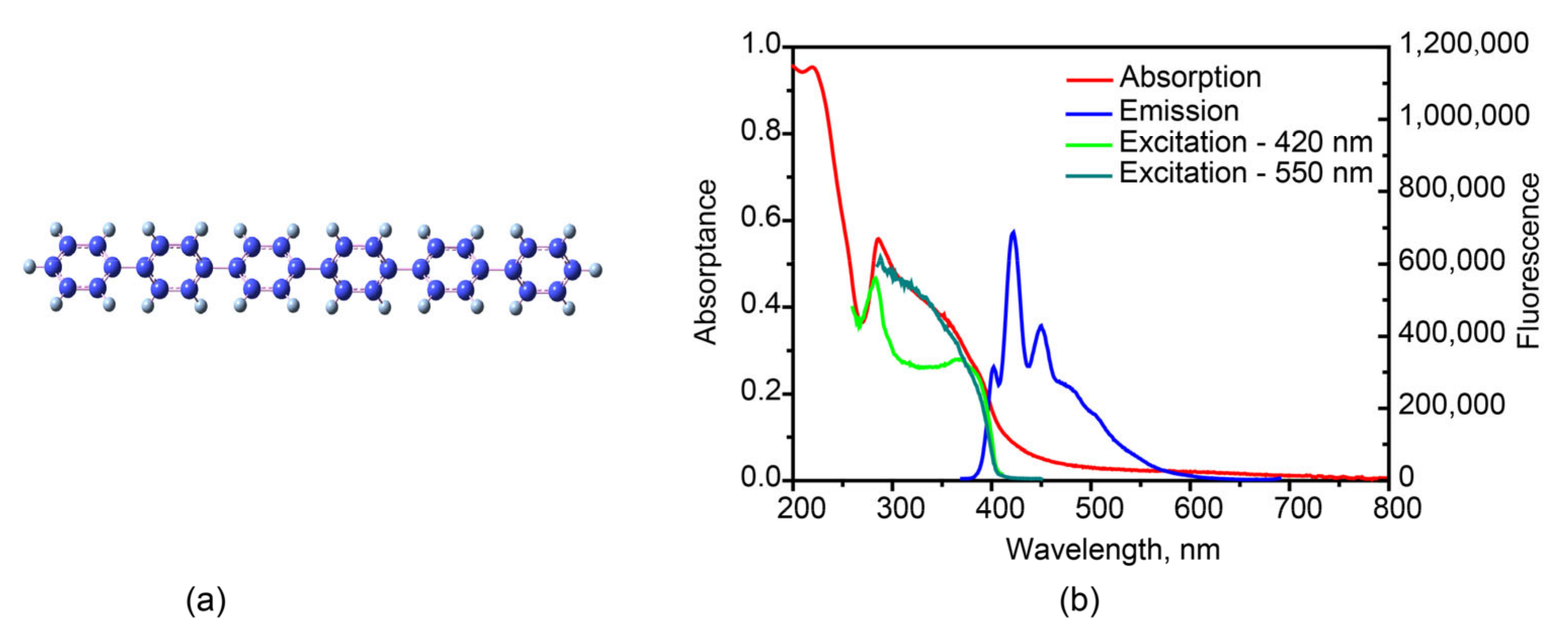
2.4. Time-Resolved Fluorescence Spectroscopy
2.5. Transient Absorption Spectroscopy
2.6. Global Analysis
3. Results and Discussion
3.1. Steady-State Absorption and Fluorescence Spectra of Hexaphenyl Film
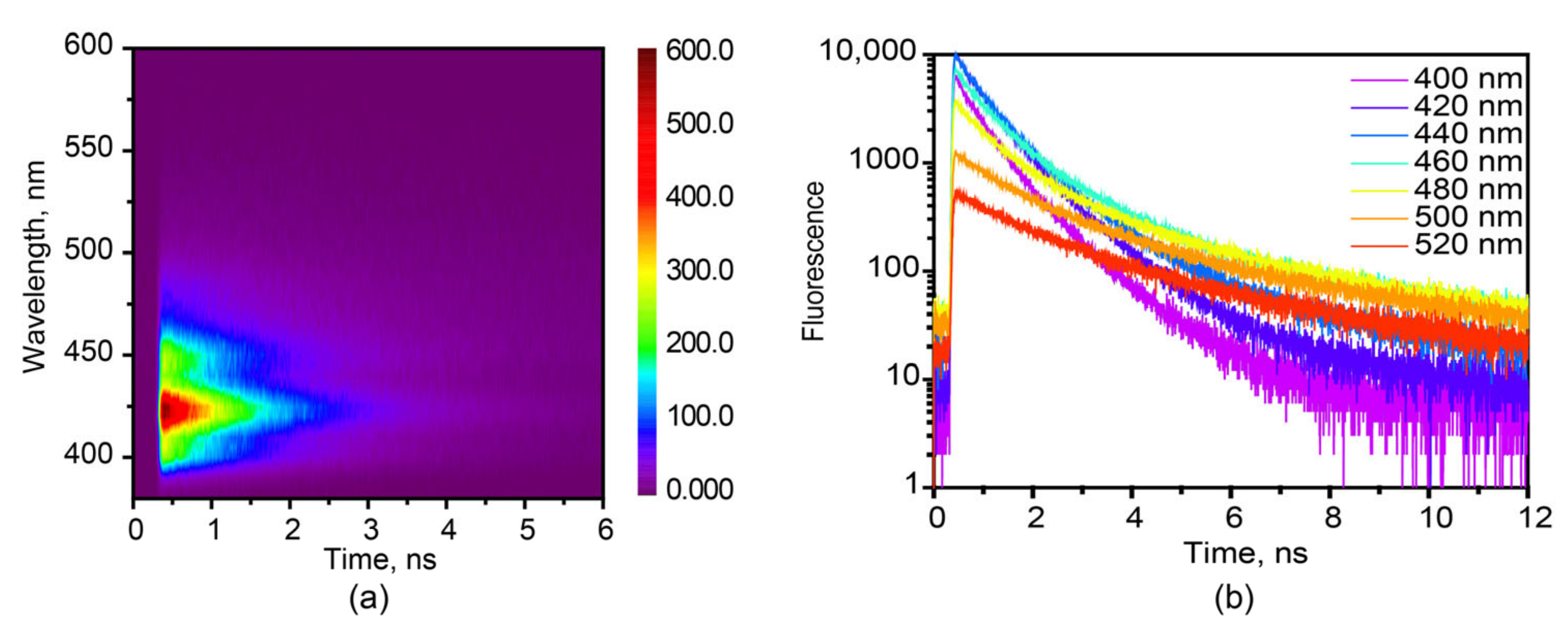
3.2. Various Emission Species in Hexaphenyl Film
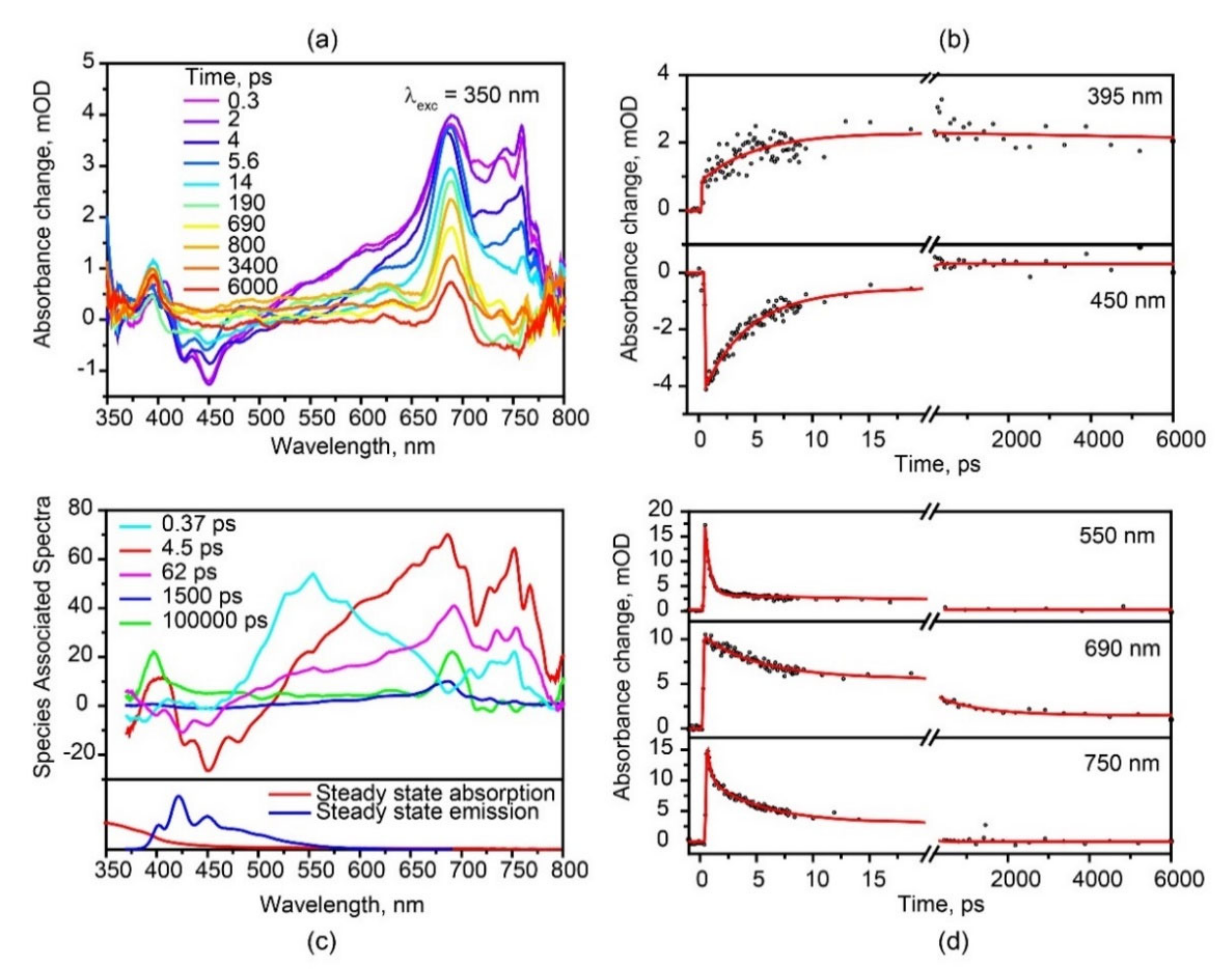
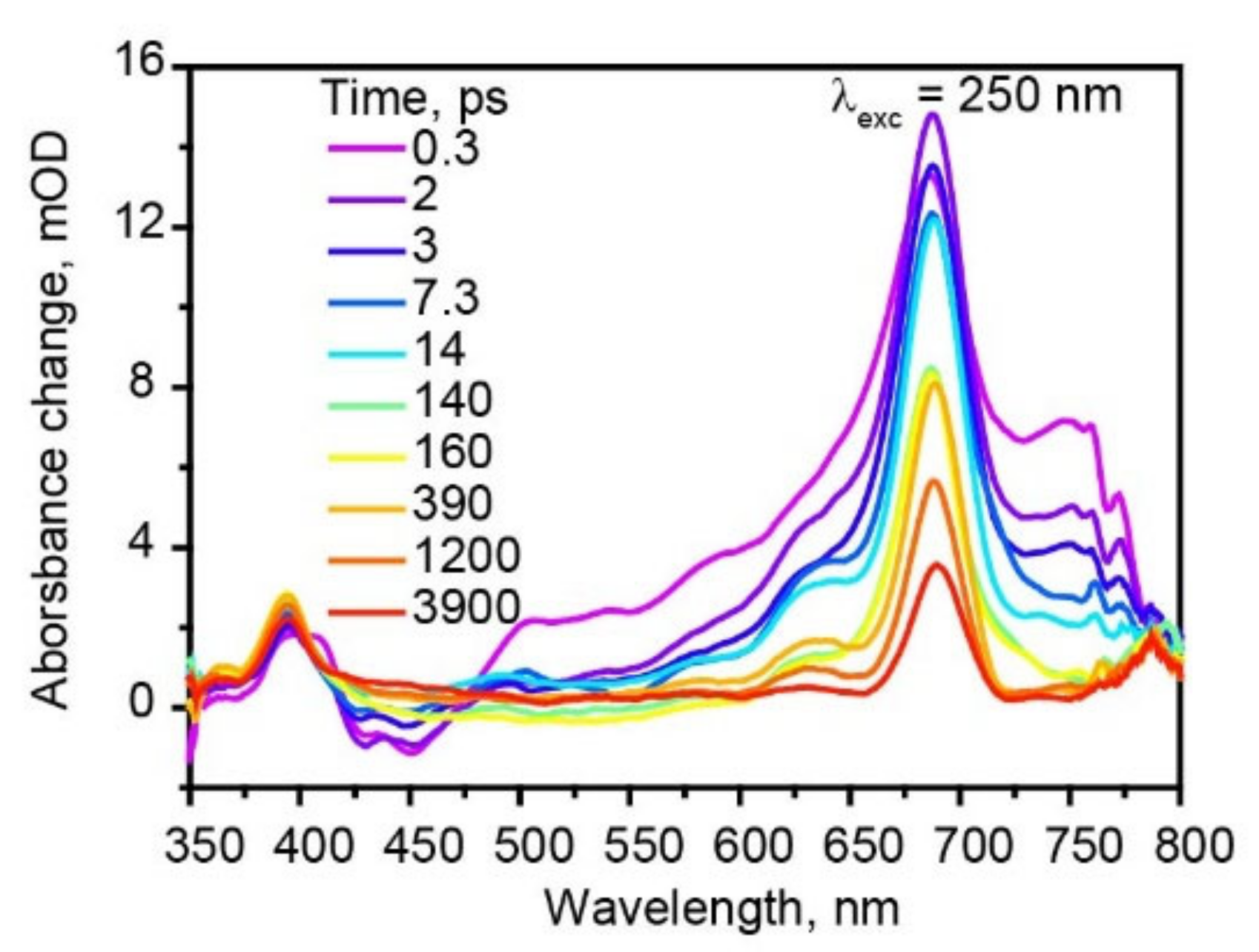
3.3. Transient Absorption Spectra of Hexaphenyl Film
3.4. Two Pathways of Triplet Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Burroughes, J.H.; Bradley, D.D.; Brown, A.R.; Marks, R.N.; Mackay, K.; Friend, R.H.; Holmes, A.B. Light-Emitting Diodes Based on Conjugated Polymers. Nature 1990, 347, 539–541. [Google Scholar] [CrossRef]
- Graupner, W.; Grem, G.; Meghdadi, F.; Paar, C.; Leising, G.; Scherf, U.; Mullen, K.; Fischer, W.; Stelzer, F. Electroluminescence with Conjugated Polymers and Oligomers. Mol. Cryst. Liq. Cryst. 1994, 256, 549–554. [Google Scholar]
- Era, M.; Tsutsui, T.; Saito, S. Polarized Electroluminescence from Oriented P-Sexiphenyl Vacuum-Deposited Film. Appl. Phys. Lett. 1995, 67, 2436–2438. [Google Scholar] [CrossRef]
- Leising, G.; Tasch, S.; Brandstatter, C.; Meghdadi, F.; Froyer, G.; Athouel, L. Red–Green–Blue Light Emission from A Thin Film Electroluminescence Device Based on Parahexaphenyl. Adv. Mater. 1997, 9, 33–36. [Google Scholar]
- Sariciftci, N.S.; Smilowitz, L.; Heeger, A.J.; Wudl, F. Photoinduced Electron Transfer from A Conducting Polymer to Buckminsterfullerene. Science 1992, 258, 1474–1476. [Google Scholar] [CrossRef]
- Leising, G.; Ekstroem, O.; Graupner, W.; Meghdadi, F.; Moser, M.; Kranzelbinder, G.; Jost, T.; Tasch, S.; Winker, B.; Athouel, L.; et al. Multicolor Electroluminescence and Stimulated Emission of Conjugated Polymers and Oligomers. Proc. SPIE—Int. Soc. Opt. Eng. 1996, 2852, 189–200. [Google Scholar]
- Piaggi, A.; Lanzani, G.; Bongiovanni, G.; Mura, A.; Graupner, W.; Meghdadi, F.; Leising, G.; Nisoli, M. Emission Properties of Para-Hexaphenyl Polycrystalline Films. Phys. Rev. B 1997, 56, 10133. [Google Scholar] [CrossRef] [Green Version]
- Piaggi, A.; Lanzani, G.; Bongiovanni, G.; Loi, M.A.; Mura, A.; Graupner, W.; Meghdadi, F.; Leising, G. Optical Properties of Polycrystalline Films. Opt. Mater. 1998, 9, 489–493. [Google Scholar]
- Frank, P.; Hlawacek, G.; Lengyel, O.; Satka, A.; Teichert, C.; Resel, R.; Winkler, A. Influence of Surface Temperature and Surface Modifications on The Initial Layer Growth of Para-Hexaphenyl on Mica (001). Surf. Sci. 2007, 601, 2152–2160. [Google Scholar] [CrossRef]
- Leising, G.; Tasch, S.; Meghdadi, F.; Athouel, L.; Froyer, G.; Scherf, U. Blue Electroluminescence with Ladder-Type Poly(Para-Phenylene) and Para-Hexaphenyl. Synth. Met. 1996, 81, 185–189. [Google Scholar] [CrossRef]
- Resel, R.; Koch, N.; Meghdadi, F.; Leising, G.; Athouel, L.; Froyer, G.; Hofer, F.A. Polymorph Crystal Structure of Hexaphenyl Observed in Thin Films. Cryst. Res. Technol. 2001, 36, 47–54. [Google Scholar]
- Baker, K.N.; Fratini, A.V.; Resch, T.; Knachel, H.C.; Adams, W.W.; Socci, E.P.; Farmer, B.L. Crystal Structures, Phase Transitions and Energy Calculations of Poly (P-Phenylene) Oligomers. Polymer 1993, 34, 1571–1587. [Google Scholar] [CrossRef]
- Athouel, L.; Froyer, G.; Riou, M.T.; Schott, M. Structural Studies of Parasexiphenyl Thin Films: Importance of The Deposition Parameters. Thin Solid Films 1996, 274, 35–45. [Google Scholar] [CrossRef]
- Congreve, D.N.; Lee, J.; Thompson, N.J.; Hontz, E.; Yost, S.R.; Reusswig, P.D.; Bahlke, M.E.; Reineke, S.; Voorhis, T.; Baldo, M.A. External Quantum Efficiency Above 100% in A Singlet-Exciton-Fission–Based Organic Photovoltaic Cell. Science 2013, 340, 334–337. [Google Scholar] [CrossRef]
- Qiao, X.; Ma, D. Nonlinear Optoelectronic Processes in Organic Optoelectronic Devices: Triplet-Triplet Annihilation and Singlet Fission. Mater. Sci. Eng. R 2020, 139, 100519. [Google Scholar]
- Rao, A.; Friend, R.H. Harnessing Singlet Exciton Fission to Break the Shockley-Queisser Limit. Nat. Rev. Mater. 2017, 2, 17063. [Google Scholar] [CrossRef]
- Futscher, M.H.; Rao, A.; Ehrler, B. The Potential of Singlet Fission Photon Multipliers as an Alternative to Silicon-Based Tandem Solar Cells. ACS Energy Lett. 2018, 3, 2587–2592. [Google Scholar] [CrossRef] [Green Version]
- MacQueen, R.W.; Liebhaber, M.; Niederhausen, J.; Mews, M.; Gersmann, C.; Jäckle, S.; Jäger, K.; Tayebjee, M.J.Y.; Schmidt, T.W.; Rech, B.; et al. Crystalline Silicon Solar Cells with Tetracene Interlayers: The Path to Silicon-Singlet Fission Heterojunction Devices. Mater. Horiz. 2018, 5, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Casillas, R.; Papadopoulos, I.; Ullrich, T.; Thiel, D.; Kunzmann, A.; Guldi, D.M. Molecular Insights and Concepts to Engineer Singlet Fission Energy Conversion Devices. Energy Environ. Sci. 2020, 13, 2741–2804. [Google Scholar] [CrossRef]
- Smith, M.B.; Michl, J. Recent Advances in Singlet Fission. Annu. Rev. Phys. Chem. 2013, 64, 361–386. [Google Scholar] [CrossRef]
- Wilson, M.W.B.; Rao, A.; Clark, J.; Kumar, R.S.S.; Brida, D.; Cerullo, G.; Friend, R.H. Ultrafast Dynamics of Exciton Fission in Polycrystalline Pentacene. J. Am. Chem. Soc. 2011, 133, 11830–11833. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, K.; Kloc, C.; Sun, H.; Michel-Beyerle, M.E.; Gurzadyan, G.G. Singlet Fission in Rubrene Single Crystal: Direct Observation by Femtosecond Pump–Probe Spectroscopy. Phys. Chem. Chem. Phys. 2012, 14, 8307–8312. [Google Scholar] [CrossRef] [PubMed]
- Pun, A.B.; Sanders, S.N.; Kumarasamy, E.; Sfeir, M.Y.; Congreve, D.N.; Campos, L.M. Triplet Harvesting from Intramolecular Singlet Fission in Polytetracene. Adv. Mater. 2017, 29, 1701416. [Google Scholar] [CrossRef] [PubMed]
- Grieco, C.; Doucette, G.S.; Munson, K.T.; Swartzfager, J.R.; Munro, J.M.; Anthony, J.E.; Dabo, I.; Asbury, J.B. Vibrational Probe of the Origin of Singlet Exciton Fission in TIPS-Pentacene Solutions. J. Chem. Phys. 2019, 151, 154701. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Bae, Y.J.; Mauck, C.M.; Mandal, A.; Young, R.M.; Wasielewski, M.R. Singlet Fission in Covalent Terrylenediimide Dimers: Probing the Nature of the Multiexciton State Using Femtosecond Mid-Infrared Spectroscopy. J. Am. Chem. Soc. 2018, 140, 9184–9192. [Google Scholar] [CrossRef]
- Ni, W.; Gurzadyan, G.G.; Zhao, J.; Che, Y.; Li, X.; Sun, L. Singlet Fission from Upper Excited Electronic States of Cofacial Perylene Dimer. J. Phys. Chem. Lett. 2019, 10, 2428–2433. [Google Scholar] [CrossRef]
- Smith, M.B.; Michl, J. Singlet Fission. Chem. Rev. 2010, 110, 6891–6936. [Google Scholar] [CrossRef]
- Graupner, W.; Meghdadi, F.; Leising, G.; Lanzani, G.; Nisoli, M.; De Silvestri, S.; Fischer, W.; Stelzer, F. Photoexcitations in Para-Hexaphenyl. Phys. Rev. B 1997, 56, 10128. [Google Scholar] [CrossRef]
- Zenz, C.; Cerullo, G.; Lanzani, G.; Graupner, W.; Meghdadi, F.; Leising, G.; De Silvestri, S. Ultrafast Photogeneration Mechanisms of Triplet States in Para-Hexaphenyl. Phys. Rev. B 1999, 59, 14336. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gong, C.; Gurzadyan, G.G.; Gelin, M.F.; Liu, J.; Sun, L. Ultrafast Relaxation Dynamics in Zinc Tetraphenylporphyrin Surface-Mounted Metal Organic Framework. J. Phys. Chem. C 2017, 122, 50–61. [Google Scholar] [CrossRef]
- Ullrich, S.; Schultz, T.; Zgierski, M.Z.; Stolow, A. Electronic Relaxation Dynamics in DNA and RNA Bases Studied by Time-Resolved Photoelectron Spectroscopy. Phys. Chem. Chem. Phys. 2004, 6, 2796–2801. [Google Scholar] [CrossRef]
- Shang, J.; Yan, S.; Cong, C.; Tan, H.; Yu, T.; Gurzadyan, G.G. Probing Near Dirac Point Electron-Phonon Interaction in Graphene. Opt. Mater. Express 2012, 2, 1713–1722. [Google Scholar] [CrossRef]
- Shang, J.; Yu, T.; Lin, J.; Gurzadyan, G.G. Ultrafast Electron-Optical Phonon Scattering and Quasiparticle Lifetime in CVD-Grown Graphene. ACS Nano 2011, 5, 3278–3283. [Google Scholar] [CrossRef]
- Snellenburg, J.J.; Laptenok, S.P.; Seger, R.; Mullen, K.M.; Van Stokkum, I.H.M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Softw. 2012, 49, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Kersting, R.; Lemmer, U.; Mahrt, R.F.; Leo, K.; Kurz, H.; Bässler, H.; Göbel, E.O. Femtosecond Energy Relaxation in π-Conjugated Polymers. Phys. Rev. Lett. 1993, 70, 3820–3823. [Google Scholar] [CrossRef] [PubMed]
- Faulques, E.; Wéry, J.; Lefrant, S.; Ivanov, V.G.; Jonusauskas, G. Transient Photoluminescence of Para-Hexaphenyl Layers. Phys. Rev. B 2002, 65, 212202. [Google Scholar] [CrossRef]
- Rzepka, E.; Jin, C.Q.; Lefrant, S.; Pelous, Y.; Froyer, G.; Siove, A. Luminescence Studies in Polyparaphenylene Samples. Synth. Met. 1989, 29, 23–28. [Google Scholar] [CrossRef]
- Kasha, M. Characterization of Electronic Transitions in Complex Molecules. Discuss. Faraday Soc. 1950, 9, 14–19. [Google Scholar] [CrossRef]
- Hestand, N.J.; Spano, F.C. Molecular Aggregate Photophysics Beyond the Kasha Model: Novel Design Principles for Organic Materials. Acc. Chem. Res. 2017, 50, 341–350. [Google Scholar] [CrossRef]
- Nakanishi, T.; Mizuno, K.I.; Matsui, A.; Nishimura, H. Reabsorption of Excitonic Luminescence in Pyrene Crystals. J. Lumin. 1991, 47, 303–308. [Google Scholar] [CrossRef]
- Kobayashi, T.; Nagakura, S. Reabsorption and High Density Excitation Effects on The Time-Resolved Fluorescence Spectra of Anthracene Crystal. Mol. Cryst. Liq. Cryst. 1974, 26, 33–43. [Google Scholar] [CrossRef]
- Louarn, G.; Athouël, L.; Froyer, G.; Buisson, J.P.; Lefrant, S. Optical Characterization of Parasexiphenyl: A Model Compound of Polyparaphenylene. Synth. Met. 1993, 57, 4762–4767. [Google Scholar] [CrossRef]
- Resel, R.; Koch, N.; Meghdadi, F.; Leising, G.; Unzog, W.; Reichmann, K. Growth and Preferred Crystallographic Orientation of Hexaphenyl thin Films. Thin Solid Films 1997, 305, 232–242. [Google Scholar] [CrossRef]
- Pope, M.; Swenberg, C.E. Electronic Processes in Organic Crystals and Polymers; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Grebner, D.; Helbig, M.; Rentsch, S. Size-Dependent Properties of Oligothiophenes by Picosecond Time-Resolved Spectroscopy. J. Phys. Chem. 1995, 99, 16991–16998. [Google Scholar] [CrossRef]
- Janssen, R.A.J.; Smilowitz, L.; Sariciftci, N.S.; Moses, D. Triplet-State Photoexcitations of Oligothiophene Films and Solutions. J. Chem. Phys. 1994, 101, 1787–1798. [Google Scholar] [CrossRef]
- Tavares, L.; Cadelano, M.; Quochi, F.; Simbrunner, C.; Schwabegger, G.; Saba, M.; Mura, A.; Bongiovanni, G.; Silva Fiho, D.A.; Cunha, W.F.; et al. Efficient Exciton Diffusion and Resonance-Energy Transfer in Multilayered Organic Epitaxial Nanofibers. J. Phys. Chem. C 2015, 119, 15689–15697. [Google Scholar] [CrossRef] [PubMed]
- Kolata, K.; Breuer, T.; Witte, G.; Chatterjee, S. Molecular Packing Determines Singlet Exciton Fission in Organic Semiconductors. ACS Nano 2014, 8, 7377–7383. [Google Scholar] [CrossRef]
- Jue Bae, Y.; Krzyaniak, M.D.; Majewski, M.B.; Desroches, M.; Morin, J.F.; Wu, Y.L.; Wasielewski, M.R. Competition Between Singlet Fission and Spin-Orbit-Induced Intersystem Crossing in Anthanthrene and Anthanthrone Derivatives. ChemPlusChem 2019, 84, 1432–1438. [Google Scholar] [CrossRef]
- Bayliss, S.L.; Kraffert, F.; Wang, R.; Zhang, C.; Bittl, R.; Behrends, J. Tuning Spin Dynamics in Crystalline Tetracene. J. Phys. Chem. Lett. 2019, 10, 1908–1913. [Google Scholar] [CrossRef]
- Wollscheid, N.; Alagna, N.; Lustres, J.L.P.; Buckup, T.; Hahn, S.; Bunz, U.H.; Motzkus, M. Unveiling the Concentration Dependent Direct Triplet Formation Via Singlet Fission in A Tetracene Derivative. EPJ Web Conf. 2019, 205, 09031. [Google Scholar] [CrossRef]
- Ito, A.; Shimizu, A.; Kishida, N.; Kawanaka, Y.; Kosumi, D.; Hashimoto, H.; Teki, Y. Excited-State Dynamics of Pentacene Derivatives with Stable Radical Substituents. Angew. Chem. Int. Ed. 2014, 126, 6833–6837. [Google Scholar] [CrossRef]
- Basel, B.S.; Zirzlmeier, J.; Hetzer, C.; Phelan, B.T.; Krzyaniak, M.D.; Reddy, S.R.; Coto, P.B.; Horwitz, N.E.; Young, R.M.; White, F.J.; et al. Unified Model for Singlet Fission Within A Non-Conjugated Covalent Pentacene Dimer. Nat. Commun. 2017, 8, 15171. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.R.; Bayliss, S.L.; Kraffert, F.; Thorley, K.J.; Anthony, J.E.; Bittl, R.; Friend, R.H.; Rao, A.; Greenham, N.C.; Behrends, J. Strongly Exchange-Coupled Triplet Pairs in an Organic Semiconductor. Nat. Phys. 2017, 13, 176–181. [Google Scholar] [CrossRef]
- Bayliss, S.L.; Weiss, L.R.; Rao, A.; Friend, R.H.; Chepelianskii, A.D.; Greenham, N.C. Spin Signatures of Exchange-Coupled Triplet Pairs Formed by Singlet Fission. Phys. Rev. B 2016, 94, 045204. [Google Scholar] [CrossRef] [Green Version]
- Tayebjee, M.J.Y.; Sanders, S.N.; Kumarasamy, E.; Campos, L.M.; Sfeir, M.Y.; McCamey, D.R. Quintet Multiexciton Dynamics in Singlet Fission. Nat. Phys. 2017, 13, 182–188. [Google Scholar] [CrossRef]
- Basel, B.S.; Young, R.M.; Krzyaniak, M.D.; Papadopoulos, I.; Hetzer, C.; Gao, Y.; La Porte, N.T.; Phelan, B.T.; Clark, T.; Tykwinski, R.R.; et al. Influence of the Heavy-Atom Effect on Singlet Fission: A Study of Platinum-Bridged Pentacene Dimers. Chem. Sci. 2019, 10, 11130–11140. [Google Scholar] [CrossRef] [Green Version]
- Miyata, K.; Kurashige, Y.; Watanabe, K.; Sugimoto, T.; Takahashi, S.; Tanaka, S.; Takeya, J.; Yanai, T.; Matsumoto, Y. Coherent Singlet Fission Activated by Symmetry Breaking. Nat. Chem. 2017, 9, 983–989. [Google Scholar] [CrossRef]
- Reddy, S.R.; Coto, P.B.; Thoss, M. Quantum Dynamical Simulation of Intramolecular Singlet Fission in Covalently Coupled Pentacene Dimers. J. Chem. Phys. 2019, 151, 044307. [Google Scholar] [CrossRef]
- Sun, K.; Xu, Q.; Chen, L.; Gelin, M.F.; Zhao, Y. Temperature Effects on Singlet Fission Dynamics Mediated by A Conical Intersection. J. Chem. Phys. 2020, 153, 194106. [Google Scholar] [CrossRef]
- Serevicius, T.; Komskis, R.; Adomėnas, P.; Adomėnienė, O.; Kreiza, G.; Jankauskas, V.; Kazlauskas, K.; Miasojedovas, A.; Jankus, V.; Monkman, A.P.; et al. Triplet-Triplet Annihilation in 9, 10-Diphenylanthracene Derivatives: The Role of Intersystem Crossing and Exciton Diffusion. J. Phys. Chem. C 2017, 121, 8515–8524. [Google Scholar] [CrossRef]
- Ford, T.A.; Avilov, I.; Beljonne, D.; Greenham, N.C. Enhanced Triplet Exciton Generation in Polyfluorene Blends. Phys. Rev. B 2005, 71, 125212. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λexc, nm | λprobe, nm | τ1, ps | A1 | τ2, ps | A2 | τ3, ps | A3 | τ4, ps | A4 |
|---|---|---|---|---|---|---|---|---|---|
| 350 | 395 | 4.5 | −0.61 | 100,000 f | 1 | ||||
| 450 | 4.5 | −0.75 | 89 | −0.25 | 100,000 f | 1 | |||
| 550 | 0.45 | 0.85 | 62 | 0.15 | |||||
| 690 | 4.6 | 0.41 | 65 | 0.18 | 1000 | 0.26 | 100,000 f | 0.15 | |
| 750 | 0.30 | 0.43 | 4.3 | 0.37 | 84 | 0.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, W.; Li, T.; Kloc, C.; Sun, L.; Gurzadyan, G.G. Singlet Fission, Polaron Generation and Intersystem Crossing in Hexaphenyl Film. Molecules 2022, 27, 5067. https://doi.org/10.3390/molecules27165067
Ni W, Li T, Kloc C, Sun L, Gurzadyan GG. Singlet Fission, Polaron Generation and Intersystem Crossing in Hexaphenyl Film. Molecules. 2022; 27(16):5067. https://doi.org/10.3390/molecules27165067
Chicago/Turabian StyleNi, Wenjun, Tianjiao Li, Christian Kloc, Licheng Sun, and Gagik G. Gurzadyan. 2022. "Singlet Fission, Polaron Generation and Intersystem Crossing in Hexaphenyl Film" Molecules 27, no. 16: 5067. https://doi.org/10.3390/molecules27165067
APA StyleNi, W., Li, T., Kloc, C., Sun, L., & Gurzadyan, G. G. (2022). Singlet Fission, Polaron Generation and Intersystem Crossing in Hexaphenyl Film. Molecules, 27(16), 5067. https://doi.org/10.3390/molecules27165067