
Design, Synthesis and Preliminary Evaluation of the Cytotoxicity and Antibacterial Activity of Novel Triphenylphosphonium Derivatives of Betulin

 , ,
, ,
Abstract
:
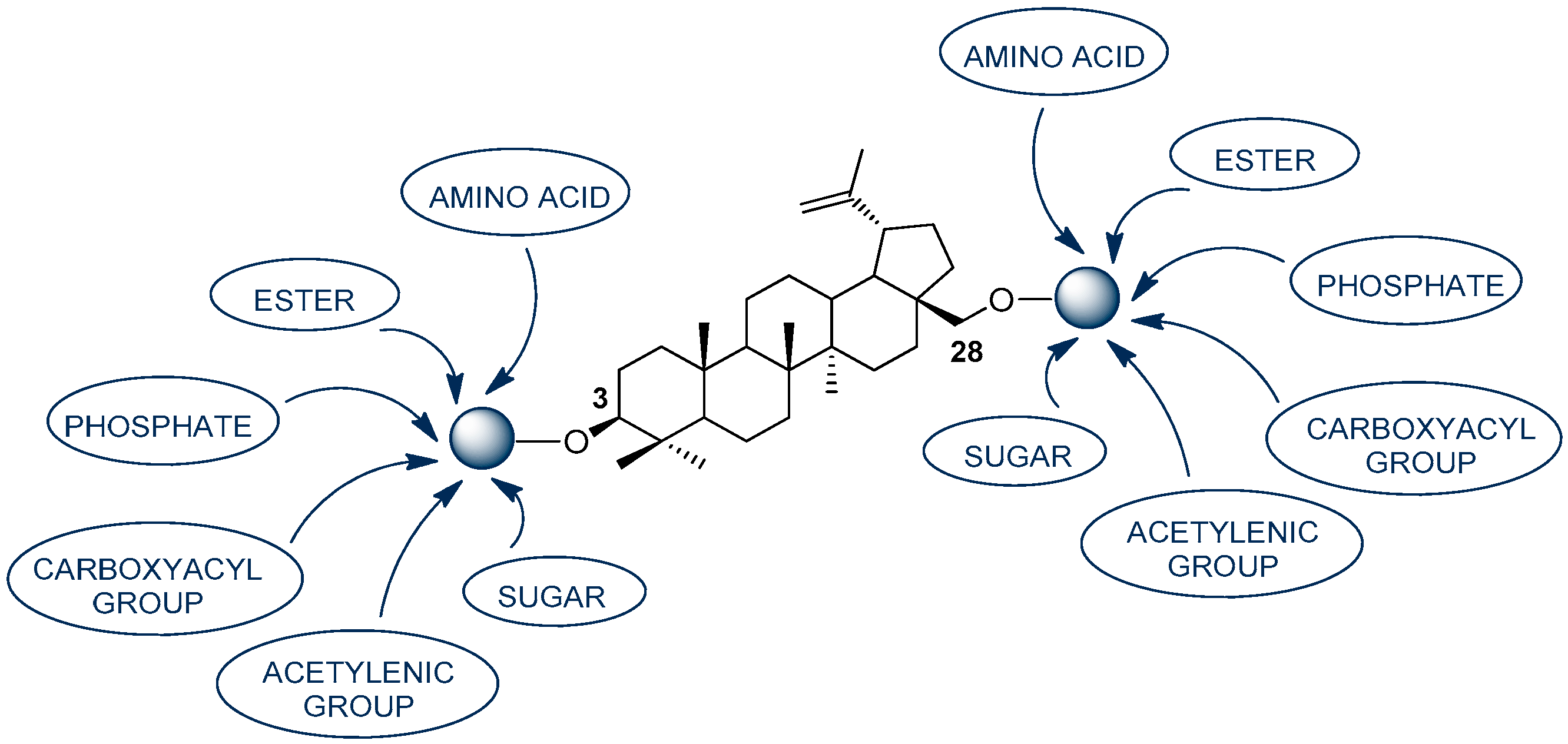
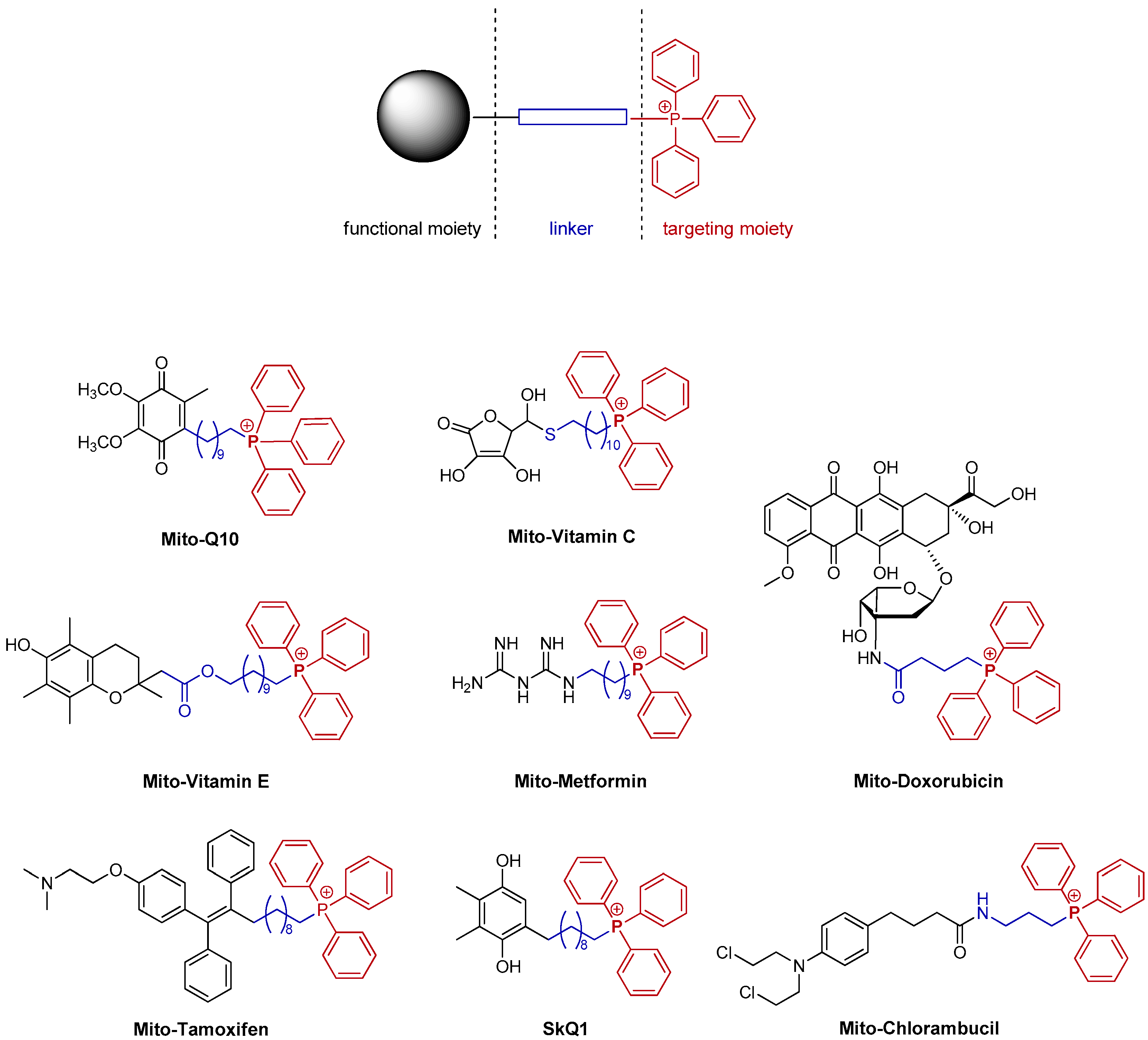
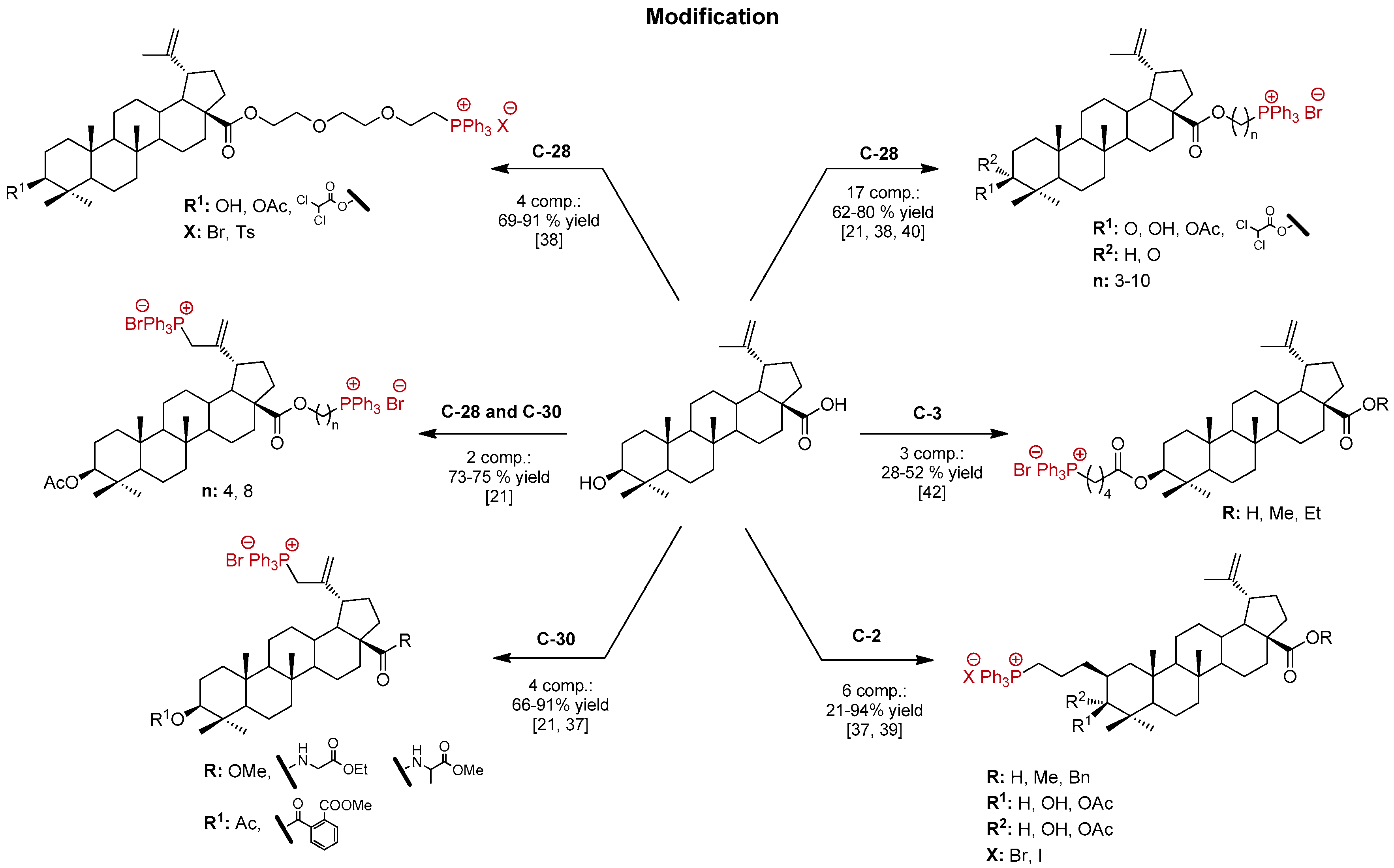
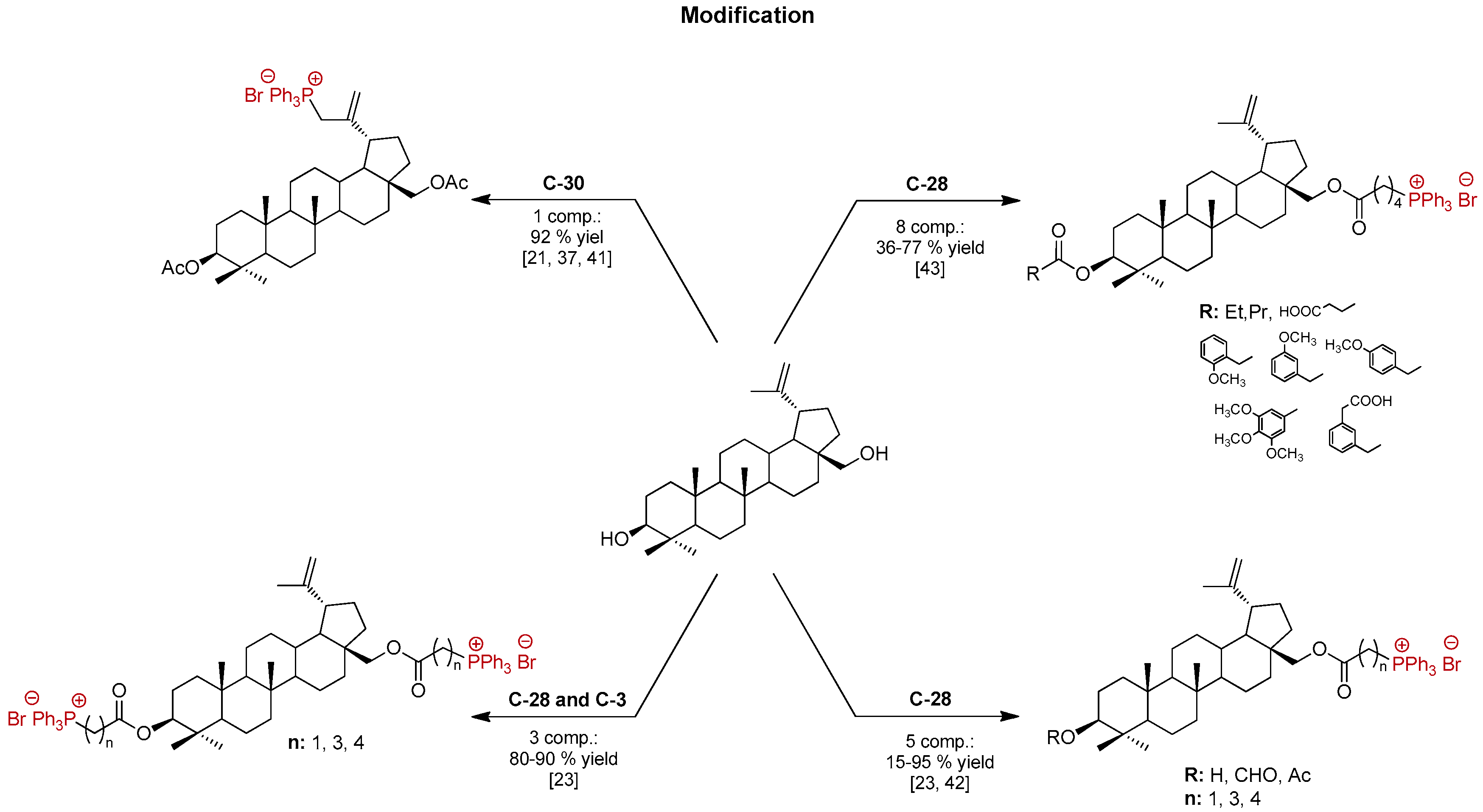
1. Introduction
2. Results and Discussion
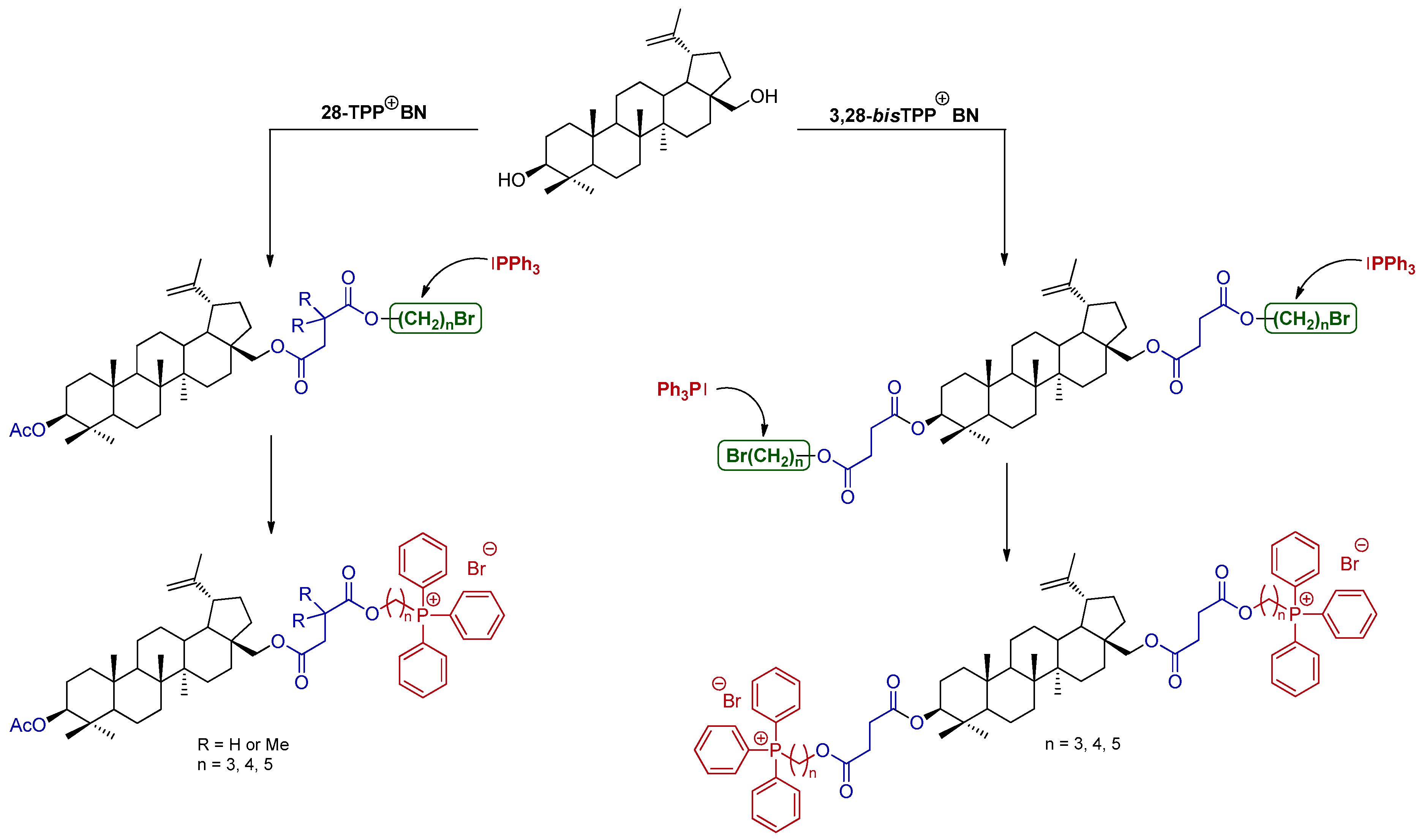
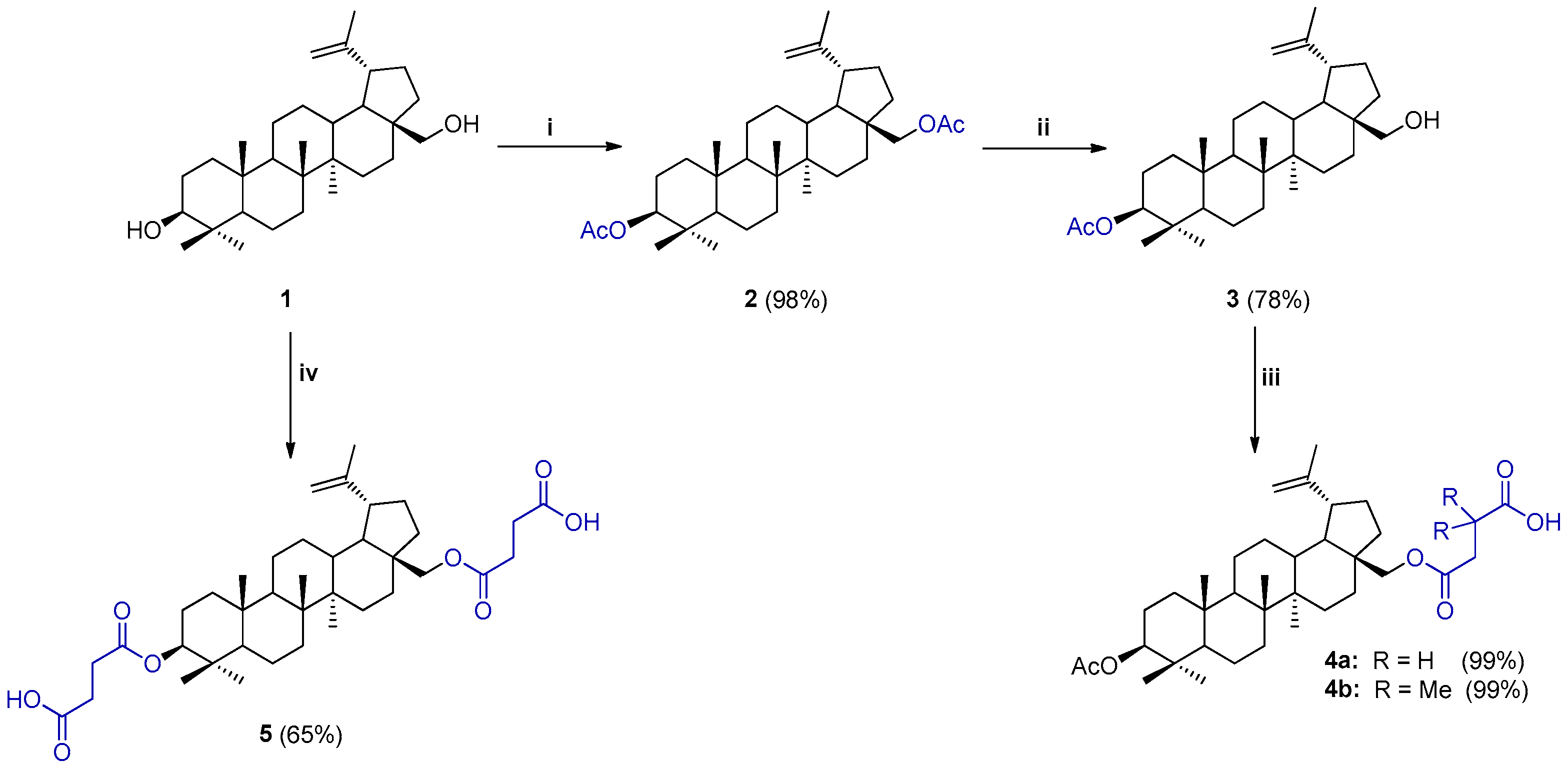
2.1. Synthesis of BN Analogs (2–5)
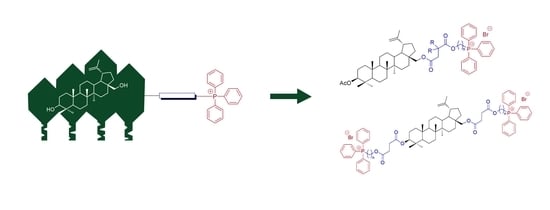
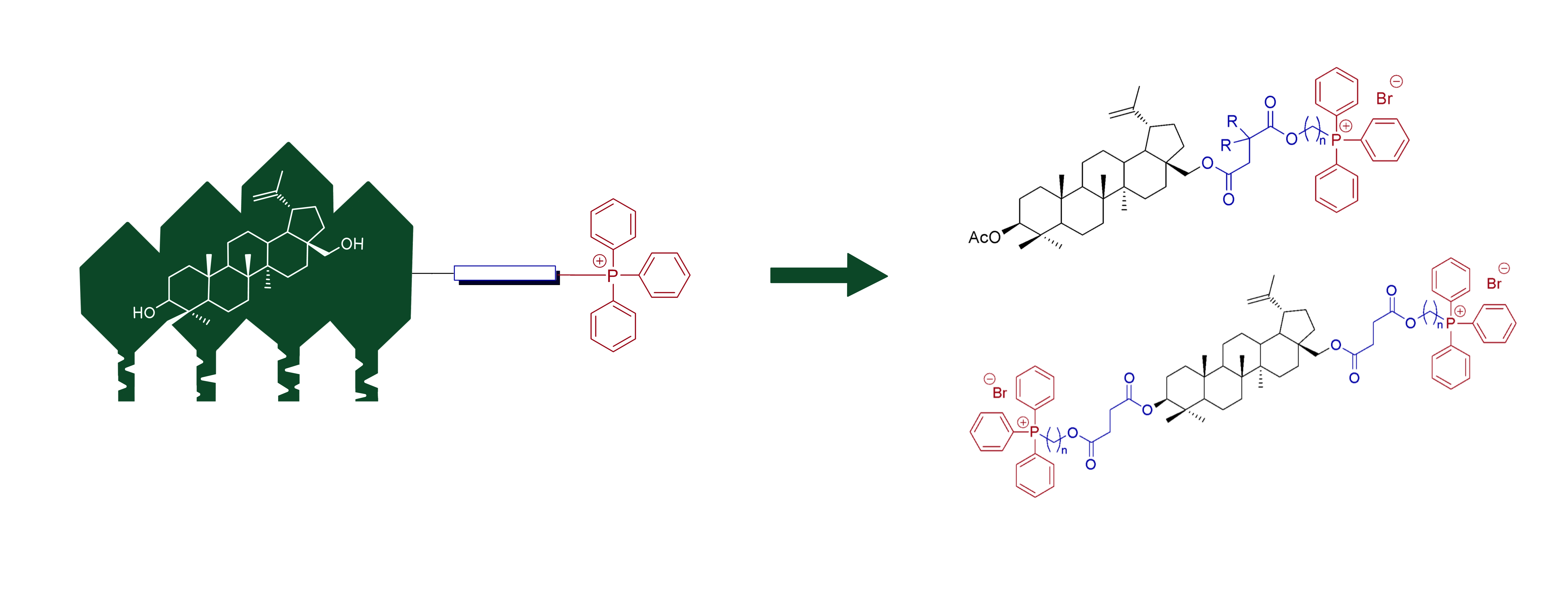
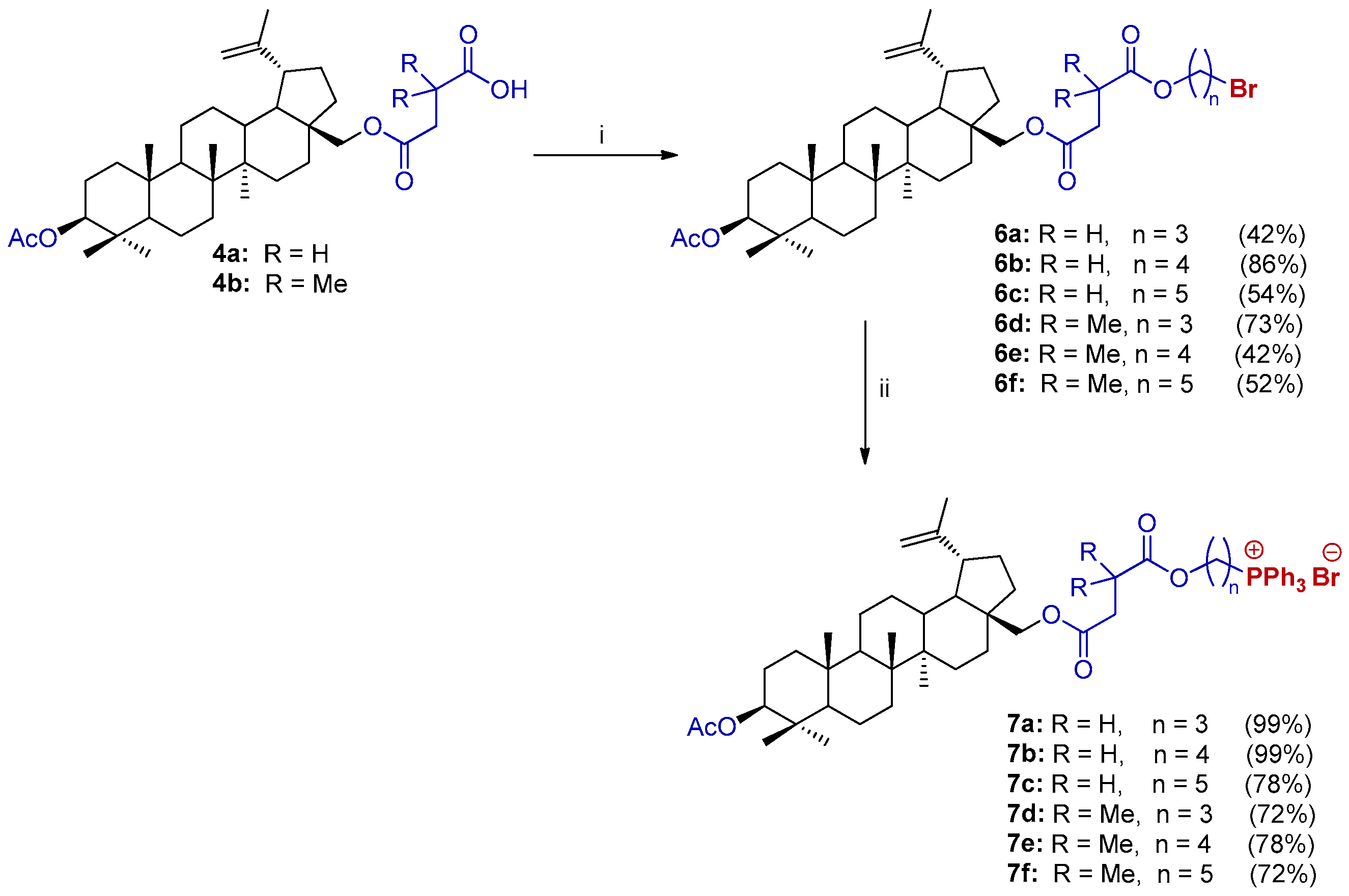
2.2. Synthesis of 28-TPP⊕-Conjugates Derivatives of BN
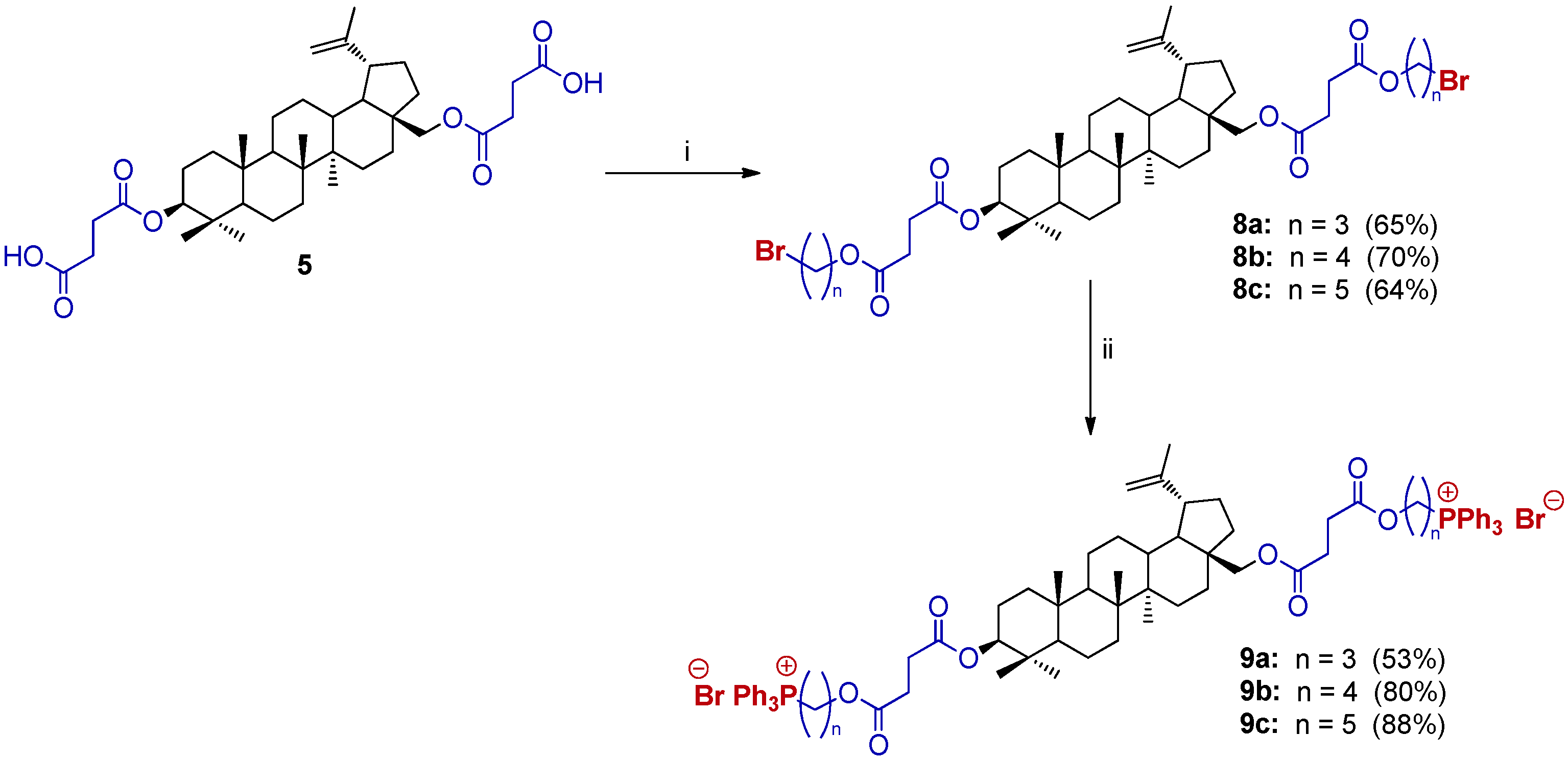
2.3. Synthesis of 3,28-bisTPP⊕-Conjugates Derivatives of BN
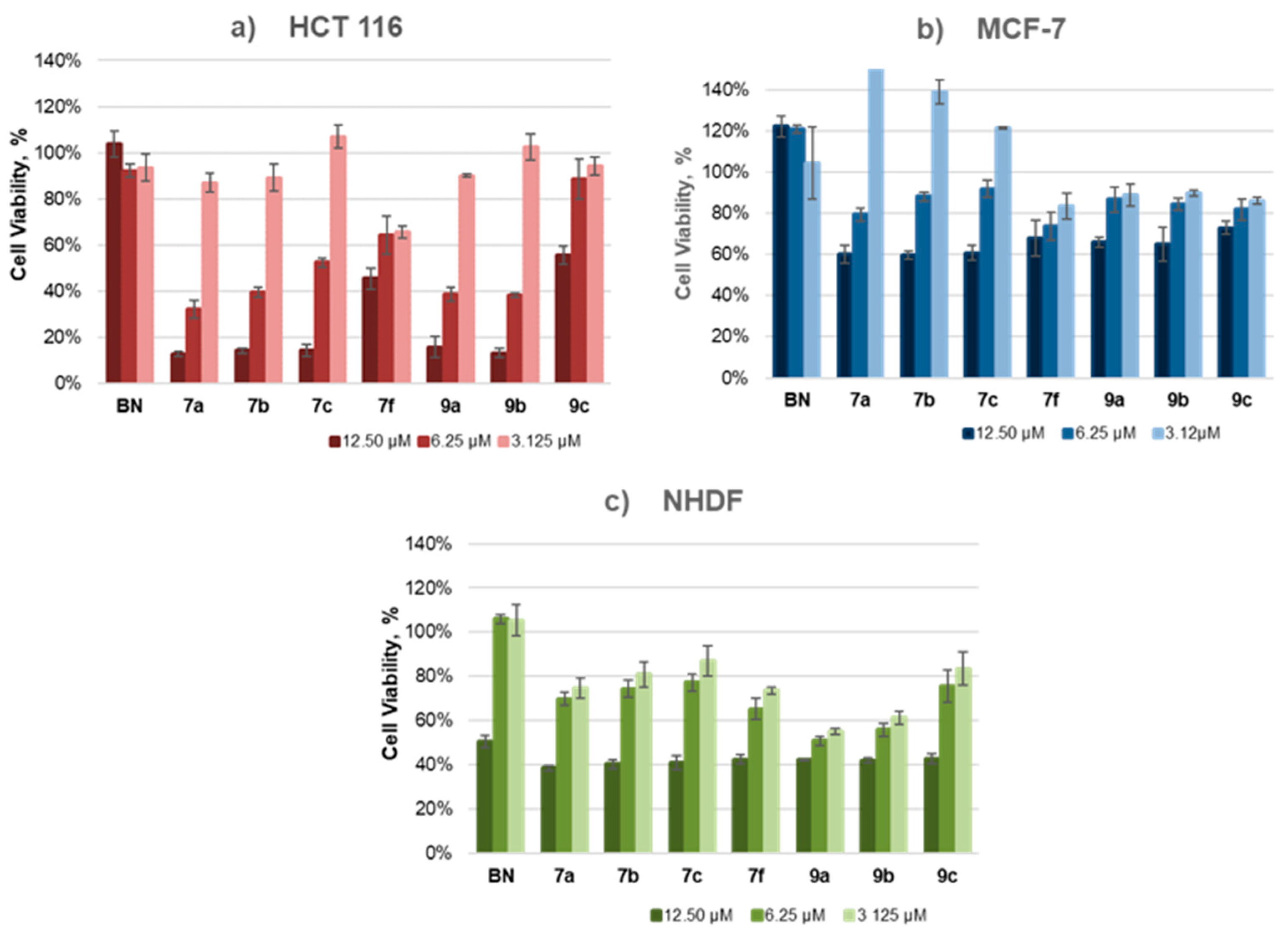
2.4. Cytotoxicity Studies
2.5. Antibacterial Studies
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of 3-O-Acetyl-28-O’-(carboxyacyl)betulin (4)
3.2.2. Synthesis of 3,28-O,O′-Bis(3′-carboxypropanoyl)betulin (5)
3.2.3. General Procedure for the Synthesis of Bromides of BN (6)
3.2.4. General Procedure for the Synthesis of Triphenylphosphonium Derivatives of BN (7, 28-TPP⊕ BN)
3.2.5. General Procedure for the Synthesis of 3,28-Bis(bromoalkoxycarbonyl)propanoyl)betulin (8)
3.2.6. General Procedure for the Synthesis of Bis(triphenylphoshonium) Derivatives of BN (9, 3,28-bisTPP⊕ BN)
3.3. Biological Evaluation
3.3.1. Cytotoxicity Assay
Cell Lines
Cell Viability Assay
3.3.2. Antibacterial Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Krasutsky, P.A. Birch Bark Research and Development. Nat. Prod. Rep. 2006, 23, 919–942. [Google Scholar] [CrossRef]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.-R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and Its Derivatives as Novel Compounds with Different Pharmacological Effects. Biotechnol. Adv. 2020, 38, 107409–107448. [Google Scholar] [CrossRef] [PubMed]
- Jonnalagadda, S.C.; Suman, P.; Morgan, D.C.; Seay, J.N. Recent Developments on the Synthesis and Applications of Betulin and Betulinic Acid Derivatives as Therapeutic Agents. Stud. Nat. Prod. Chem. 2017, 53, 45–84. [Google Scholar] [CrossRef]
- Grymel, M.; Pastuch-Gawołek, G.; Lalik, A.; Zawojak, M.; Boczek, S.; Krawczyk, M.; Erfurt, K. Glycoconjugation of Betulin Derivatives Using Copper-Catalyzed 1,3-Dipolar Azido-Alkyne Cycloaddition Reaction and a Preliminary Assay of Cytotoxicity of the Obtained Compounds. Molecules 2020, 25, 6019. [Google Scholar] [CrossRef] [PubMed]
- Hordyjewska, A.; Ostapiuk, A.; Horecka, A. Betulin and Betulinic Acid in Cancer Research. J. Pre-Clin. Clin. Res. 2018, 12, 72–75. [Google Scholar] [CrossRef]
- Drąg-Zalesińska, M.; Drąg, M.; Poręba, M.; Borska, S.; Kulbacka, J.; Saczko, J. Anticancer Properties of Ester Derivatives of Betulin in Human Metastatic Melanoma Cells (Me-45). Cancer Cell Int. 2017, 17, 4–14. [Google Scholar] [CrossRef] [PubMed]
- So, H.M.; Eom, H.J.; Lee, D.; Kim, S.; Kang, K.S.; Lee, I.K.; Baek, K.-H.; Park, J.Y.; Kim, K.H. Bioactivity Evaluations of Betulin Identified from the Bark of Betula Platyphylla Var. Japonica for Cancer Therapy. Arch. Pharm. Res. 2018, 41, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Chrobak, E.; Jastrzębska, M.; Bębenek, E.; Kadela-Tomanek, M.; Marciniec, K.; Latocha, M.; Wrzalik, R.; Kusz, J.; Boryczka, S. Molecular Structure, In Vitro Anticancer Study and Molecular Docking of New Phosphate Derivatives of Betulin. Molecules 2021, 26, 737. [Google Scholar] [CrossRef] [PubMed]
- Orchel, A.; Chodurek, E.; Jaworska-Kik, M.; Paduszyński, P.; Kaps, A.; Chrobak, E.; Bębenek, E.; Boryczka, S.; Borkowska, P.; Kasperczyk, J. Anticancer Activity of the Acetylenic Derivative of Betulin Phosphate Involves Induction of Necrotic-Like Death in Breast Cancer Cells In Vitro. Molecules 2021, 26, 615. [Google Scholar] [CrossRef]
- Karagöz, A.Ç.; Leidenberger, M.; Hahn, F.; Hampel, F.; Friedrich, O.; Marschall, M.; Kappes, B.; Tsogoeva, S.B. Synthesis of New Betulinic Acid/Betulin-Derived Dimers and Hybrids with Potent Antimalarial and Antiviral Activities. Bioorg. Med. Chem. 2019, 27, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Viszwapriya, D.; Subramenium, G.A.; Radhika, S.; Pandian, S.K. Betulin Inhibits Cariogenic Properties of Streptococcus Mutans by Targeting VicRK and Gtf Genes. Antonie Van Leeuwenhoek 2017, 110, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Salin, O.; Alakurtti, S.; Pohjala, L.; Siiskonen, A.; Maass, V.; Maass, M.; Yli-Kauhaluoma, J.; Vuorela, P. Inhibitory Effect of the Natural Product Betulin and Its Derivatives against the Intracellular Bacterium Chlamydia Pneumoniae. Biochem. Pharmacol. 2010, 80, 1141–1151. [Google Scholar] [CrossRef]
- Alakurtti, S.; Heiska, T.; Kiriazis, A.; Sacerdoti-Sierra, N.; Jaffe, C.L.; Yli-Kauhaluoma, J. Synthesis and Anti-Leishmanial Activity of Heterocyclic Betulin Derivatives. Bioorg. Med. Chem. 2010, 18, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Pęcak, P.; Orzechowska, B.; Chrobak, E.; Boryczka, S. Novel Betulin Dicarboxylic Acid Ester Derivatives as Potent Antiviral Agents: Design, Synthesis, Biological Evaluation, Structure-Activity Relationship and in-Silico Study. Eur. J. Med. Chem. 2021, 225, 113738. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, Z.; Liu, W.; Han, X.; Zhao, M. Betulin Attenuates Lung and Liver Injuries in Sepsis. Int. Immunopharmacol. 2016, 30, 50–56. [Google Scholar] [CrossRef]
- Wu, H.; Morris-Natschke, S.L.; Xu, X.; Yang, M.; Cheng, Y.; Yu, S.; Lee, K. Recent Advances in Natural Anti HIV Triterpenoids and Analogs. Med. Res. Rev. 2020, 40, 2339–2385. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Jin, J.; Hu, W.; Chen, Q.; Yang, J.; Wu, Y.; Zhou, Y.; Sun, L.; Gao, W.; Zhang, X.; et al. Betulin Alleviates the Inflammatory Response in Mouse Chondrocytes and Ameliorates Osteoarthritis via AKT/Nrf2/HO-1/NF-ΚB Axis. Front. Pharmacol. 2021, 12, 754038. [Google Scholar] [CrossRef] [PubMed]
- Laavola, M.; Haavikko, R.; Hämäläinen, M.; Leppänen, T.; Nieminen, R.; Alakurtti, S.; Moreira, V.M.; Yli-Kauhaluoma, J.; Moilanen, E. Betulin Derivatives Effectively Suppress Inflammation in Vitro and in Vivo. J. Nat. Prod. 2016, 79, 274–280. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, A.; Agnihothri, R.; McGirt, L.Y.; Bankova, L.G.; Beck, L.A. Atopic Dermatitis: A Disease Caused by Innate Immune Defects? J. Investig. Dermatol. 2009, 129, 14–30. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Bel’skii, Y.P.; Bel’skaya, N.V.; Khazanov, V.A. Triphenylphosphonium Cations of Betulinic Acid Derivatives: Synthesis and Antitumor Activity. Med. Chem. Res. 2017, 26, 518–531. [Google Scholar] [CrossRef]
- Honig, B.H.; Hubbell, W.L.; Flewelling, R.F. Electrostatic Interactions in Membranes and Proteins. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 163–193. [Google Scholar] [CrossRef] [PubMed]
- Tsepaeva, O.V.; Nemtarev, A.V.; Abdullin, T.I.; Grigor’eva, L.R.; Kuznetsova, E.V.; Akhmadishina, R.A.; Ziganshina, L.E.; Cong, H.H.; Mironov, V.F. Design, Synthesis, and Cancer Cell Growth Inhibitory Activity of Triphenylphosphonium Derivatives of the Triterpenoid Betulin. J. Nat. Prod. 2017, 80, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Lopez, M.; Joseph, J.; Zielonka, J.; Dwinell, M.B. A Review of the Basics of Mitochondrial Bioenergetics, Metabolism, and Related Signaling Pathways in Cancer Cells: Therapeutic Targeting of Tumor Mitochondria with Lipophilic Cationic Compounds. Redox Biol. 2018, 14, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Marrache, S.; Pathak, R.K.; Dhar, S. Detouring of Cisplatin to Access Mitochondrial Genome for Overcoming Resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 10444–10449. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Hartley, R.C.; Murphy, M.P. Mitochondria-Targeted Small Molecule Therapeutics and Probes. Antioxid. Redox Signal. 2011, 15, 3021–3038. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Dubinin, M.V.; Belosludtsev, K.N. Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria. J. Pers. Med. 2021, 11, 470. [Google Scholar] [CrossRef]
- Han, M.; Vakili, M.R.; Soleymani Abyaneh, H.; Molavi, O.; Lai, R.; Lavasanifar, A. Mitochondrial Delivery of Doxorubicin via Triphenylphosphine Modification for Overcoming Drug Resistance in MDA-MB-435/DOX Cells. Mol. Pharm. 2014, 11, 2640–2649. [Google Scholar] [CrossRef] [PubMed]
- Millard, M.; Gallagher, J.D.; Olenyuk, B.Z.; Neamati, N. A Selective Mitochondrial-Targeted Chlorambucil with Remarkable Cytotoxicity in Breast and Pancreatic Cancers. J. Med. Chem. 2013, 56, 9170–9179. [Google Scholar] [CrossRef] [PubMed]
- Boukalova, S.; Stursa, J.; Werner, L.; Ezrova, Z.; Cerny, J.; Bezawork-Geleta, A.; Pecinova, A.; Dong, L.; Drahota, Z.; Neuzil, J. Mitochondrial Targeting of Metformin Enhances Its Activity against Pancreatic Cancer. Mol. Cancer Ther. 2016, 15, 2875–2886. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Sachaphibulkij, K.; Stursa, J.; Bezawork-Geleta, A.; Blecha, J.; Endaya, B.; Werner, L.; Cerny, J.; Zobalova, R.; Goodwin, J.; et al. Selective Disruption of Respiratory Supercomplexes as a New Strategy to Suppress Her2 high Breast Cancer. Antioxid. Redox Signal. 2017, 26, 84–103. [Google Scholar] [CrossRef]
- Skulachev, V.P. A Biochemical Approach to the Problem of Aging: “Megaproject” on Membrane-Penetrating Ions. The First Results and Prospects. Biochem. Mosc. 2007, 72, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-Targeted Plastoquinone Derivatives as Tools to Interrupt Execution of the Aging Program. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [PubMed]
- Severin, F.F.; Severina, I.I.; Antonenko, Y.N.; Rokitskaya, T.I.; Cherepanov, D.A.; Mokhova, E.N.; Vyssokikh, M.Y.; Pustovidko, A.V.; Markova, O.V.; Yaguzhinsky, L.S.; et al. Penetrating cation/fatty acid anion pair as a mitochondria-targeted protonophore. Proc. Natl. Acad. Sci. USA 2010, 107, 663–668. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. Mitochondrial Pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Keiser, J.; Vargas, M.; Gubaidullin, R.R.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Odinokov, V.N. Synthesis and Activity of New Triphenylphosphonium Derivatives of Betulin and Betulinic Acid against Schistosoma Mansoni in Vitro and in Vivo. Bioorg. Med. Chem. 2014, 22, 6297–6304. [Google Scholar] [CrossRef] [PubMed]
- Nedopekina, D.A.; Gubaidullin, R.R.; Odinokov, V.N.; Maximchik, P.V.; Zhivotovsky, B.; Bel’skii, Y.P.; Khazanov, V.A.; Manuylova, A.V.; Gogvadze, V.; Spivak, A.Y. Mitochondria-Targeted Betulinic and Ursolic Acid Derivatives: Synthesis and Anticancer Activity. MedChemComm 2017, 8, 1934–1945. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Gubaidullin, V.N.; Odinokov, V.N.; Dzhemilev, U.M.; Bel’skii, Y.P.; Bel’skaya, N.V.; Stankevic, S.A.; et al. Synthesis of lupane triterpenoids with triphenylphosphonium substituents and studies of their antitumor activity. Russ. Chem. Bull. Int. Ed. 2013, 62, 188–198. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Nemtarev, A.V.; Salikhova, T.I.; Abdullin, T.I.; Grigor‘eva, L.R.; Khozyainova, S.A.; Mironov, V.F. Synthesis, Anticancer, and Antibacterial Activity of Betulinic and Betulonic Acid C-28-Triphenylphosphonium Conjugates with Variable Alkyl Linker Length. Anticancer Agents Med. Chem. 2020, 20, 286–300. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Mironov, V.F.; Khairutdinov, B.I.; Zue, Y.F. Reaction of 30-Bromolup-20(29)-ene-3β,28-diyl Diacetate with Triphenylphosphine. Russ. J. Org. Chem. 2014, 50, 919–920. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, T.; Yuan, H.; Li, D.; Lou, H.; Fan, P. Mitochondria-Targeted Lupane Triterpenoid Derivatives and Their Selective Apoptosis-Inducing Anticancer Mechanisms. J. Med. Chem. 2017, 60, 6353–6363. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Xu, X.; Liu, J.; Jia, Q.; Ke, C.; Zhang, H.; Xu, C.; Ou, E.; Tan, W. Mitochondria-Targeted Triphenylphosphonium Conjugated C-3 Modified Betulin: Synthesis, Antitumor Properties and Mechanism of Action. ChemMedChem 2022, 17, e202100659. [Google Scholar] [CrossRef] [PubMed]
- Grymel, M.; Zawojak, M.; Adamek, J. Triphenylphosphonium Analogues of Betulin and Betulinic Acid with Biological Activity: A Comprehensive Review. J. Nat. Prod. 2019, 82, 1719–1730. [Google Scholar] [CrossRef]
- Thibeault, D.; Gauthier, C.; Legault, J.; Bouchard, J.; Dufour, P.; Pichette, A. Synthesis and Structure–Activity Relationship Study of Cytotoxic Germanicane- and Lupane-Type 3β-O-Monodesmosidic Saponins Starting from Betulin. Bioorg. Med. Chem. 2007, 15, 6144–6157. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, O.B.; Giniyatullina, G.V.; Yamansarov, E.Y.; Tolstikov, G.A. Betulin and Ursolic Acid Synthetic Derivatives as Inhibitors of Papilloma Virus. Bioorg. Med. Chem. Lett. 2010, 20, 4088–4090. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Chiyo, J.; Ikeshiro, Y.; Nagao, T.; Okabe, H.; Cosentino, L.M.; Fowke, K.; Lee, K.H. 3,28-Di-O-(Dimethylsuccinyl)-Betulin Isomers as Anti-HIV Agents. Bioorg. Med. Chem. Lett. 2001, 11, 183–185. [Google Scholar] [CrossRef]
- Komissarova, N.G.; Dubovitskii, S.N.; Orlov, A.V.; Shitikova, O.V. New Conjugates of Betulin with 2-Aminoethanesulfonic Acid. Chem. Nat. Compd. 2019, 55, 300–304. [Google Scholar] [CrossRef]
- Krukiewicz, K.; Bednarczyk-Cwynar, B.; Turczyn, R.; Zak, J.K. EQCM Verification of the Concept of Drug Immobilization and Release from Conducting Polymer Matrix. Electrochim. Acta 2016, 212, 694–700. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 13C NMR (CDCl3, TMS, δ (ppm)/JC-P (Hz) | |||||
|---|---|---|---|---|---|
| TPP⊕ | |||||
| CH2P⊕ | Cipso | Cmeta | Cortho | Cpara | |
| 7a | 19.8/52.3 | 118.1/85.7 | 130.5/12.1 | 133.8/9.8 | 135.0/3.0 |
| 7b | 22.2/50.6 | 118.3/84.9 | 130.4/12.1 | 133.8/9.9 | 134.9/3.0 |
| 7c | 22.7/49.4 | 118.4/85.0 | 130.5/12.1 | 133.7/9.8 | 134.9/3.0 |
| 7d | 19.4/51.8 | 118.3/86.3 | 130.4/12.6 | 133.8/10.4 | 134.9/0.5 |
| 7e | 22.1/49.3 | 118.3/88.1 | 130.4/12.6 | 133.7/10.4 | 134.9/0.5 |
| 7f | 22.7/50.1 | 118.4/85.8 | 130.4/12.4 | 133.7/9.9 | 134.9/3.0 |
| 9a | 19.7/51.8 | 118.1/86.3 | 130.5/12.8 | 133.8/9.2 | 135.1/0.5 |
| 9b | 22.2/50.0 | 118.2/88.8 | 130.4/12.9 | 133.7/9.9 | 135.0/3.0 |
| 9c | 22.8/49.3 | 118.4/85.0 | 130.5/12.2 | 133.8/9.8 | 135.0/3.0 |
| δ, ppm | 19.4–22.8 | 118.1–118.4 | 130.4–130.5 | 133.7–133.8 | 134.9–135.0 |
| JC-P, Hz | 49.3–52.3 | 84.9–88.1 | 12.1–12.9 | 9.2–10.4 | 0.5–3.0 |
| No. | R | n | Activity IC50, µM a,b | ||
|---|---|---|---|---|---|
| HCT 116 | MCF-7 | NHDF | |||
| BN | − | − | neg | neg | neg |
| 7a | H | 3 | 5.56 ± 0.28 | 13.71± 0.54 | 9.68 ± 0.27 |
| 7b | H | 4 | 5.77 ± 0.27 | 14.35 ± 0.38 | 10.71 ± 0.25 |
| 7c | H | 5 | 6.48 ± 0.04 | 15.52 ± 0.92 | 11.29 ± 0.53 |
| 7f | Me | 5 | 12.71 ± 0.89 | 50.47 ± 3.92 | 10.03 ± 0.48 |
| 9a | H | 3 | 6.32 ± 0.27 | 31.30 ± 3.02 | 5.91 ± 0.33 |
| 9b | H | 4 | 7.97 ± 0.51 | 23.60 ± 0.33 | 8.02 ± 0.35 |
| 9c | H | 5 | 18.99 ± 0.51 | 53.30 ± 5.41 | 10.60 ± 0.34 |
| No. | S. aureus ATCC 25923 | S. epidermidis ATCC 12228 | Escherichia coli ATCC 25922 | |||
|---|---|---|---|---|---|---|
| 200 μM | 250 μM | 200 μM | 250 μM | 200 μM | 250 μM | |
| BN | 5.1 | 5.6 | neg | 4.7 | neg | neg |
| 7a | 0.9 | 0 | neg | 0 | neg | neg |
| 7b | 2.2 | 0 | neg | 0 | neg | neg |
| 7c | 1.0 | 0.1 | neg | 0.7 | neg | neg |
| 7d | 0 | 0 | 0 | 0 | neg | neg |
| 7e | 0 | 0 | 0 | 0 | neg | neg |
| 7f | 0 | 0 | 0 | 0 | neg | neg |
| 9a | 0 | 0 | 0 | 0 | neg | neg |
| 9b | 0 | 0 | 0 | 0 | neg | neg |
| 9c | 0 | 0 | 0 | 0 | neg | neg |
| Control | 7.5 | 7.5 | 4.1 | 4.9 | 5.1 | 5.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grymel, M.; Lalik, A.; Kazek-Kęsik, A.; Szewczyk, M.; Grabiec, P.; Erfurt, K. Design, Synthesis and Preliminary Evaluation of the Cytotoxicity and Antibacterial Activity of Novel Triphenylphosphonium Derivatives of Betulin. Molecules 2022, 27, 5156. https://doi.org/10.3390/molecules27165156
Grymel M, Lalik A, Kazek-Kęsik A, Szewczyk M, Grabiec P, Erfurt K. Design, Synthesis and Preliminary Evaluation of the Cytotoxicity and Antibacterial Activity of Novel Triphenylphosphonium Derivatives of Betulin. Molecules. 2022; 27(16):5156. https://doi.org/10.3390/molecules27165156
Chicago/Turabian StyleGrymel, Mirosława, Anna Lalik, Alicja Kazek-Kęsik, Marietta Szewczyk, Patrycja Grabiec, and Karol Erfurt. 2022. "Design, Synthesis and Preliminary Evaluation of the Cytotoxicity and Antibacterial Activity of Novel Triphenylphosphonium Derivatives of Betulin" Molecules 27, no. 16: 5156. https://doi.org/10.3390/molecules27165156
APA StyleGrymel, M., Lalik, A., Kazek-Kęsik, A., Szewczyk, M., Grabiec, P., & Erfurt, K. (2022). Design, Synthesis and Preliminary Evaluation of the Cytotoxicity and Antibacterial Activity of Novel Triphenylphosphonium Derivatives of Betulin. Molecules, 27(16), 5156. https://doi.org/10.3390/molecules27165156