
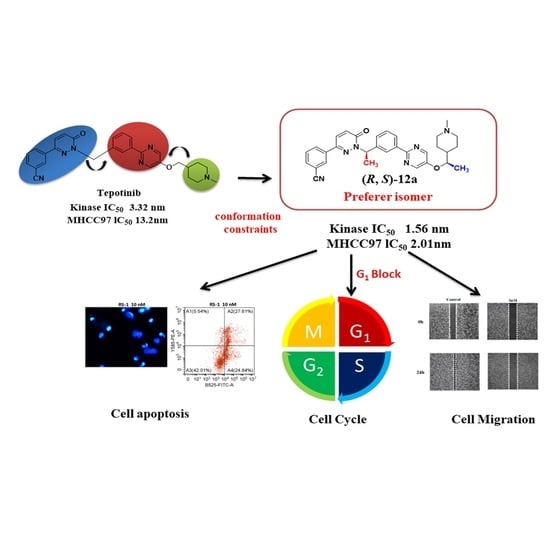
Design, Synthesis, and Evaluation of New Mesenchymal–Epithelial Transition Factor (c-Met) Kinase Inhibitors with Dual Chiral Centers


Abstract
:
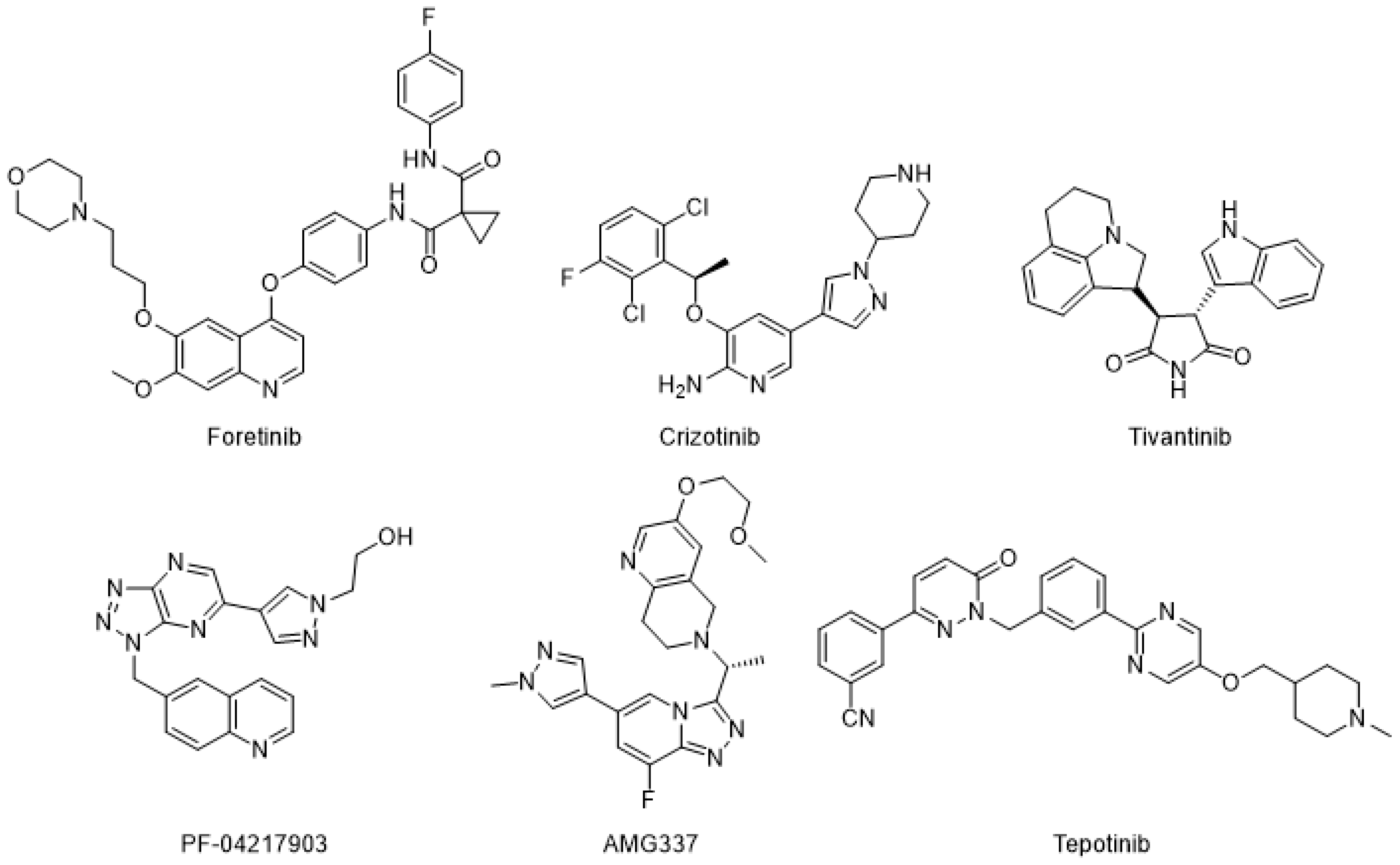
1. Introduction
2. Results and Discussion
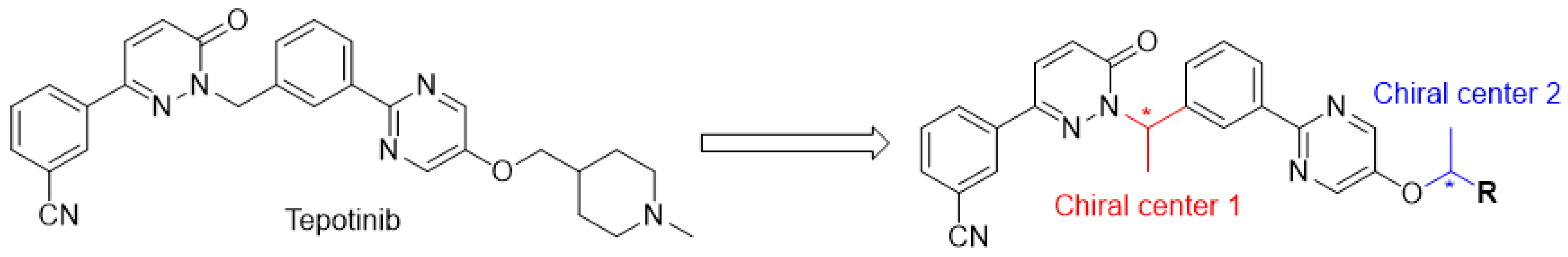
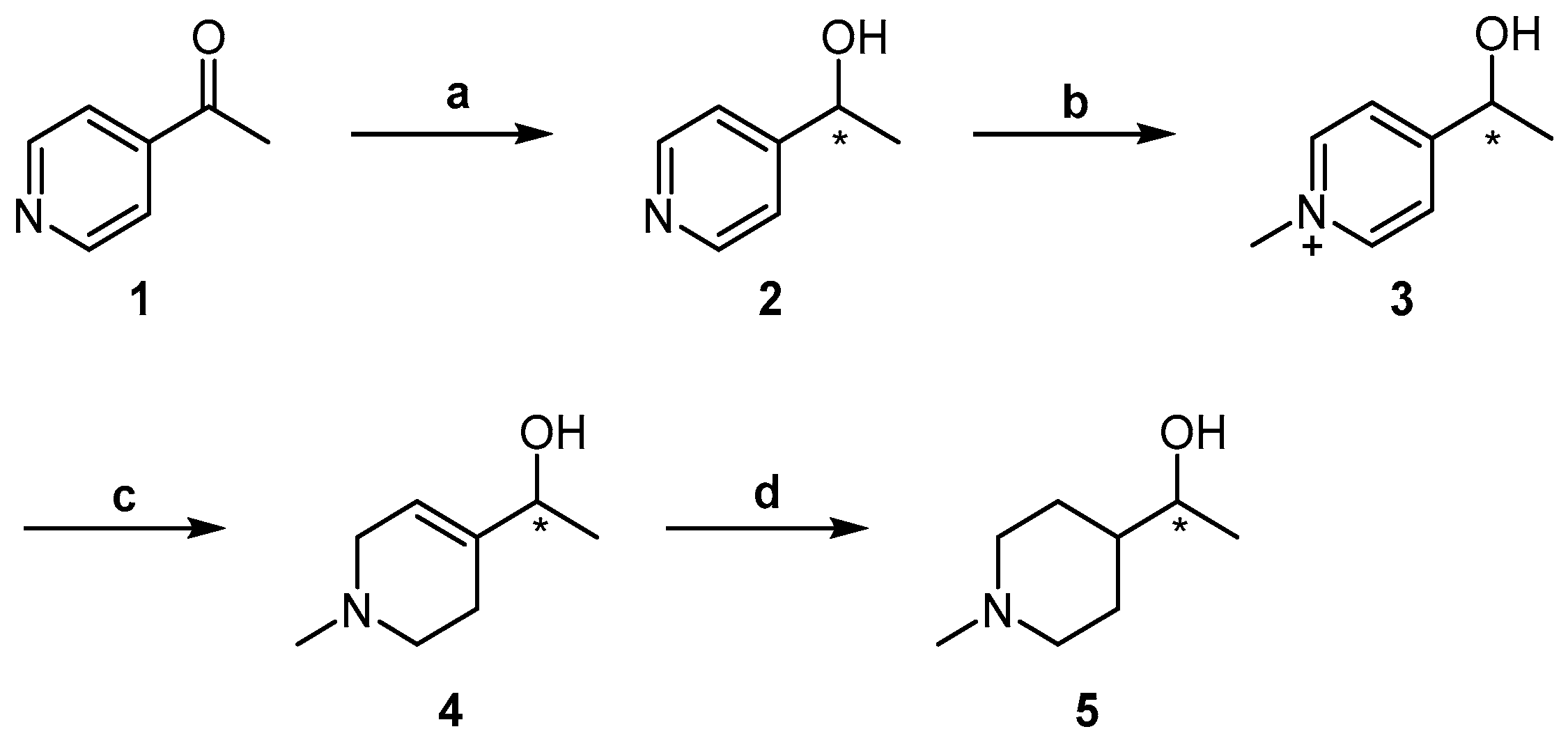
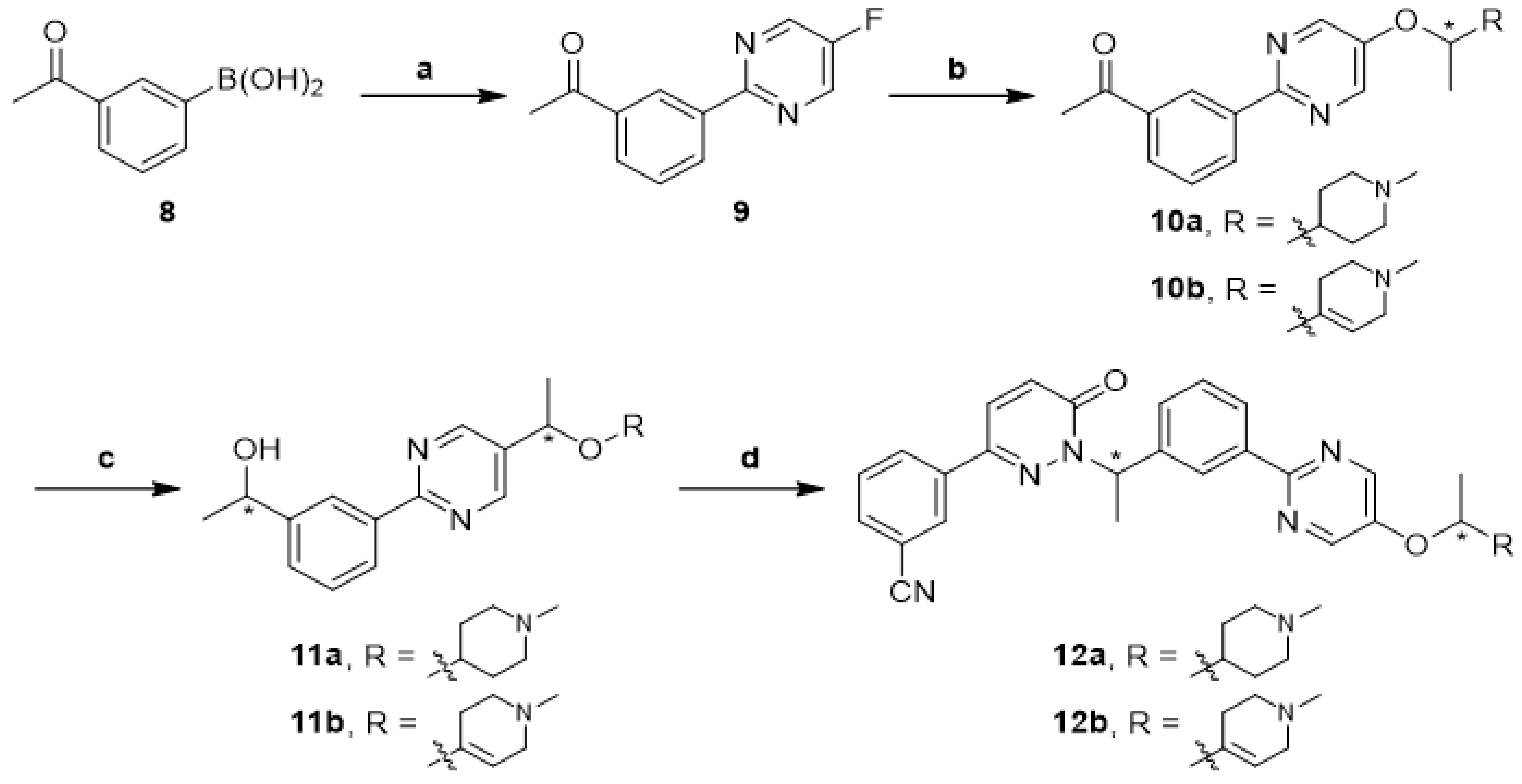
2.1. Chemistry
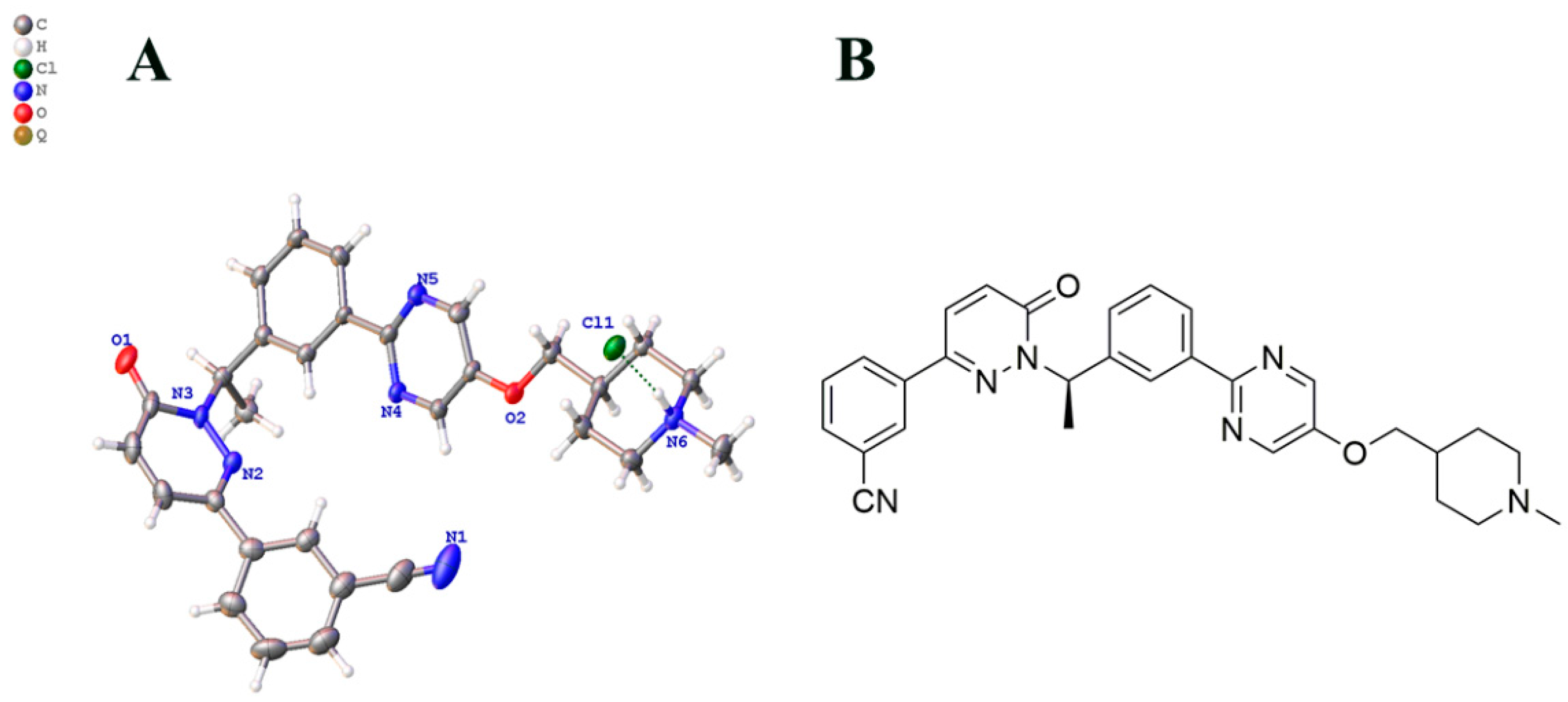
2.2. The Preperation of Monocrystal
2.3. The Result of X-ray Diffraction
2.4. In Vitro Antiproliferative Activities and SARs
2.5. Toxicity towards Normal Cells
2.6. Effects of Compound (R, S)-12a on Tumor Cell Morphology
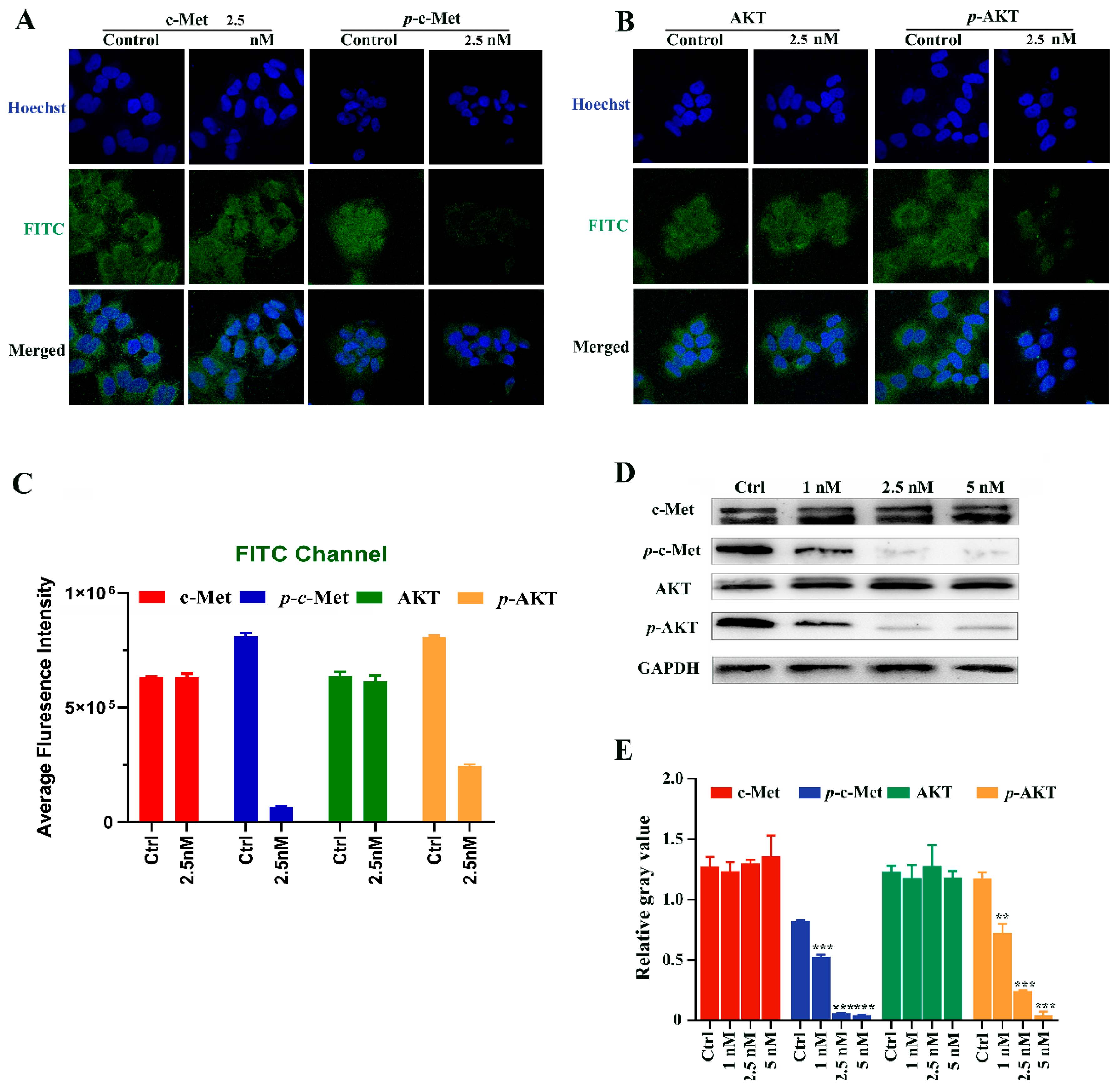
2.7. Immunofluorescence Staining and Western Blotting
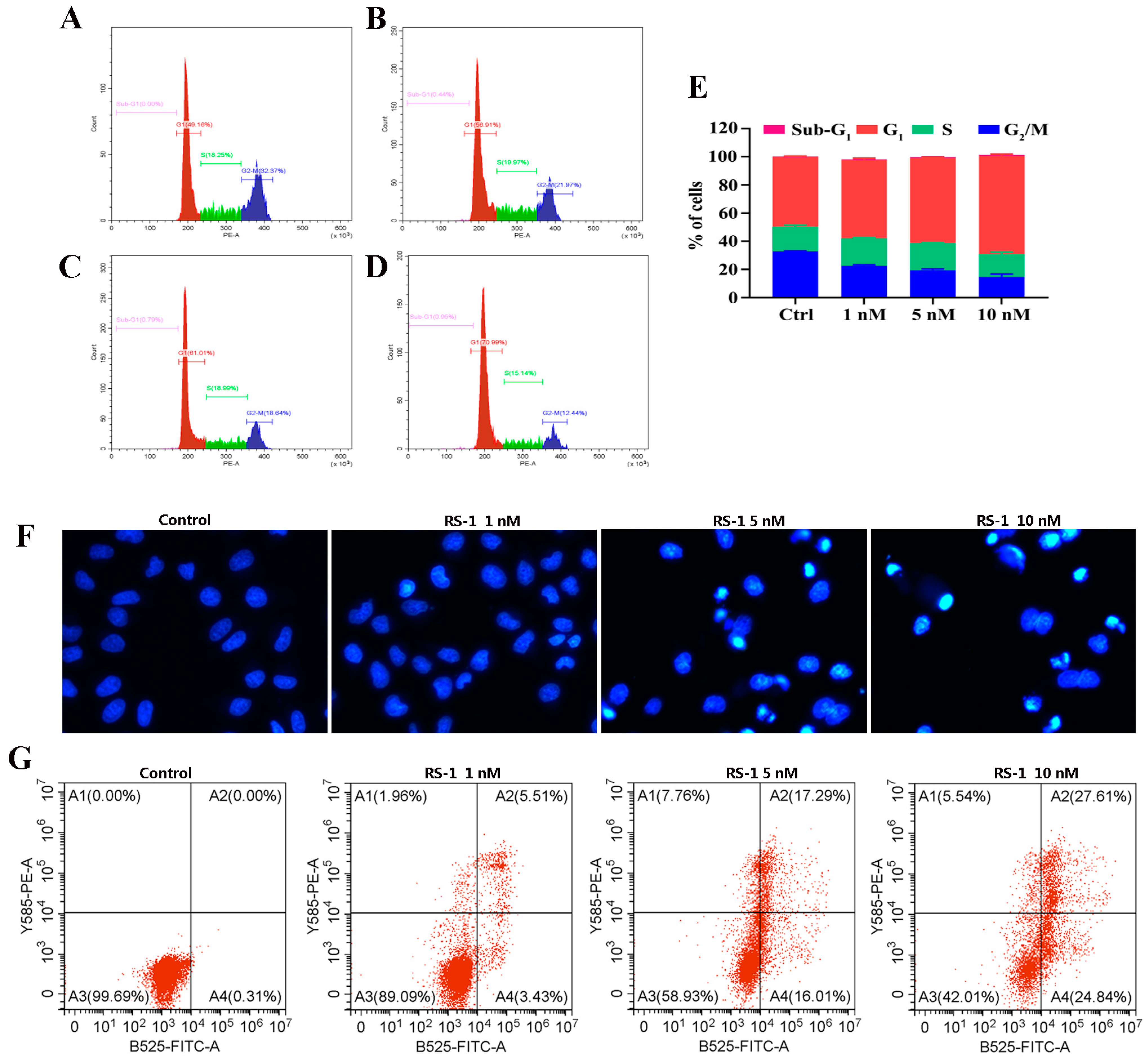
2.8. Cell Cycle Study
2.9. Cellular Apoptosis Analysis
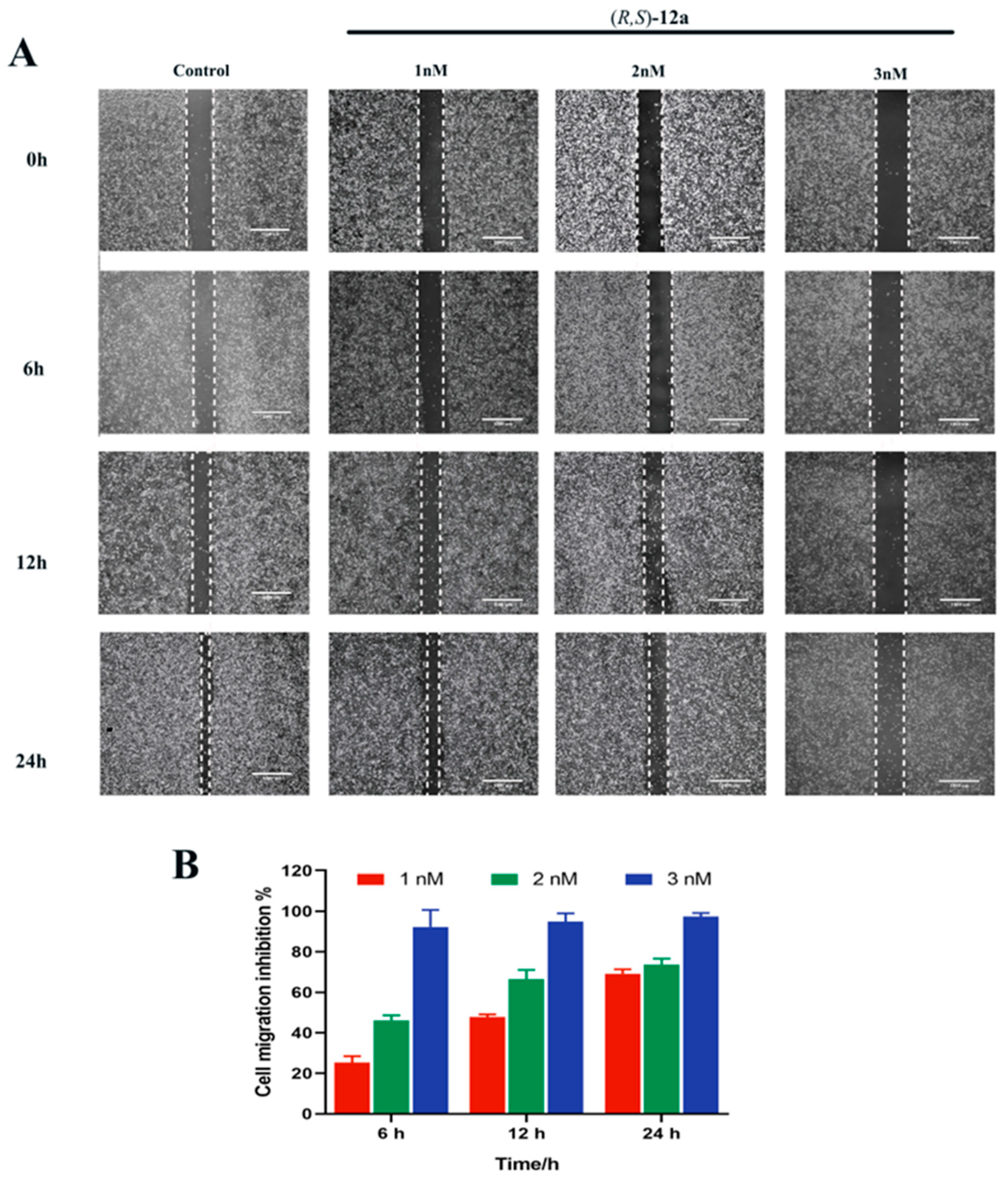
2.10. (R, S)-12a Inhibited MHCC-97H Tumor Cell Migration
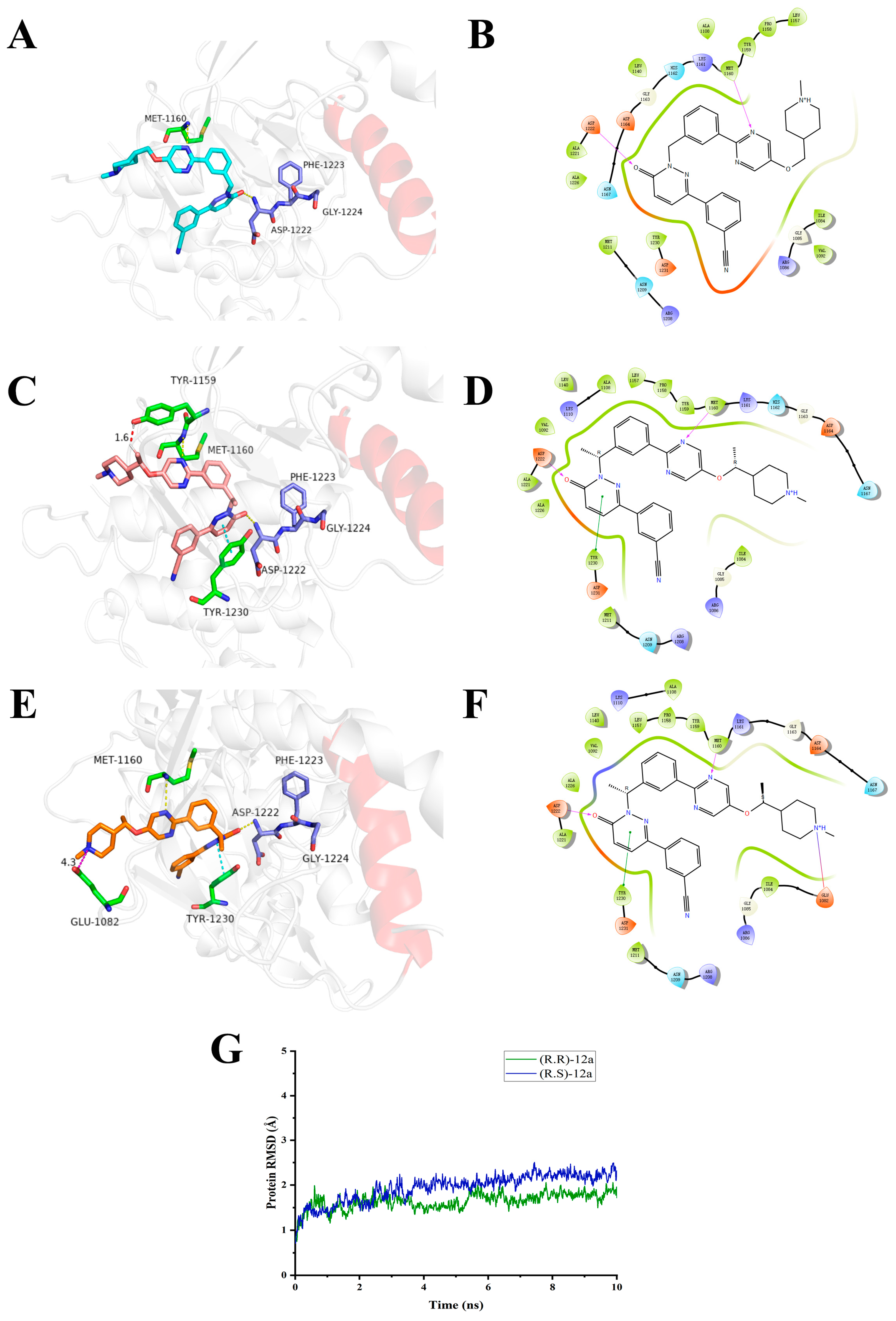
2.11. Computational Characterization of the Binding Modes between Compounds 12a and c-Met
2.12. Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Methods and Materials for Synthesis
4.1.2. General Procedures for Preparation of Compounds (R)-2 and (S)-2
4.1.3. General Procedures for Preparation of Compounds (R)-4 and (S)-4
4.1.4. General Procedures for Preparation of Compounds (R)-5 and (S)-5 [11]
1-(3-(5-Fluoropyrimidin-2-yl)phenyl)ethan-1-one (9)
(S,R)-1-(3-(5-(1-((1-Methyl-1,2,3,6-tetrahydropyridin-4-yl)oxy)ethyl)pyrimidin-2-yl)phenyl)ethan-1-ol (11)
General Procedures for Preparation of Compounds 12a-b
(R,R)-3-(1-(1-(3-(5-(1-((1-Methylpiperidin-4-yl)oxy)ethyl)pyrimidin-2-yl)phenyl)ethyl)-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile ((R, R)-12a)
(R,S)-3-(1-(1-(3-(5-(1-((1-Methylpiperidin-4-yl)oxy)ethyl)pyrimidin-2-yl)phenyl)ethyl)-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile ((R, S)-12a)
(S,R)-3-(1-(1-(3-(5-(1-((1-Methylpiperidin-4-yl)oxy)ethyl)pyrimidin-2-yl)phenyl)ethyl)-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile ((S, R)-12a)
(S,S)-3-(1-(1-(3-(5-(1-((1-Methylpiperidin-4-yl)oxy)ethyl)pyrimidin-2-yl)phenyl)ethyl)-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile ((S, S)-12a)
(R,R)-3-(1-(1-(3-(5-(1-((1-Methyl-1,2,3,6-tetrahydropyridin-4-yl)oxy)ethyl)pyrimidin-2-yl)phenyl)ethyl)-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile ((R, R)-12b)
(R,S)-3-(1-(1-(3-(5-(1-((1-Methyl-1,2,3,6-tetrahydropyridin-4-yl)oxy)ethyl)pyrimidin-2-yl)phenyl)ethyl)-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile ((R, S)-12b)
(S,R)-3-(1-(1-(3-(5-(1-((1-Methyl-1,2,3,6-tetrahydropyridin-4-yl)oxy)ethyl)pyrimidin-2-yl)phenyl)ethyl)-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile ((S, R)-12b)
(S,S)-3-(1-(1-(3-(5-(1-((1-Methyl-1,2,3,6-tetrahydropyridin-4-yl)oxy)ethyl)pyrimidin-2-yl)phenyl)ethyl)-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile ((S, S)-12b)
4.2. Biological Effects
4.2.1. Cell Culture and Cytotoxicity Assay
4.2.2. Kinase Inhibition Assay
4.2.3. Cell Cycle Analysis
4.2.4. Cellular Apoptosis Analysis [31]
4.2.5. Wound Healing Assay [34]
4.2.6. Immunofluorescence Staining
4.2.7. Western Blotting Analysis [35,36,37]
4.2.8. Molecular Docking
4.2.9. Molecular Dynamics Simulations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.-L.; Kmiecik, T.E.; Woude, G.F.V.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-Met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef]
- Wang, H.; Rao, B.; Lou, J.; Li, J.; Liu, Z.; Li, A.; Cui, G.; Ren, Z.; Yu, Z. The function of the HGF/c-Met axis in hepatocellular carcinoma. Front. Cell Dev. Biol. 2020, 8, 55–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldahl, J.; Mi, J.; Pineda, A.; Kim, W.K.; Olson, A.; Hooker, E.; He, Y.; Yu, E.J.; Le, V.; Lee, D.H.; et al. Aberrant activation of hepatocyte growth factor/MET signaling promotes β-catenin-mediated prostatic tumorigenesis. J. Biol. Chem. 2020, 295, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-dependent solid tumours–molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.-P.; Tong, X.-M.; Wang, M.-H. Oncogenic mechanism-based pharmaceutical validation of therapeutics targeting MET receptor tyrosine kinase. Ther. Adv. Med. Oncol. 2021, 13, 1–25. [Google Scholar] [CrossRef]
- Maulik, G.; Shrikhande, A.; Kijima, T.; Ma, P.C.; Morrison, P.T.; Salgia, R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002, 13, 41–59. [Google Scholar] [CrossRef]
- Benvenuti, S.; Comoglio, P.M. The MET receptor tyrosine kinase in invasion and metastasis. J. Cell. Physiol. 2007, 213, 316–325. [Google Scholar] [CrossRef]
- Knudsen, B.S.; Vande Woude, G. Showering c-MET-dependent cancers with drugs. Curr. Opin. Genet. Dev. 2008, 18, 87–96. [Google Scholar] [CrossRef]
- Bouattour, M.; Raymond, E.; Qin, S.; Cheng, A.L.; Stammberger, U.; Locatelli, G.; Faivre, S. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 2018, 67, 1132–1149. [Google Scholar] [CrossRef]
- Bradley, C.A.; Salto-Tellez, M.; Laurent-Puig, P.; Bardelli, A.; Rolfo, C.; Tabernero, J.; Khawaja, H.A.; Lawler, M.; Johnston, P.G.; Van Schaeybroeck, S. Targeting c-Met in gastrointestinal tumours: Rationale, opportunities and challenges. Nat. Rev. Clin. Oncol. 2017, 14, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, F.; Giovannetti, E.; Saso, L.; Firuzi, O. HGF/MET pathway aberrations as diagnostic, prognostic, and predictive biomarkers in human cancers. Crit. Rev. Clin. Lab. Sci. 2019, 56, 533–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, J.G.; Burrows, J.; Salgia, R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005, 225, 1–26. [Google Scholar] [CrossRef]
- Salgia, R.; Sattler, M.; Scheele, J.; Stroh, C.; Felip, E. The promise of selective MET inhibitors in non-small cell lung cancer with MET exon 14 skipping. Cancer Treat. Rev. 2020, 87, 102022–102033. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual Inhibitor of mesenchymal–epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Munshi, N.; Jeay, S.; Li, Y.; Chen, C.R.; France, D.S.; Ashwell, M.A.; Hill, J.; Moussa, M.M.; Leggett, D.S.; Li, C.J. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol. Cancer Ther. 2010, 9, 1544–1553. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Engst, S.; Yamaguchi, K.; Yu, P.; Won, K.A.; Mock, L.; Lou, T.; Tan, J.; Li, C.; Tam, D.; et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009, 69, 8009–8016. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.J.; McTigue, M.; Nambu, M.; Tran-Dubé, M.; Pairish, M.; Shen, H.; Jia, L.; Cheng, H.; Hoffman, J.; Le, P.; et al. Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-MET) protein kinase inhibitors and identification of the clinical candidate 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo [4,5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)ethanol (PF-04217903) for the treatment of cancer. J. Med. Chem. 2012, 55, 8091–8109. [Google Scholar]
- Boezio, A.A.; Copeland, K.W.; Rex, K.; Albrecht, B.K.; Bauer, D.; Bellon, S.F.; Boezio, C.; Broome, M.A.; Choquette, D.; Coxon, A.; et al. Discovery of (R)-6-(1-(8-Fluoro-6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-1,6-naphthyridin-5(6H)-one (AMG 337), a potent and selective inhibitor of MET with high unbound target coverage and robust in vivo antitumor activity. J. Med. Chem. 2016, 59, 2328–2342. [Google Scholar] [PubMed] [Green Version]
- Dorsch, D.; Schadt, O.; Stieber, F.; Meyring, M.; Grädler, U.; Bladt, F.; Friese-Hamim, M.; Knühl, C.; Pehl, U.; Blaukat, A. Identification and optimization of pyridazinones as potent and selective c-Met kinase inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Tepotinib: First Approval. Drugs 2020, 80, 829–833. [Google Scholar] [CrossRef]
- Smyth, E.C.; Sclafani, F.; Cunningham, D. Emerging molecular targets in oncology: Clinical potential of MET/hepatocyte growth-factor inhibitors. Onco Targets Ther. 2014, 7, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.H.; Park, B.H.; Hong, S.S. Progress in cancer therapy targeting c-Met signaling pathway. Arch. Pharm. Res. 2012, 35, 595–604. [Google Scholar] [CrossRef]
- Rucki, A.A.; Xiao, Q.; Muth, S.; Chen, J.; Che, X.; Kleponis, J.; Sharma, R.; Anders, R.A.; Jaffee, E.M.; Zheng, L. Dual Inhibition of Hedgehog and c-Met Pathways for Pancreatic Cancer Treatment. Mol. Cancer Ther. 2017, 16, 2399–2409. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef]
- Puccini, A.; Marín-Ramos, N.I.; Bergamo, F.; Schirripa, M.; Lonardi, S.; Lenz, H.-J.; Loupakis, F.; Battaglin, F. Safety and Tolerability of c-MET Inhibitors in Cancer. Drug Saf. 2019, 42, 211–233. [Google Scholar] [CrossRef]
- ZiJian, Z.; Li, C.; Ming, Z.; Xin, W. Research progress of c-Met kinase inhibitors. Chin. J. Med. Chem. 2020, 30, 493–501. [Google Scholar]
- MacKenzie, L.E.; Stachelek, P. The twists and turns of chiral chemistry. Nat. Chem. 2021, 13, 521–522. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B. Pharmacologically active compounds in the environment and their chirality. Chem. Soc. Rev. 2010, 39, 4466–4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, D.; Kharbanda, A.; Yan, W.; Lakkaniga, N.R.; Frett, B.; Li, H.-Y. The exploration of chirality for improved druggability within the human kinome. J. Med. Chem. 2020, 63, 441–469. [Google Scholar] [CrossRef]
- Hao, H.; Wang, G.; Sun, J. Enantioselective pharmacokinetics of ibuprofen and involved mechanisms. Drug Metab. Rev. 2005, 37, 215–234. [Google Scholar] [CrossRef]
- Kramer, C.; Ting, A.; Zheng, H.; Hert, J.; Schindler, T.; Stahl, M.; Robb, G.; Crawford, J.J.; Blaney, J.; Montague, S.; et al. Learning medicinal chemistry absorption, distribution, metabolism, excretion, and toxicity (ADMET) rules from cross-company matched molecular pairs analysis (MMPA). J. Med. Chem. 2018, 61, 3277–3292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yu, L.S.; Zeng, S. Stereoselectivity of chiral drug transport: A focus on enantiomer-transporter interaction. Drug Metab. Rev. 2014, 46, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef]
- Luo, Z.; Qin, F.; Yan, S.; Li, X. An efficient and promising method to prepare Ladostigil (TV3326) via asymmetric transfer hydrogenation catalyzed by Ru–Cs-DPEN in an HCOONa–H2O–surfactant system. Tetrahedron Asymmetry 2012, 23, 333–338. [Google Scholar] [CrossRef]
- Yao, H.; Yan, M.; Li, X.; Hu, J. Crystal structure of 4-(((2-(3-(1-(3-(3-cyanophenyl)-6-oxopyridazin-1(6H)-yl)ethyl)phenyl) pyrimidin-5-yl)oxy)methyl)-1-methylpiperidin-1-ium chloride monohydrate, C30H33N6O2Cl. Z. Für Krist. New Cryst. Struct. 2022. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; An, B.; Song, X.; Zhang, Q.; Chen, C.; Wei, S.; Fan, R.; Li, X.; Zou, Y. Design, synthesis and biological evaluation of novel 2,4-diaryl pyrimidine derivatives as selective EGFR (L858R/T790M) inhibitors. Eur. J. Med. Chem. 2021, 212, 113019–113033. [Google Scholar] [CrossRef]
- Matsumoto, K.; Umitsu, M.; De Silva, D.M.; Roy, A.; Bottaro, D.P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017, 108, 296–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Sun, G.; Chen, D.; Peng, X.; Chen, Y.L.; Su, Y.; Ji, Y.; Liang, J.; Wang, X.; Chen, L.; et al. Design and optimization of a series of 1-sulfonylpyrazolo[4,3-b]pyridines as selective c-Met inhibitors. J. Med. Chem. 2015, 58, 2513–2529. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-J.; Wu, J.-Q.; Hu, Y.; Pu, S.; Lin, Y.; Zeng, Z.; Hu, J.; Chen, W.-H. Design, synthesis and biological evaluation of indole-based [1,2,4]triazolo[4,3-a] pyridine derivatives as novel microtubule polymerization inhibitors. Eur. J. Med. Chem. 2021, 223, 113629–113643. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, Z.; Liu, W.; Chen, J.; Zheng, Z.; Duan, M. The Conformational Transition Pathways and Hidden Intermediates in DFG-Flip Process of c-Met Kinase Revealed by Metadynamics Simulations. J. Chem. Inf. Model 2022, 62, 3651–3663. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Kong, D.; Jiang, H.; Wang, J.; Cheng, M. In Silico exploration of aryl sulfonamide analogs as voltage-gated sodium channel 1.7 inhibitors by using 3D-QSAR, molecular docking study, and molecular dynamics simulations. Comput. Biol. Chem. 2018, 77, 214–225. [Google Scholar] [CrossRef]
- Song, P.L.; Wang, G.; Su, Y.; Wang, H.X.; Wang, J.; Li, F.; Cheng, M.S. Strategy and validation of a structure-based method for the discovery of selective inhibitors of PAK isoforms and the evaluation of their anti-cancer activity. Bioorg. Chem. 2019, 91, 103168. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Comp. | Config. of C1 | Config. of C2 | R1 | R2 | R3 | Kinase | MHCC97H Cell |
| IC50 (μM) a | |||||||
| (R, R)-12a | R | R | -CH3 | -CH3 |  | 0.0287 ± 0.005 | 0.030 ± 0.008 |
| (R, S)-12a | R | S | -CH3 | -CH3 | 0.00156 ± 0.001 | 0.002 ± 0.002 | |
| (S, R)-12a | S | R | -CH3 | -CH3 | 0.195 ± 0.0732 | 0.21 ± 0.095 | |
| (S, S)-12a | S | S | -CH3 | -CH3 | 0.0358 ± 0.0068 | 0.048 ± 0.018 | |
| (R, R)-12b | R | R | -CH3 | -CH3 |  | / | 0.13 ± 0.038 |
| (R, S)-12b | R | S | -CH3 | -CH3 | / | 0.009 ± 0.005 | |
| (S, R)-12b | S | R | -CH3 | -CH3 | / | 0.616 ± 0.0782 | |
| (S, S)-12b | S | S | -CH3 | -CH3 | / | 0.136 ± 0.052 | |
| (±)-5b | / | / | -CH3 | -H |  | / | 0.0353 ± 0.011 |
| (R)-5b | R | / | -CH3 | -H | / | 0.0037 ± 0.001 | |
| (S)-5b | S | / | -CH3 | -H | / | 0.1182 ± 0.056 | |
| Tepotinib | / | / | / | 0.0033 ± 0.0012 | 0.013 ± 0.002 | ||
| Compound | MHCC-97H | HaCat | Selectivity Index | LO2 | Selectivity Index |
|---|---|---|---|---|---|
| Tepotinib | 0.013 ± 0.002 μM | 1.951 ± 0.349 μM | 150.08 | 8.73 ± 0.841 μM | 671.54 |
| (R, S)-12a | 0.002 ± 0.002 μM | 1.907 ± 0.25 μM | 953.5 | 11.50 ± 1.53 μM | 5750.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, H.; Ren, Y.; Yan, J.; Liu, J.; Hu, J.; Yan, M.; Li, X. Design, Synthesis, and Evaluation of New Mesenchymal–Epithelial Transition Factor (c-Met) Kinase Inhibitors with Dual Chiral Centers. Molecules 2022, 27, 5359. https://doi.org/10.3390/molecules27175359
Yao H, Ren Y, Yan J, Liu J, Hu J, Yan M, Li X. Design, Synthesis, and Evaluation of New Mesenchymal–Epithelial Transition Factor (c-Met) Kinase Inhibitors with Dual Chiral Centers. Molecules. 2022; 27(17):5359. https://doi.org/10.3390/molecules27175359
Chicago/Turabian StyleYao, Han, Yuanyuan Ren, Jun Yan, Jiadai Liu, Jinhui Hu, Ming Yan, and Xingshu Li. 2022. "Design, Synthesis, and Evaluation of New Mesenchymal–Epithelial Transition Factor (c-Met) Kinase Inhibitors with Dual Chiral Centers" Molecules 27, no. 17: 5359. https://doi.org/10.3390/molecules27175359
APA StyleYao, H., Ren, Y., Yan, J., Liu, J., Hu, J., Yan, M., & Li, X. (2022). Design, Synthesis, and Evaluation of New Mesenchymal–Epithelial Transition Factor (c-Met) Kinase Inhibitors with Dual Chiral Centers. Molecules, 27(17), 5359. https://doi.org/10.3390/molecules27175359