
Synthesis, Structure and Antileishmanial Evaluation of Endoperoxide–Pyrazole Hybrids

 , , ,
, , ,  , and
, and 
Abstract
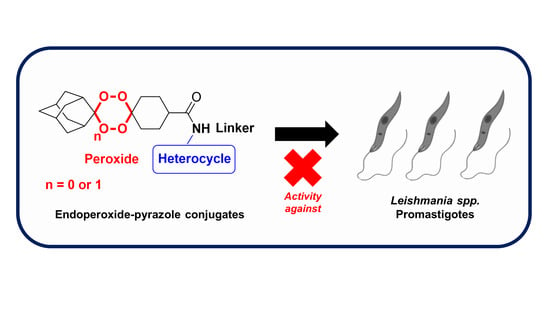
:
1. Introduction
2. Results and Discussion
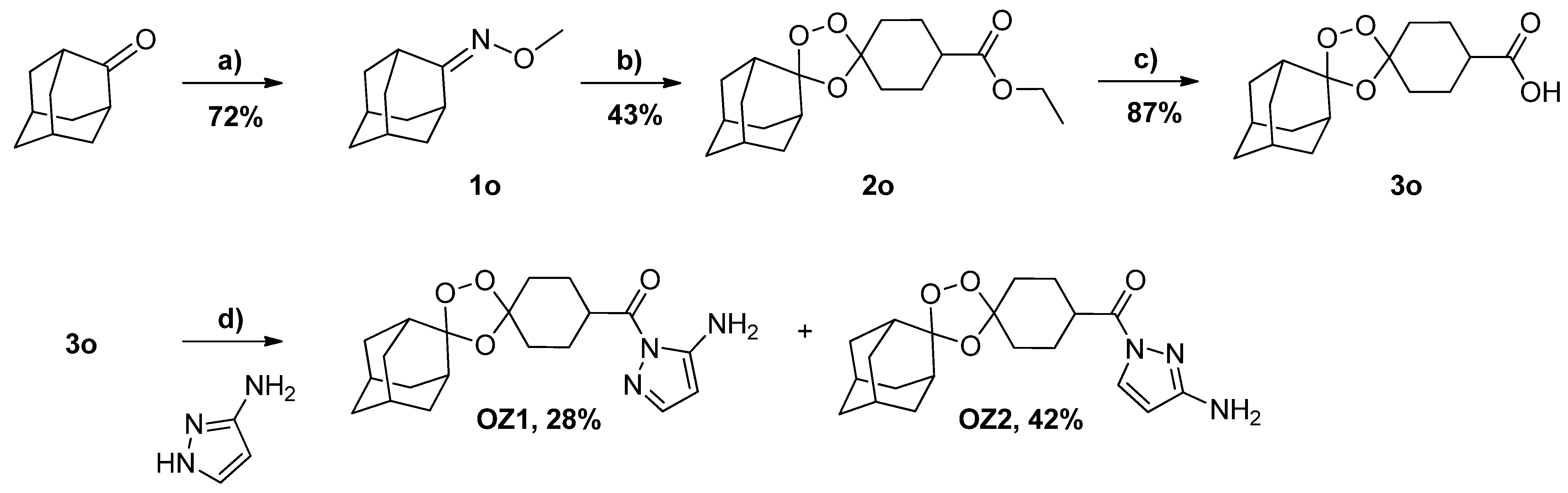
2.1. Synthesis and Structure Analysis
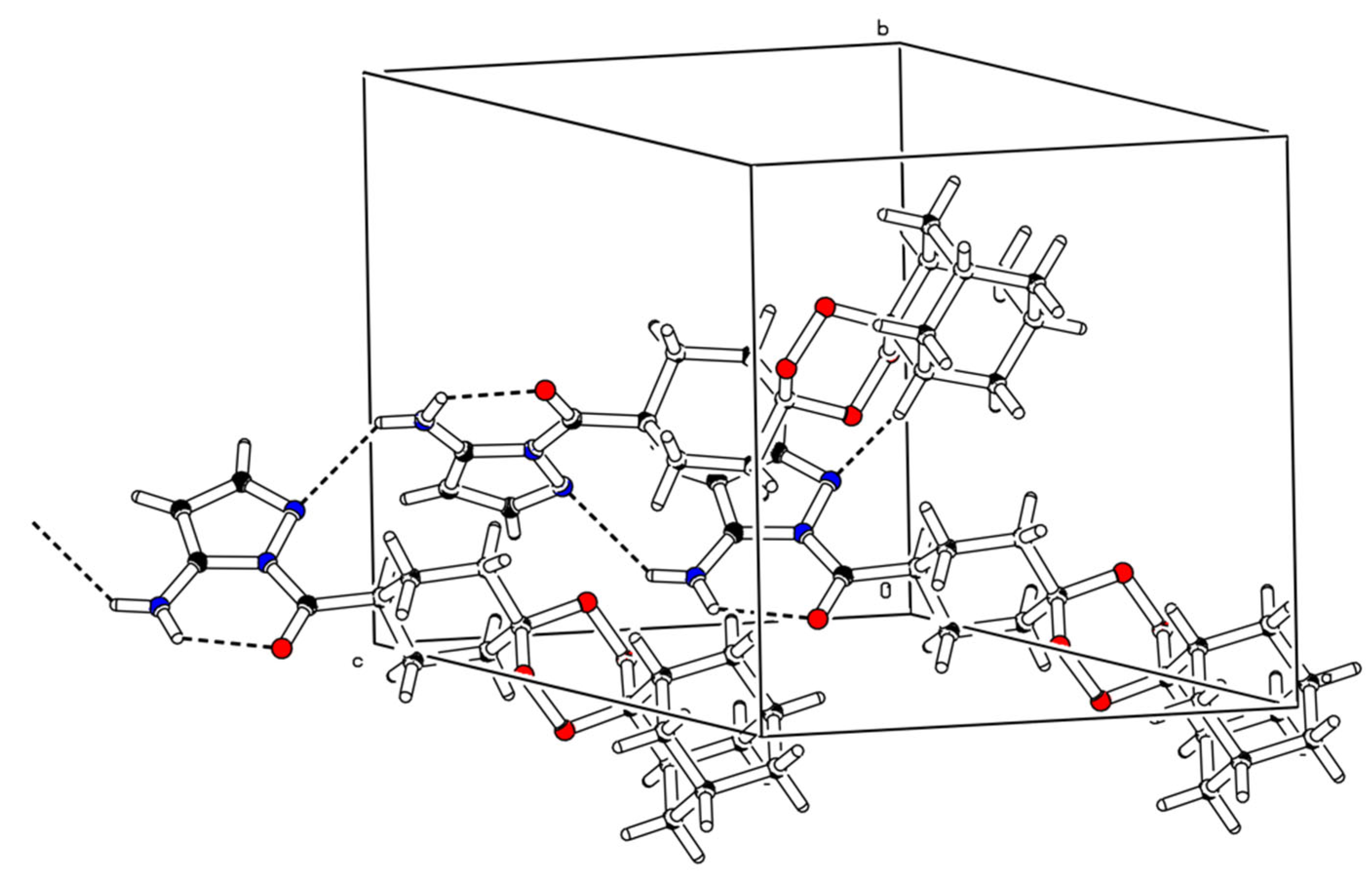
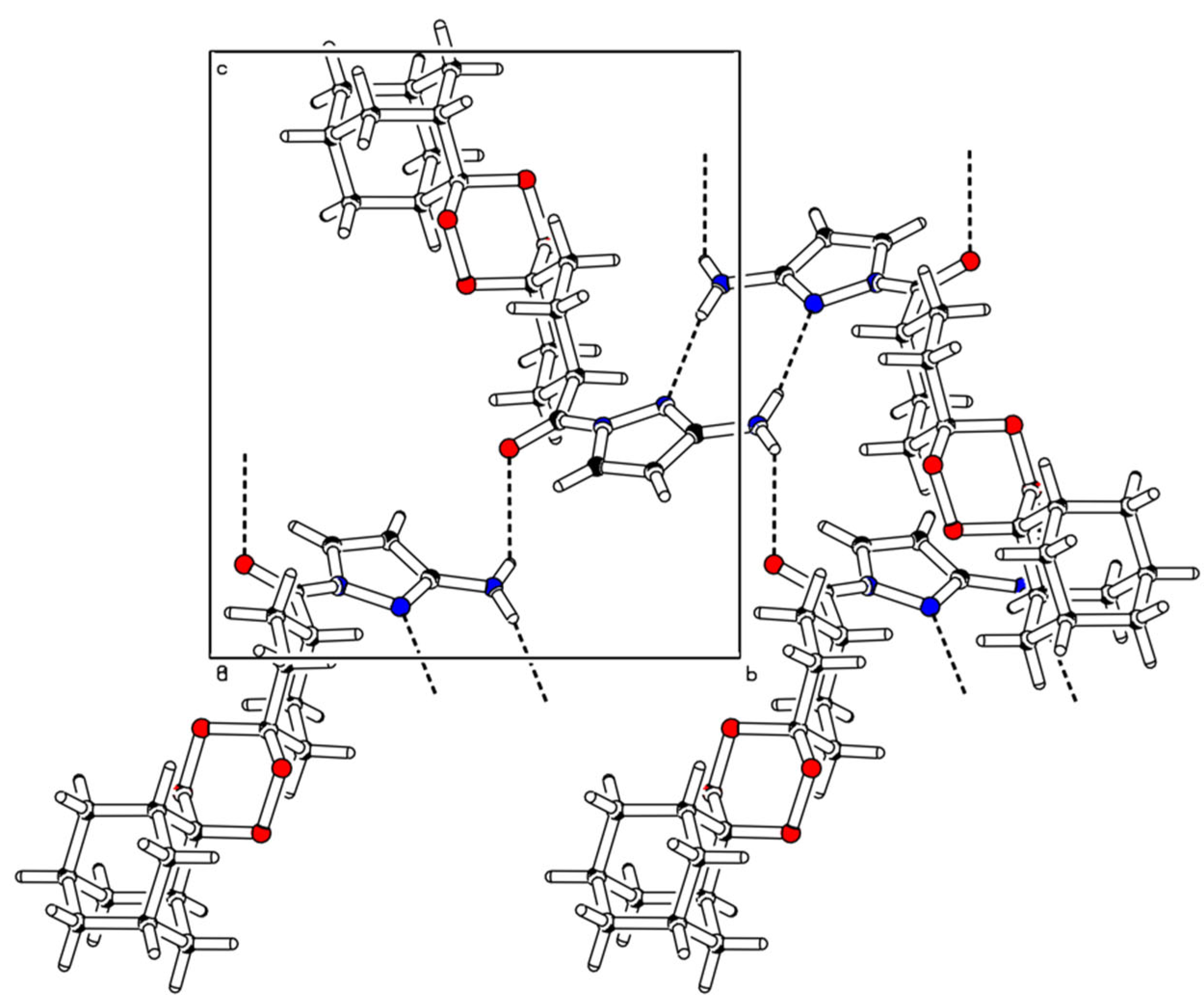
2.2. X-ray Crystal Analysis
2.3. In Vitro Susceptibility of Leishmania spp. to Endoperoxide–Pyrazole Hybrids
2.4. Effects of Endoperoxide–Pyrazole Hybrids on L. tropica Promastigotes Morphology
2.5. Evaluation of the Hybrids’ Stability upon Conversion to Their Salt Forms
3. Materials and Methods
3.1. Chemicals
3.2. Equipment
3.3. Synthesis
3.4. X-ray Crystallography
3.5. Biological Evaluation
3.5.1. Parasites and Cell Lines
3.5.2. Leishmania spp. Promastigotes In Vitro Susceptibility
3.5.3. In Vitro Cytotoxicity Assessment in THP-1 Cell Line
3.5.4. Leishmania spp. Intracellular Amastigotes Susceptibility
3.6. Software
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Cabral, L.I.L.; Pomel, S.; Cojean, S.; Amado, P.S.M.; Loiseau, P.M.; Cristiano, M.L.S. Synthesis and Antileishmanial Activity of 1,2,4,5-Tetraoxanes against Leishmania Donovani. Molecules 2020, 25, 465. [Google Scholar] [CrossRef]
- Mann, S.; Frasca, K.; Scherrer, S.; Henao-Martínez, A.F.; Newman, S.; Ramanan, P.; Suarez, J.A. A Review of Leishmaniasis: Current Knowledge and Future Directions. Curr. Trop. Med. Reports 2021, 8, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Roatt, B.M.; de Oliveira Cardoso, J.M.; De Brito, R.C.F.; Coura-Vital, W.; de Oliveira Aguiar-Soares, R.D.; Reis, A.B. Recent Advances and New Strategies on Leishmaniasis Treatment. Appl. Microbiol. Biotechnol. 2020, 104, 8965–8977. [Google Scholar] [CrossRef] [PubMed]
- Azim, M.; Khan, S.A.; Ullah, S.; Ullah, S.; Anjum, S.I. Therapeutic Advances in the Topical Treatment of Cutaneous Leishmaniasis: A Review. PLoS Negl. Trop. Dis. 2021, 15, e0009099. [Google Scholar] [CrossRef] [PubMed]
- Cortes, S.; Albuquerque, A.; Cabral, L.I.L.; Lopes, L.; Campino, L.; Cristiano, M.L.S. In Vitro Susceptibility of Leishmania Infantum to Artemisinin Derivatives and Selected Trioxolanes. Antimicrob. Agents Chemother. 2015, 59, 5032–5035. [Google Scholar] [CrossRef] [PubMed]
- Antolínez, I.V.; Barbosa, L.C.A.; Borgati, T.F.; Baldaia, A.; Ferreira, S.R.; Almeida, R.M.; Fujiwara, R.T. Tetraoxanes as New Agents Agains Leishmania Amazonensis. Chem. Biodivers. 2020, 17, e2000142. [Google Scholar] [CrossRef] [PubMed]
- Medrán, N.S.; Sayé, M.; Pereira, C.A.; Tekwani, B.L.; La-Venia, A.; Labadie, G.R. Expanding the Scope of Synthetic 1,2,4-Trioxanes towards Trypanosoma Cruzi and Leishmania Donovani. Bioorg. Med. Chem. Lett. 2020, 30, 127491. [Google Scholar] [CrossRef]
- Mendes, A.; Armada, A.; Cabral, L.I.L.; Amado, P.S.M.; Campino, L.; Cristiano, M.L.S.; Cortes, S. 1,2,4-Trioxolane and 1,2,4,5-Tetraoxane Endoperoxides against Old-World Leishmania Parasites: In Vitro Activity and Mode of Action. Pharmaceuticals 2022, 15, 446. [Google Scholar] [CrossRef]
- Opsenica, D.M.; Šolaja, B.A. Antimalarial Peroxides. J. Serbian Chem. Soc. 2009, 74, 1155–1193. [Google Scholar] [CrossRef]
- Amewu, R.K.; Chadwick, J.; Hussain, A.; Panda, S.; Rinki, R.; Janneh, O.; Ward, S.A.; Miguel, C.; Burrell-Saward, H.; Vivas, L.; et al. Synthesis and Evaluation of the Antimalarial, Anticancer, and Caspase 3 Activities of Tetraoxane Dimers. Bioorganic Med. Chem. 2013, 21, 7392–7397. [Google Scholar] [CrossRef]
- Woodley, C.M.; Amado, P.S.M.; Cristiano, M.L.S.; O’Neill, P.M. Artemisinin Inspired Synthetic Endoperoxide Drug Candidates: Design, Synthesis, and Mechanism of Action Studies. Med. Res. Rev. 2021, 41, 3062–3095. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.M.; Amewu, R.K.; Nixon, G.L.; ElGarah, F.B.; Mungthin, M.; Chadwick, J.; Shone, A.E.; Vivas, L.; Lander, H.; Barton, V.; et al. Identification of a 1,2,4,5-Tetraoxane Antimalarial Drug-Development Candidate (RKA 182) with Superior Properties to the Semisynthetic Artemisinins. Angew. Chem.-Int. Ed. 2010, 49, 5693–5697. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Sabbani, S.; Nixon, G.L.; Schnaderbeck, M.; Roberts, N.L.; Shore, E.R.; Riley, C.; Murphy, B.; Mcgillan, P.; Ward, S.A.; et al. Optimisation of the synthesis of second generation 1,2,4,5 tetraoxane antimalarials. Tetrahedron 2016, 72, 6118–6126. [Google Scholar] [CrossRef]
- Mohamed, L.W.; Shaaban, M.A.; Zaher, A.F.; Alhamaky, S.M.; Elsahar, A.M. Synthesis of New Pyrazoles and Pyrozolo [3,4-b] Pyridines as Anti-Inflammatory Agents by Inhibition of COX-2 Enzyme. Bioorg. Chem. 2019, 83, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Bennani, F.E.; Doudach, L.; Cherrah, Y.; Ramli, Y.; Karrouchi, K.; Ansar, M.; Faouzi, M.E.A. Overview of Recent Developments of Pyrazole Derivatives as an Anticancer Agent in Different Cell Line. Bioorg. Chem. 2020, 97, 103470. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, G.; Cai, X.-J.; Chen, Z.; Song, B.-A.; Bhadury, P.S.; Yang, S.; Jin, L.-H.; Xue, W.; Hu, D.-Y.; Zeng, S. Synthesis and Antiviral Activities of Pyrazole Derivatives Containing an Oxime Moiety. J. Agric. Food Chem. 2008, 56, 10160–10167. [Google Scholar] [CrossRef] [PubMed]
- Bekhit, A.A.; Saudi, M.N.; Hassan, A.M.M.; Fahmy, S.M.; Ibrahim, T.M.; Ghareeb, D.; El-Seidy, A.M.; Al-Qallaf, S.M.; Habib, H.J.; Bekhit, A.E.D.A. Synthesis, Molecular Modeling and Biological Screening of Some Pyrazole Derivatives as Antileishmanial Agents. Future Med. Chem. 2018, 10, 2325–2344. [Google Scholar] [CrossRef]
- Secrieru, A.; O’Neill, P.M.; Cristiano, M.L.S. Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules 2020, 25, 42. [Google Scholar] [CrossRef]
- Castillo, J.-C.; Portilla, J. Recent Advances in the Synthesis of New Pyrazole Derivatives. Targets Heterocycl. Syst. 2018, 22, 194–223. [Google Scholar] [CrossRef]
- Gao, C.; Chang, L.; Xu, Z.; Yan, X.-F.; Ding, C.; Zhao, F.; Wu, X.; Feng, L.S. Recent Advances of Tetrazole Derivatives as Potential Anti-Tubercular and Anti-Malarial Agents. Eur. J. Med. Chem. 2019, 163, 404–412. [Google Scholar] [CrossRef]
- Uddin, A.; Chawla, M.; Irfan, I.; Mahajan, S.; Singh, S.; Abid, M. Medicinal Chemistry Updates on Quinoline- and Endoperoxide-Based Hybrids with Potent Antimalarial Activity. RSC Med. Chem. 2021, 12, 24–42. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Singh, P.; Singh, A.K.; Awasthi, S.K. Advancement of Chimeric Hybrid Drugs to Cure Malaria Infection: An Overview with Special Emphasis on Endoperoxide Pharmacophores. Eur. J. Med. Chem. 2021, 219, 113408. [Google Scholar] [CrossRef] [PubMed]
- Tibon, N.S.; Ng, C.H.; Cheong, S.L. Current Progress in Antimalarial Pharmacotherapy and Multi-Target Drug Discovery. Eur. J. Med. Chem. 2020, 188, 111983. [Google Scholar] [CrossRef] [PubMed]
- Campino, L.; Pratlong, F.; Abranches, P.; Rioux, J.-A.; Santos-Gomes, G.; Alves-Pires, C.; Cortes, S.; Ramada, J.; Cristovão, J.M.; Afonso, M.O.; et al. Leishmaniasis in Portugal: Enzyme Polymorphism of Leishmania Infantum Based on the Identification of 213 Strains. Trop. Med. Int. Health 2006, 11, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Saik, I.E.I.; Benlabsir, C.; Fellah, H.; Lemrani, M.; Riyad, M. Transmission Patterns of Leishmania Tropica around the Mediterranean Basin: Could Morocco Be Impacted by a Zoonotic Spillover? PLoS Negl. Trop. Dis. 2022, 16, e0010009. [Google Scholar] [CrossRef]
- Amado, P.S.M.; Frija, L.M.T.; Coelho, J.A.S.; O’Neill, P.M.; Cristiano, M.L.S. Synthesis of Non-Symmetrical Dispiro-1,2,4,5-Tetraoxanes and Dispiro-1,2,4-Trioxanes Catalyzed by Silica Sulfuric Acid. J. Org. Chem. 2021, 86, 10608–10620. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Vennerstrom, J.L. Differentiation between 1,2,4,5-Tetraoxanes and 1,2,4,5,7,8- Hexaoxonanes Using 1H and 13C NMR Analyses. J. Heterocycl. Chem. 2001, 38, 463–466. [Google Scholar] [CrossRef]
- Secrieru, A.; Lopes, S.; Cristiano, M.L.S.; Fausto, R. UV-Induced Tautomerization in Argon Matrix. Molecules 2021, 26, 4299. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. A General Definition of Ring Puckering Coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Cortes, S.; Sousa, C.B.; Morais, T.; Lago, J.; Campino, L. Potential of the natural products against leishmaniasis in Old World-a review of in-vitro studies. Pathog. Glob. Health 2020, 114, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Madusanka, R.K.; Silva, H.; Karunaweera, N.D. Treatment of Cutaneous Leishmaniasis and Insights into Species-Specific Responses: A Narrative Review. Infect. Dis. Ther. 2022, 11, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Maurício, I.L. Leishmania Taxonomy. In The Leishmaniases: Old Neglected Tropical Diseases; Springer: Cham, Switzerland, 2018; pp. 15–30. [Google Scholar] [CrossRef]
- Machín, L.; Nápoles, R.; Gille, L.; Monzote, L. Leishmania Amazonensis Response to Artemisinin and Derivatives. Parasitol. Int. 2021, 80, 102218. [Google Scholar] [CrossRef] [PubMed]
- Woodley, C.M.; Nixon, G.L.; Basilico, N.; Parapini, S.; Hong, W.D.; Ward, S.A.; Biagini, G.A.; Leung, S.C.; Taramelli, D.; Onuma, K.; et al. Enantioselective Synthesis and Profiling of Potent, Nonlinear Analogues of Antimalarial Tetraoxanes E209 and N205. ACS Med. Chem. Lett. 2021, 12, 1077–1085. [Google Scholar] [CrossRef]
- Brown, R.J.C.; Brown, R.F.C. Melting Point and Molecular Symmetry. J. Chem. Educ. 2000, 77, 724. [Google Scholar] [CrossRef]
- Gautam, A.; Ahmed, T.; Paliwal, J.; Batra, V. Pharmacokinetics and Pharmacodynamics of Endoperoxide Antimalarials. Curr. Drug Metab. 2009, 10, 289–306. [Google Scholar] [CrossRef]
- Adebayo, J.O.; Tijjani, H.; Adegunloye, A.P.; Ishola, A.A.; Balogun, E.A.; Malomo, S.O. Enhancing the antimalarial activity of artesunate. Parasitol. Res. 2020, 119, 2749–2764. [Google Scholar] [CrossRef]
- Bruno de Sousa, C.; Lago, J.H.G.; Macridachis, J.; Oliveira, M.; Brito, L.; Vizetto-Duarte, C.; Florindo, C.; Hendrickx, S.; Maes, L.; Morais, T.; et al. Report of in Vitro Antileishmanial Properties of Iberian Macroalgae. Nat. Prod. Res. 2019, 33, 1778–1782. [Google Scholar] [CrossRef]
- Hendrickx, S.; Guerin, P.J.; Caljon, G.; Croft, S.L.; Maes, L. Evaluating drug resistance in visceral leishmaniasis: The challenges. Parasitology 2018, 145, 453–463. [Google Scholar] [CrossRef]
- Jiricek, J.; Kondreddi, R.R.; Smith, P.W. Indole Carboxamide Derivatives and Uses Thereof. International Patent WO2014037900A1, March 2014. [Google Scholar]
- Stec, J.; Onajole, O.K.; Lun, S.; Guo, H.; Merenbloom, B.; Vistoli, G.; Bishai, W.R.; Kozikowski, A.P. Indole-2-Carboxamide-Based MmpL3 Inhibitors Show Exceptional Antitubercular Activity in an Animal Model of Tuberculosis Infection. J. Med. Chem. 2016, 59, 6232–6247. [Google Scholar] [CrossRef]
- Vennerstrom, J.L.; Dong, Y.; Chollet, J.; Matile, H.; Wang, X.; Spiraghavan, K.; Chapman, W.N. Spiro and Dispiro 1,2,4-Trioxolane Antimalarials. U.S. Patent US 7 371 778 B2, May 2008. [Google Scholar]
- Lis, L.; Cabral, L.I.L.; Sena, M.I.; Guerreiro, B.; Rodrigues, A.S.; De Andrade-Neto, V.F.; Cristiano, M.L.S.; Nogueira, F. New endoperoxides highly active in vivo and in vitro against artemisinin-resistant Plasmodium Falciparum. Malar. J. 2018, 17, 145. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal- Structure Determination. Acta Crystallogr. Sect. A 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Yamamoto, E.S.; Campos, B.L.S.; Jesus, J.A.; Laurenti, M.D.; Ribeiro, S.P.; Kallás, E.G.; Rafael-Fernandes, M.; Santos-Gomes, G.; Silva, M.S.; Sessa, D.P.; et al. The Effect of Ursolic Acid on Leishmania (Leishmania) amazonensis Is Related to Programed Cell Death and Presents Therapeutic Potential in Experimental Cutaneous Leishmaniasis. PLoS ONE 2015, 10, e0144946. [Google Scholar] [CrossRef] [PubMed]
- Bruker. APEX3; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Bruker. SAINT V8.38A; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. D 2009, D65, 148–155. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Isomer OZ1 | Isomer OZ2 | ||||||
| 1H, δ (ppm) | 13C, δ (ppm) | 1H, δ (ppm) | 13C, δ (ppm) | ||||
| COSY | HSQC | HMBC | COSY | HSQC | HMBC | ||
| 1.67 | 1.97 | 36.85 | 26.75, 35.05, 111.73 | 1.67 | 1.72, 1.80, 1.81 | 30.20, 36.85 | 26.75, 34.95, 111.49, 111.73 |
| 1.69 | 1.97 | 34.85 | 111.73 | 1.68 | 1.73 | 34.85 | 26.79, 111.49 |
| 1.72 | 2.00, 1.97 | 34.85 | 26.75 | 1.72 | 1.67 | 35.82 | - |
| 1.79 | 2.01, 2.02, 3.57 | 26.55 | 26.50 | 1.73 | 1.67, 1.68, 2.02 | 34.85 | - |
| 1.81 | 3.57 | 33.52 | 36.76, 108.07 | 1.75 | 1.97, 2.02 | 33.69 | - |
| 1.82 | 3.57 | 27.05, 26.22 | 33.32, 33.10, 40.91 | 1.77 | 1.67, 1.90, 1.96 | 26.55 | 26.53, 39.69 |
| 1.97 | 1.67, 1.69, 1.72, 3.57 | 36.51 | 36.76, 36.50, 111.73 | 1.80 | 1.67, 1.90 | 33.36 | 36.89, 39.70, 108.07 |
| 2.00 | 1.72, 3.57 | 34.85 | 34.81 | 1.81 | 1.67, 1.90 | 26.38 | 39.73 |
| 2.01 | 1.79, 3.57 | 26.38 | 1.82 | 1.72 | 27.05 | 26.50, 39.73 | |
| 2.02 | 1.79, 3.57 | 33.52 | 26.26, 33.34, 40.91 | 1.90 | 1.81 | 34.85 | 26.58, 33.84, 37.02 |
| 3.57 | 1.79, 1.81, 1.82, 1.97, 2.00, 2.01 | 40.83 | - | 1.96 | 1.67, 1.77 | 26.38. 33.52 | 39.8, 108.07, 173.12 |
| 5.19 | - | -NH | 89.02 | 1.97 | 1.75, 1.77, 1.81 | 34.85, 36.51 | 26.53, 36.85, 111.49, 173.12 |
| 5.37 | 5.54, 7.36 | 88.02 | 143.97,150.32 | 2.02 | 1.73, 1.75 | 33.52 | 26.50, 33.40, 39.69, 108.07 |
| 5.54 | 5.37, 7.36 | -NH | 89.02 | 3.98 | - | -NH2 | 100.99, 157.40 |
| 7.36 | 5.37, 5.54 | 143.97 | 89.02, 150.32 | 5.88 | 7.98 | 100.99 | 130.18, 157.40 |
| - | - | - | - | 7.98 | 5.88 | 130.18 | 100.99; 157.40 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Isomer T1 | Isomer T2 | ||||||
| 1H, δ (ppm) | 13C, δ (ppm) | 1H, δ (ppm) | 13C, δ (ppm) | ||||
| COSY | HSQC | HMBC | COSY | HSQC | HMBC | ||
| 1.60 | 1.62, 1.70, 1.73, 1.86 | 33.19 | - | 1.59 | 1.69 | 33.19 | - |
| 1.62 | 1.60, 1.70, 1.73, | 30.47 | - | 1.69 | 1.59, 1.71 | 37.01 | 33.34 |
| 1.70 | 1.73 | 37.01 | 33.34 | 1.71 | 3.14, 1.76 | 37.01 | 26.99, 33.34 |
| 1.73 | 1.70, 3.16 | 33.19 | 26.99 | 1.76 | 1.69, 1.71, 3.14, 1.97 | 33.19 | - |
| 1.86 | 1.97, 3.16, 3.65 | 30.56, 33.19 | - | 1.83 | 3.48 | 27.21 | - |
| 1.97 | 1.73, 3.65 | 27.05 | - | 1.87 | 1.76, 2.15, 3.48 | 28.21 | 30.17, 34.07 |
| 3.16 | 1.73 | 30.2 | - | 1.97 | 1.76, 2.15 | 33.19 | 172.78 |
| 3.65 | 1.86, 1.97 | 41.33 | 178.1 | 2.02 | 1.87. 1.97 | 33.19 | - |
| 5.19 | - | -NH | 88.92 | 3.14 | 1.71, 1.76 | - | - |
| 5.37 | 7.36, 5.54 | 88.92 | 143.97, 150.32 | 3.48 | 1.85, 1.87 | 40.00 | 172.78 |
| 5.54 | 5.37, 7.36 | -NH | 88.92 | 3.97 | 8.00 | -NH2 | 101.04, 157.40 |
| 7.36 | 5.37; 5.54 | 144.04 | 89.02, 150.32 | 5.88 | 8.00 | 101.04 | 130.14, 157.40 |
| - | - | - | - | 8.00 | 3.97, 5.88 | 130.14 | 101.04, 157.40 |
| T1: D–H…A | D–H (Å) | H…A (Å) | D…A (Å) | <D–H…A (°) |
|---|---|---|---|---|
| N3–H3A…N2 a | 0.88 | 2.55 | 3.528 (11) | 137 |
| N3–H3B…O5 | 0.88 | 2.15 | 2.755 (11) | 125 |
| T2: D–H…A | D–H (Å) | H…A (Å) | D…A (Å) | <D–H…A (°) |
| N3–H3A…N2 b | 0.868 (18) | 2.192 (18) | 3.0432 (16) | 166.7 (17) |
| N3–H3B…O5 c | 0.830 (18) | 2.513 (18) | 3.1726 (17) | 137.2 (16) |
| ID | Structures | ClogP a | ClogS a | Cytotoxicity CC50 ± SEM b (µΜ) | Promastigotes Susceptibility IC50 ± SEM b (µΜ) | Intracellular Amastigote Susceptibility IC50 ± SEM c (µΜ) | SI d | |
|---|---|---|---|---|---|---|---|---|
| L. tropica | L. infantum | L. infantum | ||||||
| 2o |  | 2.61 | 1.38 | 381 ± 172 | 156 ± 18 | 123 ± 1 | 330 ± 48 | 1.2 |
| 2t |  | 2.38 | 1.21 | 374 ± 119 | 143 ± 9 | 128 ± 27 | 45 ± 28 | 8.4 |
| OZ1 |  | 2.01 | 1.30 | 664 ± 163 | 219 ± 25 | >400 | ND | ND |
| OZ2 |  | 2.01 | 1.30 | 202 ± 116 | 161 ± 19 | 219 ± 44 | ND | ND |
| T1 |  | 1.79 | 1.18 | >800 | >400 | >400 | ND | ND |
| T2 |  | 1.79 | 1.18 | >800 | ND | ND | ND | ND |
| PIR |  | −0.51 | 5.59 | 202 ± 10 | >400 | >400 | ND | ND |
| AmB | Amphotericin B | −1.19 | 2.48 | 23 ± 3 | 0.30 ± 0.1 | 0.20 ± 0.01 | 0.08 ± 0.05 | 290 |
| Compounds | Promastigotes Susceptibility IC50 ± SEM a (µΜ) | Cytotoxicity CC50 ± SEM a (µΜ) | Intracellular Amastigotes Susceptibility IC50 ± SEM b (µΜ) | SI c | |
|---|---|---|---|---|---|
| L. tropica | L. infantum | L. infantum | |||
| OZ1•TsOH | 199 ± 9 | 181 ± 35 | 510 ± 109 | 246 ± 32 | 2.1 |
| OZ1•MsOH | 198 ± 24 | 183 ± 48 | 336 ± 196 | >400 | <0.8 |
| OZ1•HCl | 135 ± 36 | 164 ± 20 | 509 ± 155 | 87 ± 39 | 5.9 |
| OZ2•TsOH | 260 ± 82 | 217 ± 47 | 479 ± 83 | ND | ND |
| OZ2•HCl | >400 | 258 ± 91 | 425 ± 104 | ND | ND |
| T1•TsOH | >400 | >400 | 331 ± 126 | ND | ND |
| T1•HCl | >400 | >400 | 529 ± 108 | ND | ND |
| T2•HCl | 266 ± 8 | 313 ± 40 | 444 ± 22 | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amado, P.S.M.; Costa, I.C.C.; Paixão, J.A.; Mendes, R.F.; Cortes, S.; Cristiano, M.L.S. Synthesis, Structure and Antileishmanial Evaluation of Endoperoxide–Pyrazole Hybrids. Molecules 2022, 27, 5401. https://doi.org/10.3390/molecules27175401
Amado PSM, Costa ICC, Paixão JA, Mendes RF, Cortes S, Cristiano MLS. Synthesis, Structure and Antileishmanial Evaluation of Endoperoxide–Pyrazole Hybrids. Molecules. 2022; 27(17):5401. https://doi.org/10.3390/molecules27175401
Chicago/Turabian StyleAmado, Patrícia S. M., Inês C. C. Costa, José A. Paixão, Ricardo F. Mendes, Sofia Cortes, and Maria L. S. Cristiano. 2022. "Synthesis, Structure and Antileishmanial Evaluation of Endoperoxide–Pyrazole Hybrids" Molecules 27, no. 17: 5401. https://doi.org/10.3390/molecules27175401
APA StyleAmado, P. S. M., Costa, I. C. C., Paixão, J. A., Mendes, R. F., Cortes, S., & Cristiano, M. L. S. (2022). Synthesis, Structure and Antileishmanial Evaluation of Endoperoxide–Pyrazole Hybrids. Molecules, 27(17), 5401. https://doi.org/10.3390/molecules27175401