
pH-Responsive PEGylated Niosomal Nanoparticles as an Active-Targeting Cyclophosphamide Delivery System for Gastric Cancer Therapy


 , , and
, , and 
Abstract
:1. Introduction
2. Result and Discussion
2.1. Optimization Studies of Cyclophosphamide-Loaded Niosomes
2.1.1. Analysis of Particle Size
2.1.2. Polydispersity Index Analysis
2.1.3. Entrapment Efficiency (EE%) Analysis
2.1.4. Accuracy and Validity of the Model
2.1.5. Data Optimization
2.2. Size Distribution and Morphological Characterization
2.3. Fourier Transform Infrared Spectroscopy (FTIR) Spectroscopy Analysis
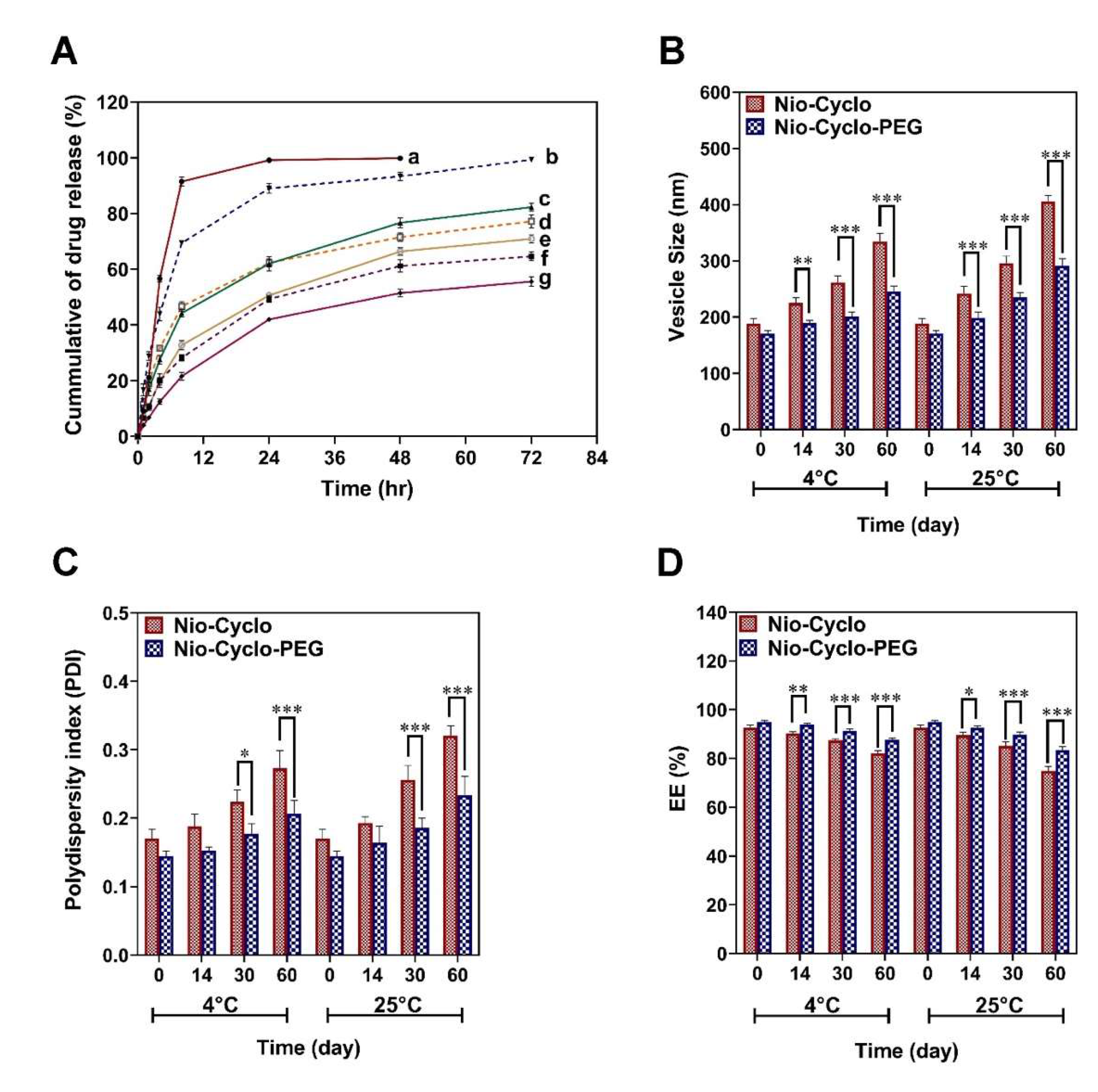
2.4. Drug Release and Kinetic Modelling
2.5. Physical Stability Examination
2.6. Cytotoxicity Study
2.7. Gene Expression Analysis
2.8. Apoptosis Analysis
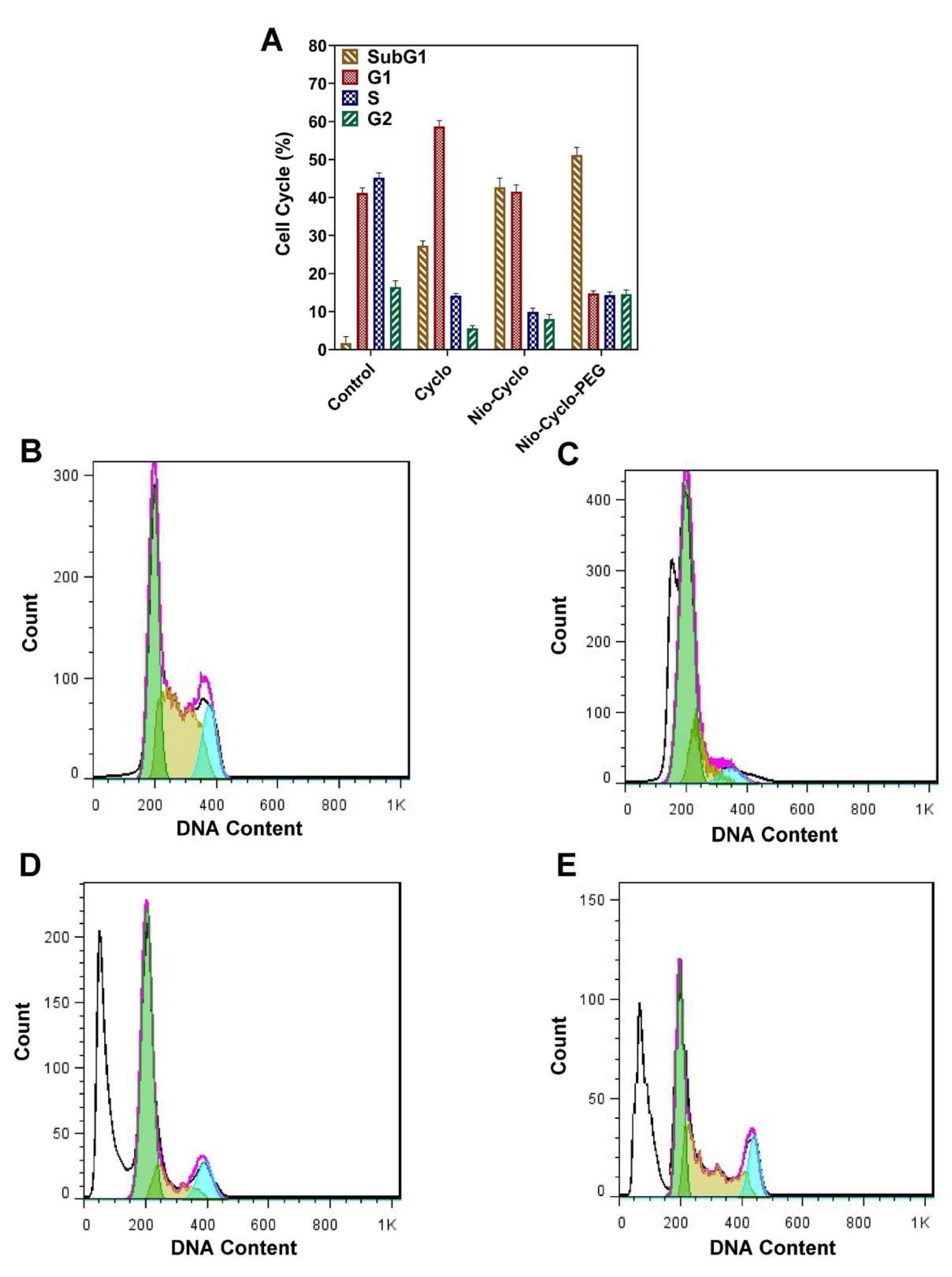
2.9. Cell Cycle
2.10. Scratch Assay
3. Conclusions
4. Experimental Section
4.1. Chemicals
4.2. Optimization of Niosomal Formulations Using Response Surface Methodology
4.3. Synthesis of Cyclophosphamide-Loaded Niosomes and PEGylated-Nio-Cyclo
4.4. Characterizations of Niosomal Formulation
4.5. Analysis of Entrapment Efficiency
4.6. In Vitro Drug Release Study
4.7. Release Kinetic Modelling
4.8. Stability Study
4.9. Cytotoxicity Study
4.10. Apoptotic Gene Expression Analysis
4.11. Apoptosis Analysis
4.12. Cell Cycle
4.13. Scratch Assay
4.14. Statistical Analysis and Curve Fitting
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tang, J.; Lee, D.; Tice, T.R.; Schwendeman, S.P.; Prausnitz, M.R. Clinical translation of long-acting drug delivery formulations. Nat. Rev. Mater. 2022, 7, 406–420. [Google Scholar] [CrossRef]
- Shukla, M.K.; Dubey, A.; Pandey, S.; Singh, S.K.; Gupta, G.; Prasher, P.; Chellappan, D.K.; Oliver, B.G.; Kumar, D.; Dua, K. Managing Apoptosis in Lung Diseases Using Nano-Assisted Drug Delivery System. Curr. Pharm. Des. 2022. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-j.; De Geest, B.G. Nanomedicine and Cancer Immunotherapy; Nature Publishing Group: Berlin, Germany, 2020. [Google Scholar]
- Zhang, R.X.; Li, J.; Zhang, T.; Amini, M.A.; He, C.; Lu, B.; Ahmed, T.; Lip, H.; Rauth, A.M.; Wu, X.Y. Importance of integrating nanotechnology with pharmacology and physiology for innovative drug delivery and therapy–an illustration with firsthand examples. Acta Pharmacol. Sin. 2018, 39, 825–844. [Google Scholar] [CrossRef]
- Ma, X.; Feng, H.; Liang, C.; Liu, X.; Zeng, F.; Wang, Y.J.J. Mesoporous silica as micro/nano-carrier: From passive to active cargo delivery, a mini review. J. Mater. Sci. Technol. 2017, 33, 1067–1074. [Google Scholar] [CrossRef]
- Akbarzadeh, I.; Tavakkoli Yaraki, M.; Ahmadi, S.; Chiani, M.; Nourouzian, D. Folic acid-functionalized niosomal nanoparticles for selective dual-drug delivery into breast cancer cells: An in-vitro investigation. Adv. Powder Technol. 2020, 31, 4064–4071. [Google Scholar] [CrossRef]
- Fernandes, N.; Rodrigues, C.F.; Moreira, A.F.; Correia, I.J. Overview of the application of inorganic nanomaterials in cancer photothermal therapy. Biomater. Sci. 2020, 8, 2990–3020. [Google Scholar] [CrossRef]
- Jiang, C.-C.; Cao, Y.-K.; Xiao, G.-Y.; Zhu, R.-F.; Lu, Y.-P. A review on the application of inorganic nanoparticles in chemical surface coatings on metallic substrates. RSC Adv. 2017, 7, 7531–7539. [Google Scholar] [CrossRef]
- Barani, M.; Mirzaei, M.; Torkzadeh-Mahani, M.; Lohrasbi-Nejad, A.; Nematollahi, M.H. A new formulation of hydrophobin-coated niosome as a drug carrier to cancer cells. Mater. Sci. Eng. C 2020, 113, 110975. [Google Scholar] [CrossRef]
- Shukla, M.K.; Singh, S.K.; Pandey, S.; Gupta, P.K.; Choudhary, A.; Jindal, D.K.; Dua, K.; Kumar, D. Potential Immunomodulatory Activities of Plant Products. South Afr. J. Bot. 2022, in press. [Google Scholar] [CrossRef]
- Yadavar-Nikravesh, M.-S.; Ahmadi, S.; Milani, A.; Akbarzadeh, I.; Khoobi, M.; Vahabpour, R.; Bolhassani, A.; Bakhshandeh, H. Construction and characterization of a novel Tenofovir-loaded PEGylated niosome conjugated with TAT peptide for evaluation of its cytotoxicity and anti-HIV effects. Adv. Powder Technol. 2021, 32, 3161–3173. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Kim, J.-K.; Park, J.-S.; Byun, Y.; Kim, C.-K. The use of PEGylated liposomes to prolong circulation lifetimes of tissue plasminogen activator. Biomaterials 2009, 30, 5751–5756. [Google Scholar] [CrossRef]
- Lawrence, M.; Chauhan, S.; Lawrence, S.; Barlow, D. The formation, characterization and stability of non-ionic surfactant vesicles. STP Pharma Sci. 1996, 6, 49–60. [Google Scholar]
- Li, Y.-P.; Pei, Y.-Y.; Zhang, X.-Y.; Gu, Z.-H.; Zhou, Z.-H.; Yuan, W.-F.; Zhou, J.-J.; Zhu, J.-H.; Gao, X.-J. PEGylated PLGA nanoparticles as protein carriers: Synthesis, preparation and biodistribution in rats. J. Control. Release 2001, 71, 203–211. [Google Scholar] [CrossRef]
- Akbarzadeh, I.; Shayan, M.; Bourbour, M.; Moghtaderi, M.; Noorbazargan, H.; Eshrati Yeganeh, F.; Saffar, S.; Tahriri, M. Preparation, Optimization and In-Vitro Evaluation of Curcumin-Loaded Niosome@ calcium Alginate Nanocarrier as a New Approach for Breast Cancer Treatment. Biology 2021, 10, 173. [Google Scholar] [CrossRef]
- Karimifard, S.; Rezaei, N.; Jamshidifar, E.; Moradi Falah Langeroodi, S.; Abdihaji, M.; Mansouri, A.; Hosseini, M.; Ahmadkhani, N.; Rahmati, Z.; Heydari, M.J.A.A.N.M. pH-Responsive Chitosan-Adorned Niosome Nanocarriers for Co-Delivery of Drugs for Breast Cancer Therapy. ACS Appl. Nano Mater. 2022, 5, 8811–8825. [Google Scholar] [CrossRef]
- Rezaei, T.; Rezaei, M.; Karimifard, S.; Beram, F.M.; Dakkali, M.S.; Heydari, M.; Afshari-Behbahanizadeh, S.; Mostafavi, E.; Bokov, D.O.; Ansari, M.J.J.F. Folic Acid-Decorated pH-Responsive Nanoniosomes With Enhanced Endocytosis for Breast Cancer Therapy: In Vitro Studies. Front. Pharmacol. 2022, 13, 851242. [Google Scholar]
- Bourbour, M.; Khayam, N.; Noorbazargan, H.; Yaraki, M.T.; Lalami, Z.A.; Akbarzadeh, I.; Yeganeh, F.E.; Dolatabadi, A.; Rad, F.M.; Tan, Y.N.J.M.S.D.; et al. Evaluation of Anti-Cancer and Anti-Metastatic Effects of Folate-PEGylated Niosome for Co-Delivery of Letrozole and Ascorbic Acid on Breast Cancer Cells. Mol. Syst. Des. Eng. 2022. Advance Article. [Google Scholar] [CrossRef]
- Sahrayi, H.; Hosseini, E.; Karimifard, S.; Khayam, N.; Meybodi, S.M.; Amiri, S.; Bourbour, M.; Farasati Far, B.; Akbarzadeh, I.; Bhia, M.J.P. Co-Delivery of Letrozole and Cyclophosphamide via Folic Acid-Decorated Nanoniosomes for Breast Cancer Therapy: Synergic Effect, Augmentation of Cytotoxicity, and Apoptosis Gene Expression. Pharmaceuticals 2021, 15, 6. [Google Scholar] [CrossRef]
- Baj, J.; Korona-Głowniak, I.; Forma, A.; Maani, A.; Sitarz, E.; Rahnama-Hezavah, M.; Radzikowska, E.; Portincasa, P. Mechanisms of the Epithelial–Mesenchymal Transition and Tumor Microenvironment in Helicobacter pylori-Induced Gastric Cancer. Cells 2020, 9, 1055. [Google Scholar] [CrossRef]
- Yusefi, A.R.; Lankarani, K.B.; Bastani, P.; Radinmanesh, M.; Kavosi, Z. Risk factors for gastric cancer: A systematic review. Asian Pac. J. Cancer Prev. APJCP 2018, 19, 591. [Google Scholar]
- Mehta, S. Anti-cancer drugs targeting using nanocarrier niosomes-a review. TMR Cancer 2020, 3, 169–174. [Google Scholar]
- Wang, W.; Sang, L.; Zhao, Y.; Wei, Z.; Qi, M.; Li, Y.J.J. Inherently radiopaque polyurethane beads as potential multifunctional embolic agent in hepatocellular carcinoma therapy. J. Mater. Sci. Technol. 2021, 63, 106–114. [Google Scholar] [CrossRef]
- Jensen, M.-B.; Lænkholm, A.-V.; Nielsen, T.O.; Eriksen, J.O.; Wehn, P.; Hood, T.; Ram, N.; Buckingham, W.; Ferree, S.; Ejlertsen, B. The Prosigna gene expression assay and responsiveness to adjuvant cyclophosphamide-based chemotherapy in premenopausal high-risk patients with breast cancer. Breast Cancer Res. 2018, 20, 79. [Google Scholar] [CrossRef] [Green Version]
- Sucic, M.; Luetic, K.; Jandric, I.; Drmic, D.; Sever, A.Z.; Vuletic, L.B.; Halle, Z.B.; Strinic, D.; Kokot, A.; Seiwerth, R.S. Therapy of the rat hemorrhagic cystitis induced by cyclophosphamide. Stable gastric pentadecapeptide BPC 157, L-arginine, L-NAME. Eur. J. Pharmacol. 2019, 861, 172593. [Google Scholar] [CrossRef] [PubMed]
- Hemati, M.; Haghiralsadat, F.; Jafary, F.; Moosavizadeh, S.; Moradi, A. Targeting cell cycle protein in gastric cancer with CDC20siRNA and anticancer drugs (doxorubicin and quercetin) co-loaded cationic PEGylated nanoniosomes. Int. J. Nanomed. 2019, 14, 6575. [Google Scholar] [CrossRef]
- Alemi, A.; Zavar Reza, J.; Haghiralsadat, F.; Zarei Jaliani, H.; Haghi Karamallah, M.; Hosseini, S.A.; Haghi Karamallah, S. Paclitaxel and curcumin coadministration in novel cationic PEGylated niosomal formulations exhibit enhanced synergistic antitumor efficacy. J. Nanobiotechnol. 2018, 16, 28. [Google Scholar] [CrossRef]
- Tavano, L.; Aiello, R.; Ioele, G.; Picci, N.; Muzzalupo, R.J.C.; Biointerfaces, S.B. Niosomes from glucuronic acid-based surfactant as new carriers for cancer therapy: Preparation, characterization and biological properties. Colloids Surf B Biointerfaces 2014, 118, 7–13. [Google Scholar] [CrossRef]
- Abdelbary, A.A.; AbouGhaly, M.H. Design and optimization of topical methotrexate loaded niosomes for enhanced management of psoriasis: Application of Box–Behnken design, in-vitro evaluation and in-vivo skin deposition study. Int. J. Pharm. 2015, 485, 235–243. [Google Scholar] [CrossRef]
- Zidan, A.S.; Mokhtar Ibrahim, M.; Megrab, N.A.E. Optimization of methotrexate loaded niosomes by Box–Behnken design: An understanding of solvent effect and formulation variability. Drug Dev. Ind. Pharm. 2017, 43, 1450–1459. [Google Scholar] [CrossRef]
- Gabizon, A.; Shmeeda, H.; Grenader, T. Pharmacological basis of pegylated liposomal doxorubicin: Impact on cancer therapy. Eur. J. Pharm. Sci. 2012, 45, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Haghiralsadat, F.; Amoabediny, G.; Helder, M.N.; Naderinezhad, S.; Sheikhha, M.H.; Forouzanfar, T.; Zandieh-doulabi, B. A comprehensive mathematical model of drug release kinetics from nano-liposomes, derived from optimization studies of cationic PEGylated liposomal doxorubicin formulations for drug-gene delivery. Artif. Cells Nanomed. Biotechnol. 2018, 46, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Hajizadeh, M.R.; Maleki, H.; Barani, M.; Fahmidehkar, M.A.; Mahmoodi, M.; Torkzadeh-Mahani, M. In vitro cytotoxicity assay of D-limonene niosomes: An efficient nano-carrier for enhancing solubility of plant-extracted agents. Res. Pharm. Sci. 2019, 14, 448. [Google Scholar]
- Shad, P.M.; Karizi, S.Z.; Javan, R.S.; Mirzaie, A.; Noorbazargan, H.; Akbarzadeh, I.; Rezaie, H. Folate conjugated hyaluronic acid coated alginate nanogels encapsulated oxaliplatin enhance antitumor and apoptosis efficacy on colorectal cancer cells (HT29 cell line). Toxicol. Vitr. 2020, 65, 104756. [Google Scholar] [CrossRef]
- De Silva, L.; Fu, J.-Y.; Htar, T.T.; Muniyandy, S.; Kasbollah, A.; Kamal, W.H.B.W.; Chuah, L.-H. Characterization, optimization, and in vitro evaluation of Technetium-99m-labeled niosomes. Int. J. Nanomed. 2019, 14, 1101. [Google Scholar] [CrossRef] [Green Version]
- Shaker, D.S.; Shaker, M.A.; Hanafy, M.S. Cellular uptake, cytotoxicity and in-vivo evaluation of Tamoxifen citrate loaded niosomes. Int. J. Pharm. 2015, 493, 285–294. [Google Scholar] [CrossRef]
- Rinaldi, F.; Del Favero, E.; Rondelli, V.; Pieretti, S.; Bogni, A.; Ponti, J.; Rossi, F.; Di Marzio, L.; Paolino, D.; Marianecci, C. pH-sensitive niosomes: Effects on cytotoxicity and on inflammation and pain in murine models. J. Enzym. Inhib. Med. Chem. 2017, 32, 538–546. [Google Scholar] [CrossRef]
- Tila, D.; Yazdani-Arazi, S.N.; Ghanbarzadeh, S.; Arami, S.; Pourmoazzen, Z. pH-sensitive, polymer modified, plasma stable niosomes: Promising carriers for anti-cancer drugs. EXCLI J. 2015, 14, 21. [Google Scholar]
- Kishore, R.S.; Pappenberger, A.; Dauphin, I.B.; Ross, A.; Buergi, B.; Staempfli, A.; Mahler, H.-C. Degradation of polysorbates 20 and 80: Studies on thermal autoxidation and hydrolysis. J. Pharm. Sci. 2011, 100, 721–731. [Google Scholar] [CrossRef]
- Naderinezhad, S.; Amoabediny, G.; Haghiralsadat, F. Co-delivery of hydrophilic and hydrophobic anticancer drugs using biocompatible pH-sensitive lipid-based nano-carriers for multidrug-resistant cancers. RSC Adv. 2017, 7, 30008–30019. [Google Scholar] [CrossRef]
- Davarpanah, F.; Yazdi, A.K.; Barani, M.; Mirzaei, M.; Torkzadeh-Mahani, M. Magnetic delivery of antitumor carboplatin by using PEGylated-Niosomes. DARU J. Pharm. Sci. 2018, 26, 57–64. [Google Scholar] [CrossRef]
- Cortesi, R.; Ravani, L.; Rinaldi, F.; Marconi, P.; Drechsler, M.; Manservigi, M.; Argnani, R.; Menegatti, E.; Esposito, E.; Manservigi, R. Intranasal immunization in mice with non-ionic surfactants vesicles containing HSV immunogens: A preliminary study as possible vaccine against genital herpes. Int. J. Pharm. 2013, 440, 229–237. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar]
- Uchegbu, I.F.; Vyas, S.P. Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int. J. Pharm. 1998, 172, 33–70. [Google Scholar] [CrossRef]
- Hofland, H.; Bouwstra, J.; Verhoef, J.; Buckton, G.; Chowdry, B.; Ponec, M.; Junginger, H. Safety aspects of non-ionic surfactant vesicles: A toxicity study related to the physicochemical characteristics of non-ionic surfactants. J. Pharm. Pharmacol. 1992, 44, 287–294. [Google Scholar] [CrossRef]
- Akbarzadeh, I.; Saremi Poor, A.; Yaghmaei, S.; Norouzian, D.; Noorbazargan, H.; Saffar, S.; Ahangari Cohan, R.; Bakhshandeh, H. Niosomal delivery of simvastatin to MDA-MB-231 cancer cells. Drug Dev. Ind. Pharm. 2020, 46, 1535–1549. [Google Scholar] [CrossRef]
- Moazeni, E.; Gilani, K.; Sotoudegan, F.; Pardakhty, A.; Najafabadi, A.R.; Ghalandari, R.; Fazeli, M.R.; Jamalifar, H. Formulation and in vitro evaluation of ciprofloxacin containing niosomes for pulmonary delivery. J. Microencapsul. 2010, 27, 618–627. [Google Scholar] [CrossRef]
- Balasubramaniam, A.; Anil Kumar, V.; Sadasivan Pillai, K. Formulation and in vivo evaluation of niosome-encapsulated daunorubicin hydrochloride. Drug Dev. Ind. Pharm. 2002, 28, 1181–1193. [Google Scholar] [CrossRef]
- Eskandari, Z.; Bahadori, F.; Celik, B.; Onyuksel, H. Targeted nanomedicines for cancer therapy, from basics to clinical trials. J. Pharm. Pharm. Sci. 2020, 23, 132–157. [Google Scholar] [CrossRef]
- Rajpoot, K. Lipid-based nanoplatforms in cancer therapy: Recent advances and applications. Curr. Cancer Drug Targets 2020, 20, 271–287. [Google Scholar] [CrossRef]
- Fasehee, H.; Dinarvand, R.; Ghavamzadeh, A.; Esfandyari-Manesh, M.; Moradian, H.; Faghihi, S.; Ghaffari, S.H. Delivery of disulfiram into breast cancer cells using folate-receptor-targeted PLGA-PEG nanoparticles: In vitro and in vivo investigations. J. Nanobiotechnol. 2016, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Sahoo, S.K. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 170–183. [Google Scholar] [CrossRef]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Boice, A.; Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2020, 1867, 118688. [Google Scholar] [CrossRef]
- Nisha, R.; Kumar, P.; Gautam, A.K.; Bera, H.; Bhattacharya, B.; Parashar, P.; Saraf, S.A.; Saha, S. Assessments of in vitro and in vivo antineoplastic potentials of β-sitosterol-loaded PEGylated niosomes against hepatocellular carcinoma. J. Liposome Res. 2020, 31, 1–12. [Google Scholar] [CrossRef]
- Zhang, H.-P.; Li, S.-Y.; Wang, J.-P.; Lin, J. Clinical significance and biological roles of cyclins in gastric cancer. OncoTargets Ther. 2018, 11, 6673. [Google Scholar] [CrossRef] [Green Version]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Huang, L.; Ren, F.; Tang, R.; Feng, Z.; Chen, G. Prognostic value of expression of cyclin E in gastrointestinal cancer: A systematic review and meta-analysis. Technol. Cancer Res. Treat. 2016, 15, 12–19. [Google Scholar] [CrossRef]
- Li, H.; Qiu, Z.; Li, F.; Wang, C. The relationship between MMP-2 and MMP-9 expression levels with breast cancer incidence and prognosis. Oncol. Lett. 2017, 14, 5865–5870. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yuan, T.; Wang, H.; Yao, S.; Zhao, Y.; Liu, Y.; Jin, S.; Chu, J.; Xu, Y.; Zhou, W. MMP-2 together with MMP-9 overexpression correlated with lymph node metastasis and poor prognosis in early gastric carcinoma. Tumor Biol. 2017, 39, 1010428317700411. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, U.; Weisova, P.; Schmid, J.; Bernas, T.; Huber, H.J.; Düssmann, H.; Connolly, N.M.; Prehn, J.H. Defining external factors that determine neuronal survival, apoptosis and necrosis during excitotoxic injury using a high content screening imaging platform. PLoS ONE 2017, 12, e0188343. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.-M. Tumor necrosis factor. Cancer Lett. 2013, 328, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, I.; Keramati, M.; Azadi, A.; Afzali, E.; Shahbazi, R.; Norouzian, D.; Bakhshandeh, H. Optimization, physicochemical characterization, and antimicrobial activity of a novel simvastatin nano-niosomal gel against E. coli and S. aureus. Chem. Phys. Lipids 2021, 234, 105019. [Google Scholar] [CrossRef]
- Muzzalupo, R.; Trombino, S.; Iemma, F.; Puoci, F.; La Mesa, C.; Picci, N. Preparation and characterization of bolaform surfactant vesicles. Colloids Surf. B: Biointerfaces 2005, 46, 78–83. [Google Scholar] [CrossRef]
- Rochani, A.K.; Balasubramanian, S.; Girija, A.R.; Raveendran, S.; Borah, A.; Nagaoka, Y.; Nakajima, Y.; Maekawa, T.; Kumar, D.S. Dual mode of cancer cell destruction for pancreatic cancer therapy using Hsp90 inhibitor loaded polymeric nano magnetic formulation. Int. J. Pharm. 2016, 511, 648–658. [Google Scholar] [CrossRef]
- Akbari, J.; Saeedi, M.; Enayatifard, R.; Morteza-Semnani, K.; Hashemi, S.M.H.; Babaei, A.; Rahimnia, S.M.; Rostamkalaei, S.S.; Nokhodchi, A. Curcumin Niosomes (curcusomes) as an alternative to conventional vehicles: A potential for efficient dermal delivery. J. Drug Deliv. Sci. Technol. 2020, 60, 102035. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Bettini, R.; Catellani, P.; Santi, P.; Massimo, G.; Peppas, N.; Colombo, P. Translocation of drug particles in HPMC matrix gel layer: Effect of drug solubility and influence on release rate. J. Control. Release 2001, 70, 383–391. [Google Scholar] [CrossRef]
- Lee, E.; Jeon, H.; Lee, M.; Ryu, J.; Kang, C.; Kim, S.; Jung, J.; Kwon, Y. Molecular origin of AuNPs-induced cytotoxicity and mechanistic study. Sci. Rep. 2019, 9, 2494. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Levels of Independent Variables | Dependent Variables | |||||
|---|---|---|---|---|---|---|---|
| Lipid: Drug (mol Ratio) | Surfactant: Cholesterol (mol Ratio) | Surfactant Type | Average Particle Size (nm) | Polydispersity Index | Entrapment Efficiency (%) | Loading Capacity (%) | |
| 1 | −1 | 1 | Span60 | 214.8 | 0.184 | 88.75 | 5.24 |
| 2 | 0 | 0 | Span60 | 155.3 | 0.162 | 92.35 | 2.86 |
| 3 | 0 | 0 | Span60 | 168.4 | 0.189 | 93.79 | 2.36 |
| 4 | 1 | 0 | Span20 | 239.8 | 0.275 | 84.45 | 1.96 |
| 5 | −1 | −1 | Span20 | 286.4 | 0.287 | 75.42 | 4.93 |
| 6 | 0 | −1 | Span20 | 294.1 | 0.346 | 81.08 | 2.74 |
| 7 | 1 | 1 | Span20 | 275.3 | 0.293 | 92.75 | 2.19 |
| 8 | 0 | 0 | Span20 | 193.4 | 0.189 | 82.19 | 2.83 |
| 9 | 0 | −1 | Span60 | 249.5 | 0.255 | 83.34 | 2.63 |
| 10 | 1 | −1 | Span20 | 326.1 | 0.329 | 79.92 | 1.82 |
| 11 | 1 | −1 | Span60 | 243.4 | 0.346 | 82.43 | 1.75 |
| 12 | 0 | 0 | Span20 | 198.2 | 0.205 | 79.78 | 2.74 |
| 13 | −1 | 0 | Span60 | 162.1 | 0.231 | 83.79 | 5.03 |
| 14 | 1 | 0 | Span60 | 224.2 | 0.218 | 95.83 | 2.00 |
| 15 | 1 | 1 | Span60 | 286.4 | 0.315 | 97.74 | 2.00 |
| 16 | 0 | 0 | Span60 | 173.5 | 0.193 | 95.14 | 2.95 |
| 17 | −1 | 0 | Span20 | 185.7 | 0.285 | 84.61 | 5.53 |
| 18 | 0 | 1 | Span20 | 258.6 | 0.341 | 85.70 | 3.00 |
| 19 | 0 | 0 | Span20 | 192.1 | 0.211 | 77.24 | 2.66 |
| 20 | 0 | 1 | Span60 | 230.7 | 0.196 | 86.49 | 3.99 |
| 21 | −1 | 1 | Span20 | 232.5 | 0.194 | 84.63 | 5.73 |
| 22 | −1 | −1 | Span60 | 279.3 | 0.307 | 81.92 | 5.01 |
| Source | f-Value | p-Value Prob > f |
|---|---|---|
| Particle Size (nm) | ||
| Model | 20.00 | <0.0001 |
| A | 16.22 | 0.0014 |
| B | 9.62 | 0.0084 |
| C | 11.17 | 0.0053 |
| AB | 5.42 | 0.0368 |
| AC | 0.017 | 0.8981 |
| BC | 2.95 | 0.1098 |
| A^2 | 4.57 | 0.0521 |
| B^2 | 91.05 | <0.0001 |
| PDI | ||
| Model | 3.57 | 0.0206 |
| A | 3.63 | 0.0792 |
| B | 5.27 | 0.0390 |
| C | 3.07 | 0.1031 |
| AB | 1.46 | 0.2490 |
| AC | 0.030 | 0.8661 |
| BC | 0.27 | 0.6101 |
| A^2 | 2.00 | 0.1812 |
| B^2 | 9.46 | 0.0089 |
| EE (%) | ||
| Model | 4.90 | 0.0058 |
| A | 6.11 | 0.0280 |
| B | 14.27 | 0.0023 |
| C | 15.70 | 0.0016 |
| AB | 1.16 | 0.3009 |
| AC | 0.44 | 0.5207 |
| BC | 9.921 × 10−3 | 0.9222 |
| A^2 | 0.25 | 0.6262 |
| B^2 | 1.47 | 0.2472 |
| Models |
|---|
| Particle Size (Span20) = +194.86 + 20.17 × A − 23.37 × B + 13.83 × A × B + 15.96 × A2 + 71.23 × B2 Particle Size (Span60) = +170.91 + 18.90 × A − 6.72 × B +13.82 × A × B + 15.96 × A2+ 71.23 × B2 |
| PDI (Span20) = +0.221 + 0.022 × A − 0.022 × B + 0.019 × A × B + 0.027 × A2 + 0.060 × B2 PDI (Span60) = +0.189 + 0.026 × A − 0.035 × B + 0.019 × A × B + 0.027 × A2 + 0.060 × B2 |
| EE (Span20) = +83.21 + 2.08 × A + 4.44 × B + 1.51 × A × B + 0.88 × A2 − 2.14 × B2 EE (Span60) = +89.92 + 3.59 × A + 4.22 × B + 1.51 × A × B +0.88 × A2 − 2.14 × B2 |
| Response | R-Squared | Adj R-Squared | Adeq Precision | Lack of Fit |
|---|---|---|---|---|
| Particle size | 0.9249 | 0.8786 | 13.378 | 0.0314 |
| PDI | 0.6872 | 0.4946 | 5.201 | 0.0128 |
| EE (%) | 0.7509 | 0.5976 | 8.407 | 0.0637 |
| Parameter | Predicted by RSM | Experimental Data (Nio-Cyclo) | Nio-Cyclo-PEG | Niosome (Nio) |
|---|---|---|---|---|
| Average size (nm) | 171.656 | 188.100 ± 9.200 | 170.600 ± 5.530 | 145.540 ± 6.200 |
| polydispersity index (PDI) | 0.179 | 0.170 ± 0.014 | 0.145 ± 0.007 | 0.191 ± 0.012 |
| Entrapment efficiency (EE) (%) | 90.337 | 92.580 ± 1.120 | 94.870 ± 0.720 | - |
| Kinetic Model | Zero-Order | First-Order | Higuchi | Korsmeyer–Peppas | ||
|---|---|---|---|---|---|---|
| Ct = C0 + K0t | LogC = LogC0 + Kt/2.303 | Mt/Mꝏ = Kt.tn | ||||
| r2 | r2 | r2 | r2 | n * | ||
| Free Cyclophosphamide (Cyclo) | pH 7.4 | 0.4854 | 0.9305 | 0.6586 | 0.7666 | 0.5135 |
| Nio-Cyclo | pH 7.4 | 0.8508 | 0.9133 | 0.9599 | 0.9679 | 0.5384 |
| pH 5.4 | 0.8306 | 0.9467 | 0.9468 | 0.9556 | 0.4829 | |
| pH 1.2 | 0.6929 | 0.9588 | 0.8476 | 0.9058 | 0.3996 | |
| Nio-Cyclo-PEG | pH 7.4 | 0.8609 | 0.9082 | 0.9639 | 0.9730 | 0.6304 |
| pH 5.4 | 0.8644 | 0.9393 | 0.9666 | 0.9682 | 0.5519 | |
| pH 1.2 | 0.7809 | 0.8991 | 0.9133 | 0.9353 | 0.4288 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khodabakhsh, F.; Bourbour, M.; Yaraki, M.T.; Bazzazan, S.; Bakhshandeh, H.; Ahangari Cohan, R.; Tan, Y.N. pH-Responsive PEGylated Niosomal Nanoparticles as an Active-Targeting Cyclophosphamide Delivery System for Gastric Cancer Therapy. Molecules 2022, 27, 5418. https://doi.org/10.3390/molecules27175418
Khodabakhsh F, Bourbour M, Yaraki MT, Bazzazan S, Bakhshandeh H, Ahangari Cohan R, Tan YN. pH-Responsive PEGylated Niosomal Nanoparticles as an Active-Targeting Cyclophosphamide Delivery System for Gastric Cancer Therapy. Molecules. 2022; 27(17):5418. https://doi.org/10.3390/molecules27175418
Chicago/Turabian StyleKhodabakhsh, Farnaz, Mahsa Bourbour, Mohammad Tavakkoli Yaraki, Saina Bazzazan, Haleh Bakhshandeh, Reza Ahangari Cohan, and Yen Nee Tan. 2022. "pH-Responsive PEGylated Niosomal Nanoparticles as an Active-Targeting Cyclophosphamide Delivery System for Gastric Cancer Therapy" Molecules 27, no. 17: 5418. https://doi.org/10.3390/molecules27175418
APA StyleKhodabakhsh, F., Bourbour, M., Yaraki, M. T., Bazzazan, S., Bakhshandeh, H., Ahangari Cohan, R., & Tan, Y. N. (2022). pH-Responsive PEGylated Niosomal Nanoparticles as an Active-Targeting Cyclophosphamide Delivery System for Gastric Cancer Therapy. Molecules, 27(17), 5418. https://doi.org/10.3390/molecules27175418