
Weak Coordinating Character of Organosulfonates in Oriented Silica Films: An Efficient Approach for Immobilizing Cationic Metal-Transition Complexes



Abstract
:1. Introduction
2. Results and Discussion
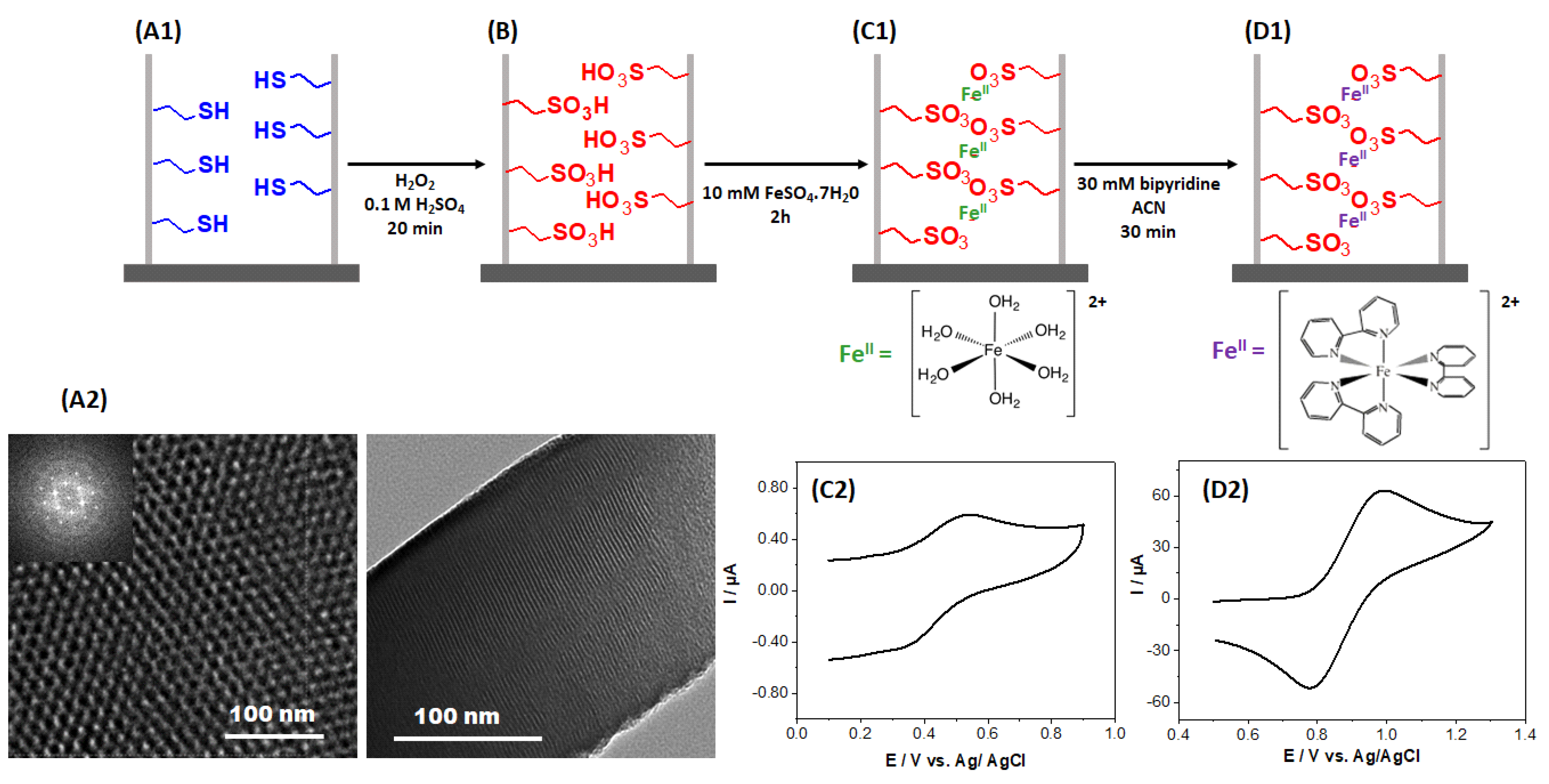
2.1. Generation of Mesoporous Films and Monitoring of the Successive Functionalization Steps
2.2. Structural Characterization of [Fe(bpy)3]2+ Complexes Formed in the Films
2.3. Electrochemical Response of the [Fe(bpy)3]2+SO3−@SiO2 Films
2.4. Optical Properties of Fe(bpy)32+ Modified Sulfonated Silica Films
3. Materials and Methods
3.1. Apparatus
3.2. Chemicals and Reagents
3.3. Preparation of Thiol and Sulfonate Functionalized Silica Films
3.4. Preparation of Fe(bpy)32+SO3−@SiO2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barton, M.R.; Zhang, Y.; Atwood, J.D. Mono-Sulfonated Derivatives of Triphenylphosphine, [NH4]TPPMS and M(TPPMS)2 (TPPMS = P(Ph)2(m-C6H4SO3−); M = Mn2+, Fe2+, Co2+ and Ni2+). Crystal Structure Determinations for [NH4][TPPMS]·½H2O, [Fe(H2O)5(TPPMS)]TPPMS, [Co(H2O)5TPPMS]TPPMS and [Ni(H2O)6] (TPPMS)4·H2O. J. Coord. Chem. 2002, 55, 969–983. [Google Scholar]
- Li, F.-F.; Ma, J.-F.; Song, S.-Y.; Yang, J.; Liu, Y.-Y.; Su, Z.-M. Influence of Neutral Ligands on the Structures of Silver(I) Sulfonates. Inorg. Chem. 2005, 44, 9374–9383. [Google Scholar]
- Forbes, T.Z.; Sevov, S.C. Metal-Organic Frameworks with Direct Transition Metal-Sulfonate Interactions and Charge-Assisted Hydrogen Bonds. Inorg. Chem. 2009, 48, 6873–6878. [Google Scholar] [CrossRef]
- Dalrymple, S.A.; Parvez, M.; Shimizu, G.K.H. Supramolecular Encapsulation of Hexaaquo Metal Ions by Second Sphere Coordination. Chem. Commun. 2001, 24, 2672–2673. [Google Scholar] [CrossRef]
- Holman, K.T.; Pivovar, A.M.; Swift, J.A.; Ward, M.D. Metric Engineering of soft Molecular Host Frameworks. Acc. Chem. Res. 2001, 34, 107–118. [Google Scholar] [CrossRef]
- Holman, K.T.; Martin, S.M.; Parker, D.P.; Ward, M.D. The Generality of Architectural Isomerism in Designer Inclusion Frameworks. J. Am. Chem. Soc. 2001, 123, 4421–4431. [Google Scholar] [CrossRef]
- Horner, M.J.; Holman, K.T.; Ward, M.D. Architectural Diversity and elastic Networks in Hydrogen-Bonded Host Frameworks: From Molecular Jaws to Cylinders. J. Am. Chem. Soc. 2007, 129, 14640–14660. [Google Scholar] [CrossRef]
- Davies, J.A.; Hockensmith, C.A.; Kukushkin, V.Y.; Kukushkin, Y.A. Synthetic Coordination chemistry: Principles and Practice; World Scientific: London, UK, 1996. [Google Scholar]
- Cote, A.P.; Shimizu, G.H.K. The Supramolecular Chemistry of the Sulfonate Group in Extended Solids. Coord. Chem. Rev. 2003, 245, 49–64. [Google Scholar] [CrossRef]
- Shimizu, G.H.K.; Vaidhyanathan, R.; Taylor, J.M. Phosphonate and Sulfonate Metal Organic Frameworks. Chem. Soc. Rev. 2009, 38, 1430–1449. [Google Scholar] [CrossRef]
- Wang, X.Y.; Sevov, S.C. Hydrogen-Bonded Metal-Complex Sulfonate (MCS) Inclusion compounds: Effect of the Guest Molecule on the Host Framework. Inorg. Chem. 2007, 46, 4626–4631. [Google Scholar] [CrossRef]
- Reddy, S.D.; Duncan, S.; Shimizu, G.K.H. A Family of supramolecular Inclusion solids Based Upon Second-Sphere Interactions. Angew. Chem. Int. Ed. 2003, 115, 1398–1402. [Google Scholar] [CrossRef]
- Dalrymple, S.A.; Shimizu, G.K.H. An Open Channel Coordination Framework Sustained by Cooperative Primary and Secondary Sphere Interactions. Chem. Commun. 2002, 19, 2224–2225. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, S.A.; Shimizu, G.K.H. Selective Guest Inclusion in a Non-Porous H-Bonded Host. Chem. Commun. 2006, 9, 956–958. [Google Scholar] [CrossRef] [PubMed]
- Feinle, A.; Lavoie-Cardinal, F.; Akbarzadeh, J.; Peterlik, H.; Adlung, M.; Wickleder, C.; Huesing, N. Novel Sol–Gel Precursors for Thin Mesoporous Eu3+-Doped Silica Coatings as Efficient Luminescent Materials. Chem. Mater. 2012, 24, 3674. [Google Scholar] [CrossRef]
- Walcarius, A. Mesoporous Materials and Electrochemistry. Chem. Soc. Rev. 2013, 42, 4098–4140. [Google Scholar] [CrossRef]
- Yamashita, H.; Mori, K.; Kuwahara, Y.; Kamegawa, T.; Wen, M.; Verma, P.; Che, M. Single-Site and Nano-Confined Photocatalysts Designed in Porous Materials for Environmental Uses and Solar Fuels. Chem. Soc. Rev. 2018, 47, 8072–8096. [Google Scholar] [CrossRef]
- Laskowski, L.; Laskowska, M.; Vilà, N.; Schabikowski, M.; Walcarius, A. Mesoporous Silica-Based Materials for Electronics-Oriented Applications. Molecules 2019, 24, 2395. [Google Scholar] [CrossRef]
- Walcarius, A.; Sibottier, E.; Etienne, M.; Ghanbaja, J. Electrochemically-Assisted Self-Assembly of Mesoporous Silica Thin films. Nat. Mater. 2007, 6, 602–608. [Google Scholar] [CrossRef]
- Goux, A.; Etienne, M.; Aubert, E.; Lecomte, C.; Ghanbaja, J.; Walcarius, A. Oriented Mesoporous Silica Films Obtained by Electro-Assisted Self-Assembly. Chem. Mater. 2009, 21, 731–741. [Google Scholar] [CrossRef]
- Walcarius, A. Electroinduced Surfactant Self-Assembly Driven to Vertical Growth of Oriented Mesoporous Films. Acc. Chem. Res. 2021, 54, 3563–3575. [Google Scholar] [CrossRef]
- Ziarani, G.M.; Hassanzadeh, Z.; Gholamzadeh, P.; Asadi, S.; Badiei, A. Advances in click chemistry for silica-based material construction. RSC Adv. 2016, 6, 21979–22006. [Google Scholar] [CrossRef]
- Cattoen, X.; Noureddine, A.; Croissant, J.; Moitra, N.; Burglova, K.; Hodacova, J.; de los Cobos, O.; Lejeune, M.; Rossignol, F.; Toulemon, D.; et al. Click Approaches in Sol-Gel Chemistry. J. Sol-Gel Sci. Technol. 2014, 70, 245–253. [Google Scholar] [CrossRef]
- Vilà, N.; Walcarius, A. Bis(terpyridine) Iron(II) Functionalized Vertically-Oriented Nanostructured Silica Films: Toward Electrochromic Materials. Front. Chem. 2020, 8, 830. [Google Scholar] [CrossRef]
- Ahoulou, S.; Vilà, N.; Pillet, S.; Carteret, C.; Schaniel, D.; Walcarius, A. Multi-Stimuli Photo and Redox Active Nanostructured Mesoporous Silica films on transparent electrodes. ChemPhysChem 2021, 22, 2464–2477. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.A., Jr.; Baldo, J.B. The Behavior of Zeta Potential of Silica Suspensions. New J. Glass Ceram. 2014, 4, 29–37. [Google Scholar]
- Zhou, P.; Yao, L.; Chen, K.; Su, B. Silica Nanochannel Membranes for Electrochemical Analysis Molecular sieving: A Comprehensive Review. Crit. Rev. Anal. Chem. 2020, 50, 424. [Google Scholar] [CrossRef]
- Basnig, D.; Vilà, N.; Herzog, G.; Walcarius, A. Voltammetric Behavior of Cationic Redox Probes at Mesoporous Silica film electrodes. J. Electroanal. Chem. 2020, 872, 113993. [Google Scholar] [CrossRef]
- Etienne, M.; Goux, A.; Sibottier, E.; Walcarius, A. Oriented Mesoporous Organosilica Films on electrode: A New Class of Nanomaterials for Sensing. J. Nanosci. Nanotechnol. 2009, 9, 2398–2406. [Google Scholar] [CrossRef]
- Vilà, N.; Ghanbaja, J.; Walcarius, A. Clickable Bifunctional and Vertically Aligned Mesoporous Silica Films. Adv. Mater. Interfaces 2016, 3, 1500440. [Google Scholar] [CrossRef]
- Wilson, K.; Lee, A.F.; Macquarrie, D.J.; Clark, J.H. Structure and Reactivity of Sol-Gel Sulphonic Acid Silicas. Appl. Catal. A 2002, 228, 127–133. [Google Scholar] [CrossRef]
- Shen, J.G.C.; Herman, R.G.; Klier, K. Sulfonic Acid-Functionalized Mesoporous Silica: Synthesis, characterization, and Catalytic Reaction of Alcohol Coupling to Ethers. J. Phys. Chem. B 2002, 106, 9975. [Google Scholar] [CrossRef]
- Dijs, I.J.; van Ochten, H.L.F.; van Walree, C.A.; Geus, J.W.; Jenneskens, L.W. Alkyl Sulphonic Acid Surface-Functionalized Silica as Heterogeneous Acid Catalyst in the Solvent Free Liquid-Phase Addition of Acetic Acid to Camphene. J. Mol. Catal. A 2002, 188, 209–224. [Google Scholar] [CrossRef]
- Greczynski, G.; Hultman, L. X-Ray Photoelectron Spectroscopy: Towards reliable binding Energy Referencing. Prog. Mater. Sci. 2020, 107, 100590. [Google Scholar] [CrossRef]
- Hanazaki, I.; Nagakura, S. Electronic Structure of the Tris(¦Á,¦Á’-dipyridyl)iron(II) Ion. Inorg. Chem. 1969, 8, 648–654. [Google Scholar] [CrossRef]
- Alexander, B.D.; Dines, T.J.; Longhurst, R.W. DFT Calculations of the Structures and Vibrational Spectra of the [Fe(bpy)3]2+ and (Ru(bpy)3]2+ Complexes. Chem. Phys. 2008, 352, 19–27. [Google Scholar] [CrossRef]
- Mallick, P.K.; Danzer, G.D.; Strommen, D.P.; Kincaid, J.R. Vibrational Spectra and Normal-Coordinate Analysis of Tris(bipyridine)ruthenium(II). J. Phys. Chem. 1988, 92, 5628–5634. [Google Scholar] [CrossRef]
- Dines, T.J.; Peacock, R.D. Surface-Enhanced Resonance Raman Spectroscopy of Iron(II) and Ruthenium(II) Bipyridyl Complexes adsorbed on Silver Sols. J. Chem. Soc. Faraday Trans. 1988, 84, 3445–3457. [Google Scholar] [CrossRef]
- Berger, R.M.; McMillin, D.R. Ultraviolet and Visible Resonance Raman spectroscopy of Tris-(2,2’-bipyridyl)iron(II): Intensity Considerations and Band Assignments. Inorg. Chim. Acta 1990, 177, 65–69. [Google Scholar] [CrossRef]
- Caswell, D.S.; Spiro, T.G. Resonance Raman Spectra of the Ground and Charge-Transfer Excited State of Pentammine(4,4’-bipyridine)ruthenium(II) and Pentammine(4,4’-bipyridinium)ruthenium(II). Inorg. Chem. 1987, 26, 18–22. [Google Scholar] [CrossRef]
- Tuchagues, J.P.; Bousseksou, A.; Molnar, G.; McGarvey, J.J.; Varret, F. The Role of Molecular Vibrations in the Spin Crossover Phenomenon. Top. Curr. Chem. 2004, 235, 85–103. [Google Scholar]
- Bousseksou, A.; McGarvey, J.J.; Varret, F.; Real, J.A.; Tuchagues, J.-P.; Deniis, A.C.; Boillot, M.L. Raman Spectroscop of the High- and Low-Spin States of the Spin Crossover complex Fe(phen)2(NCS)2: An Initial Approach to Estimation of vibrational contributions to the Associated Entropy Change. Chem. Phys. Lett. 2000, 318, 409–416. [Google Scholar] [CrossRef]
- Takemoto, J.H.; Hutchinson, B. Low-Frequency Infrared Spectra of complexes which Exhibit Magnetic Crossover. I. Iton(II) complexes of 1,10-Phenanthroline and 2,2’-bipyridine. Inorg. Chem. 1973, 12, 705–708. [Google Scholar] [CrossRef]
- König, E.; Madeja, K.; Watson, K.J. Reversible Quintet-Singlet Transition in Dithiocyanato-bis(2,2’-dipyridyl)iron(II). J. Am. Chem. Soc. 1968, 90, 1146–1153. [Google Scholar] [CrossRef]
- van der Westhuizen, D.; von Eschwege, K.G.; Conradie, J. Electrochemistry and Spectroscopy of Substituted [Ru(phen)3]2+ and [Ru(bpy)3]2+ Complexes. Electrochim. Acta 2019, 320, 134540. [Google Scholar] [CrossRef]
- Vilà, N.; Walcarius, A. Electrochemical Response of Vertically-aligned, Ferrocene-Functionalized Mesoporous Silica Films: Effect of the Supporting Electrolyte. Electrochim. Acta 2015, 179, 304–314. [Google Scholar] [CrossRef]
- Chae, D.J.; Kim, D.Y.; Kim, T.G.; Sung, Y.M.; Kim, M.D. AlGaN-based Ultraviolet Light-emitting Diodes using Fluorine-doped Indium Tin Oxide Electrodes. Appl. Phys. Lett. 2012, 100, 8–11. [Google Scholar]
- Kober, E.M.; Meyer, T.J. Concerning the Absorption Spectra of the Ions M(bpy)32+ (M = Fe, Ru, Os; bpy = 2,2’-bipyridine). Inorg. Chem. 1982, 21, 3967–3977. [Google Scholar] [CrossRef]
- Decurtins, S.; Giidel, H.U.; Ludi, A.; Felix, F.; Ferguson, J. The Electronic spectrum of Tris(2,2’-bipyridine)ruthenium(II). J. Am. Chem. Soc. 1980, 102, 4096–4102. [Google Scholar]
- Auböck, G.; Chergui, M. Sub-50-fs-Photoinduced Spin Crossover in [Fe(bpy)3]2+. Nat. Chem. 2015, 7, 629–633. [Google Scholar] [CrossRef]
- Azad, U.P.; Ganesan, V. Efficient Sensing of Nitrite by [Fe(bpy)3]2+ Immobilized Nafion Modified Electrodes. Chem. Commun. 2010, 46, 6156–6158. [Google Scholar] [CrossRef]
- Vishnu, N.; Kumar, A.S. Development of Prussian blue and [Fe(bpy)3]2+ Hybrid Modified Pencil Graphte Electrodes Utilizing its Intrinsic Iron for electroanalytical Applications. J. Electroanal. Chem. 2017, 786, 145. [Google Scholar] [CrossRef]
- De Graaf, C.; Sousa, C. Study of the Light-Induced Spin-Crossover Process of the [Fe(bpy)3]2+ Complex. Chem. Eur. J. 2010, 16, 4550–4556. [Google Scholar] [CrossRef] [PubMed]
- Ahoulou, S.; Vilà, N.; Pillet, S.; Schaniel, D.; Walcarius, A. Coordination Polymers as Template for Mesoporous Silica Films: A Novel Composite Material Fe(HTrz)3@SiO2 with Remarkable Electrochemical Properties. Chem. Mater. 2019, 31, 5796–5807. [Google Scholar] [CrossRef]
- Ganesan, V.; Walcarius, A. surfactant templated sulfonic Acid Functionalized Silica Microspheres as New Efficient Ion Exchangers and Electrode Modifiers. Langmuir 2004, 20, 3632. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Films Composition | Si2p Signal | S2p Signal | Fe2p Signal | N1s Signal | Atomic Ratios | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| %At | BE/Ev | %At | BE/Ev | %At | BE/Ev | %At | BE/Ev | S/Si | S/Fe | N/Fe | |
| MCTMS/TEOS | |||||||||||
| 5/95 | 95.8 | 102.0 | 4.2 | 161.7 | - - - - | - - - - | - - - - | - - - - | 0.04 | - - - - | - - - - |
| 10/90 | 90.5 | 102.0 | 9.5 | 161.7 | - - - - | - - - - | - - - - | - - - - | 0.11 | - - - - | - - - - |
| 20/80 | 81.2 | 102.0 | 18.8 | 161.8 | - - - - | - - - - | - - - - | - - - - | 0.23 | - - - - | - - - - |
| 30/70 | 72.3 | 102.0 | 27.7 | 161.7 | - - - - | - - - - | - - - - | - - - - | 0.38 | - - - - | - - - - |
| SO3−/TEOS | |||||||||||
| 5/95 | 96.0 | 102.0 | 4.0 | 167.1 | - - - - | - - - - | - - - - | - - - - | 0.04 | - - - - | - - - - |
| 10/90 | 96.7 | 102.0 | 9.3 | 167.2 | - - - - | - - - - | - - - - | - - - - | 0.10 | - - - - | - - - - |
| 20/80 | 81.6 | 102.0 | 18.4 | 167.2 | - - - - | - - - - | - - - - | - - - - | 0.23 | - - - - | - - - - |
| 30/70 | 71.5 | 102.0 | 28.5 | 167.2 | - - - - | - - - - | - - - - | - - - - | 0.40 | - - - - | - - - - |
| Fe2+SO3−/TEOS | |||||||||||
| 5/95 | 94.3 | 102.0 | 3.8 | 167.7 | 1.9 | 710.7/723.4 | - - - - | - - - - | 0.04 | 2.00 | - - - - |
| 10/90 | 88.1 | 102.0 | 8.5 | 167.7 | 3.7 | 710.8/723.5 | - - - - | - - - - | 0.10 | 2.30 | - - - - |
| 20/80 | 79.9 | 102.0 | 14.2 | 167.8 | 5.9 | 710.7/723.4 | - - - - | - - - - | 0.18 | 2.41 | - - - - |
| 30/70 | 69.9 | 102.0 | 23.1 | 167.8 | 7.0 | 710.7/723.4 | - - - - | - - - - | 0.33 | 3.30 | - - - - |
| [Fe(bpy)3]2+ SO3−/TEOS | |||||||||||
| 5/95 | 87.5 | 102.0 | 2.9 | 167.5 | 1.4 | 710.7/723.4 | 8.2 | 399.9 | 0.03 | 2.10 | 5.86 |
| 10/90 | 74.7 | 102.0 | 5.9 | 167.4 | 2.9 | 710.8/723.4 | 16.5 | 399.8 | 0.08 | 2.03 | 5.69 |
| 20/80 | 48.5 | 102.0 | 11.1 | 167.4 | 5.5 | 710.9/723.3 | 31.9 | 399.7 | 0.23 | 2.02 | 5.80 |
| 30/70 | 65.3 | 102.0 | 22.5 | 167.4 | 6.1 | 710.9/723.4 | 6.1 | 399.8 | 0.34 | 3.69 | 1.00 |
| Peak Position (cm−1) | Assignment |
|---|---|
| 155 | δ (N-Fe-N); benzene cycle bending modes |
| 369 | ν (Fe-N) |
| 662 | Ring deformation |
| 766 | Ring deformation |
| 1022 | Ring deformation |
| 1275 | δ (C-H) |
| 1322 | δ (C-H) |
| 1489 | ν (C=C) |
| 1562 | δ (C-H) |
| 1605 | ν (C=N) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahoulou, S.; Richart, C.; Carteret, C.; Pillet, S.; Vilà, N.; Walcarius, A. Weak Coordinating Character of Organosulfonates in Oriented Silica Films: An Efficient Approach for Immobilizing Cationic Metal-Transition Complexes. Molecules 2022, 27, 5444. https://doi.org/10.3390/molecules27175444
Ahoulou S, Richart C, Carteret C, Pillet S, Vilà N, Walcarius A. Weak Coordinating Character of Organosulfonates in Oriented Silica Films: An Efficient Approach for Immobilizing Cationic Metal-Transition Complexes. Molecules. 2022; 27(17):5444. https://doi.org/10.3390/molecules27175444
Chicago/Turabian StyleAhoulou, Samuel, Clara Richart, Cédric Carteret, Sébastien Pillet, Neus Vilà, and Alain Walcarius. 2022. "Weak Coordinating Character of Organosulfonates in Oriented Silica Films: An Efficient Approach for Immobilizing Cationic Metal-Transition Complexes" Molecules 27, no. 17: 5444. https://doi.org/10.3390/molecules27175444
APA StyleAhoulou, S., Richart, C., Carteret, C., Pillet, S., Vilà, N., & Walcarius, A. (2022). Weak Coordinating Character of Organosulfonates in Oriented Silica Films: An Efficient Approach for Immobilizing Cationic Metal-Transition Complexes. Molecules, 27(17), 5444. https://doi.org/10.3390/molecules27175444