
Molecular Genomic Study of Inhibin Molecule Production through Granulosa Cell Gene Expression in Inhibin-Deficient Mice


 , ,
, , 
Abstract
:
1. Introduction
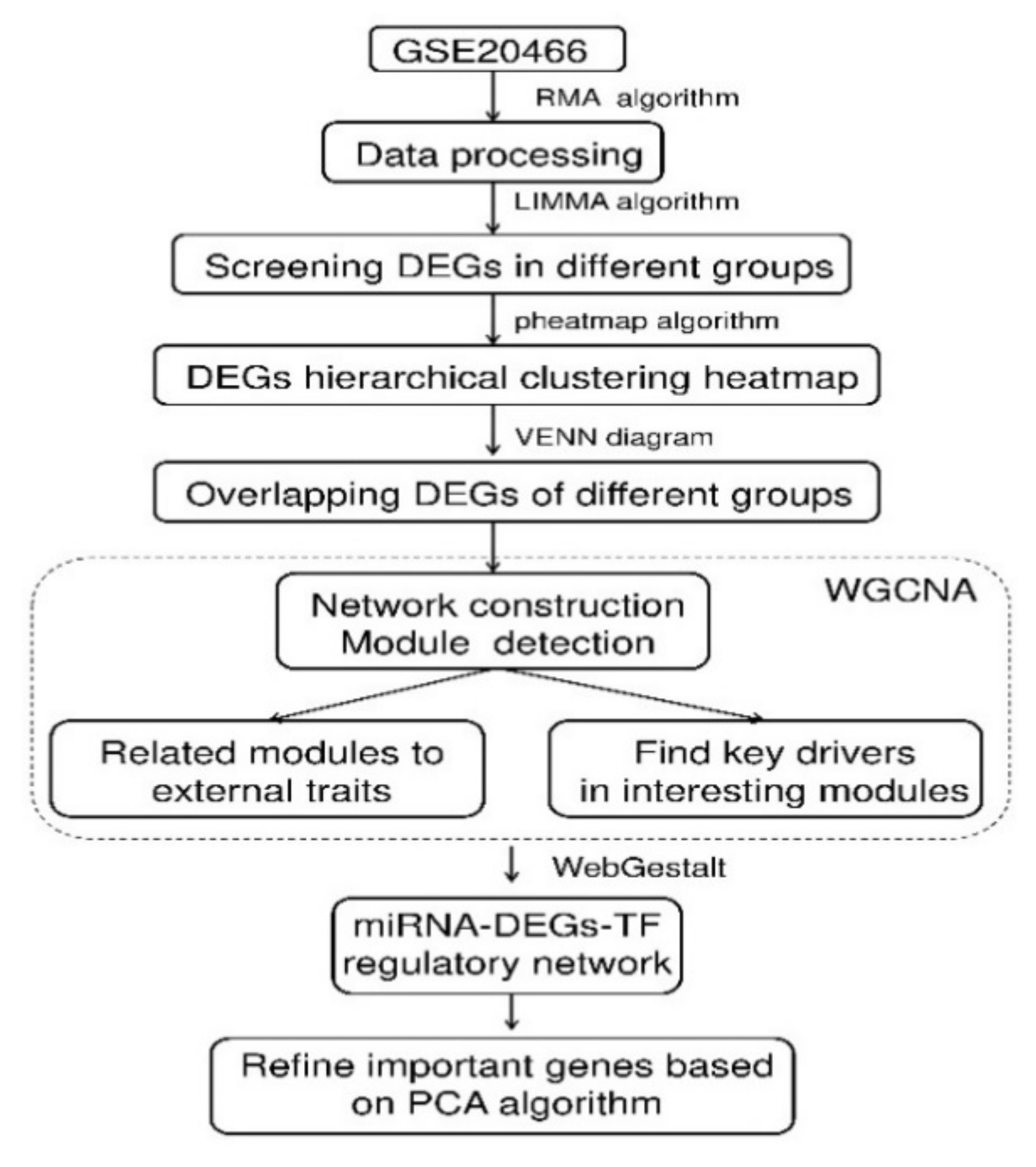
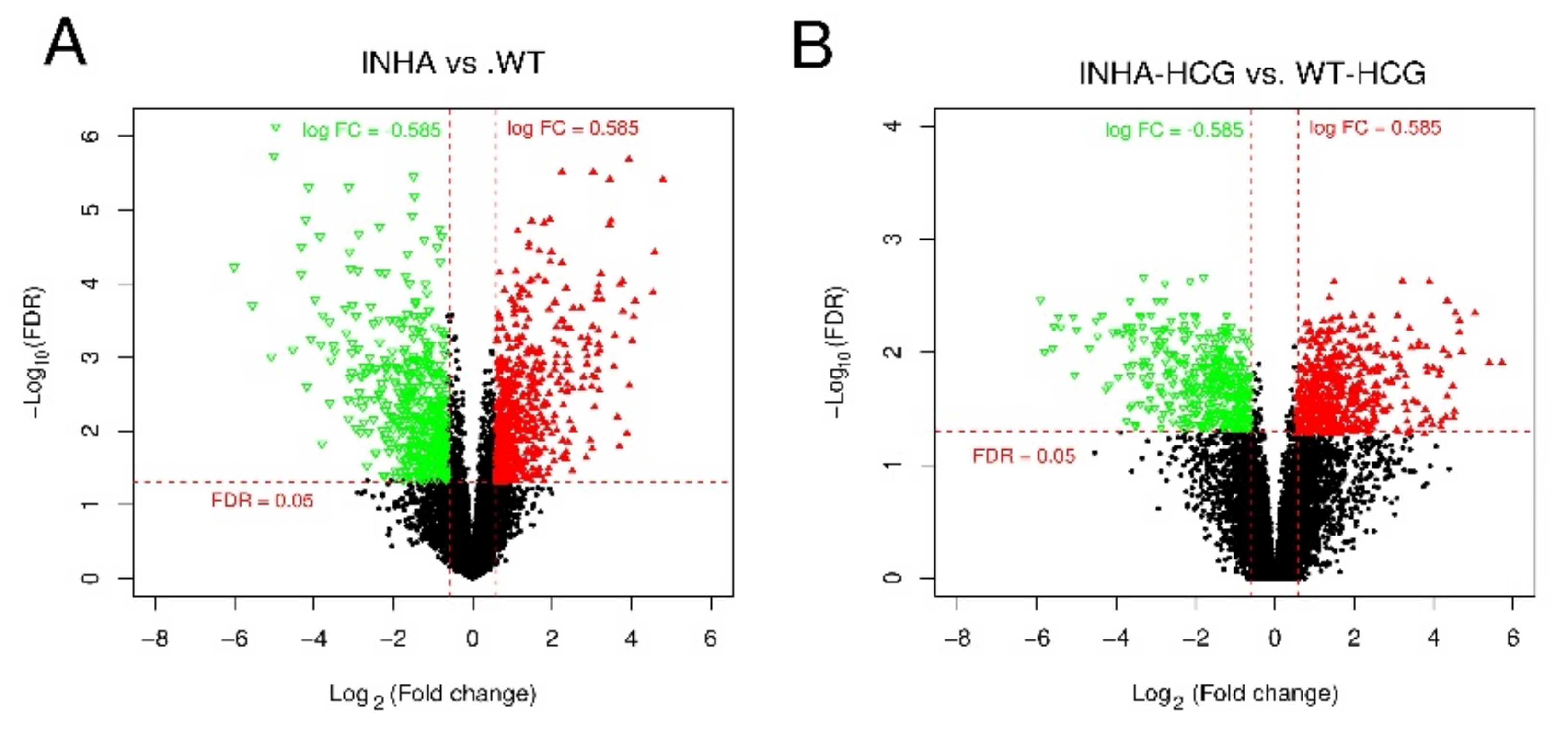
2. Results
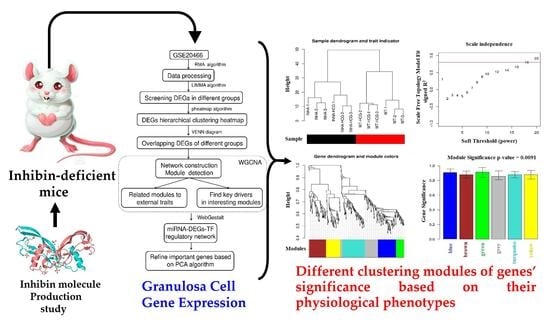
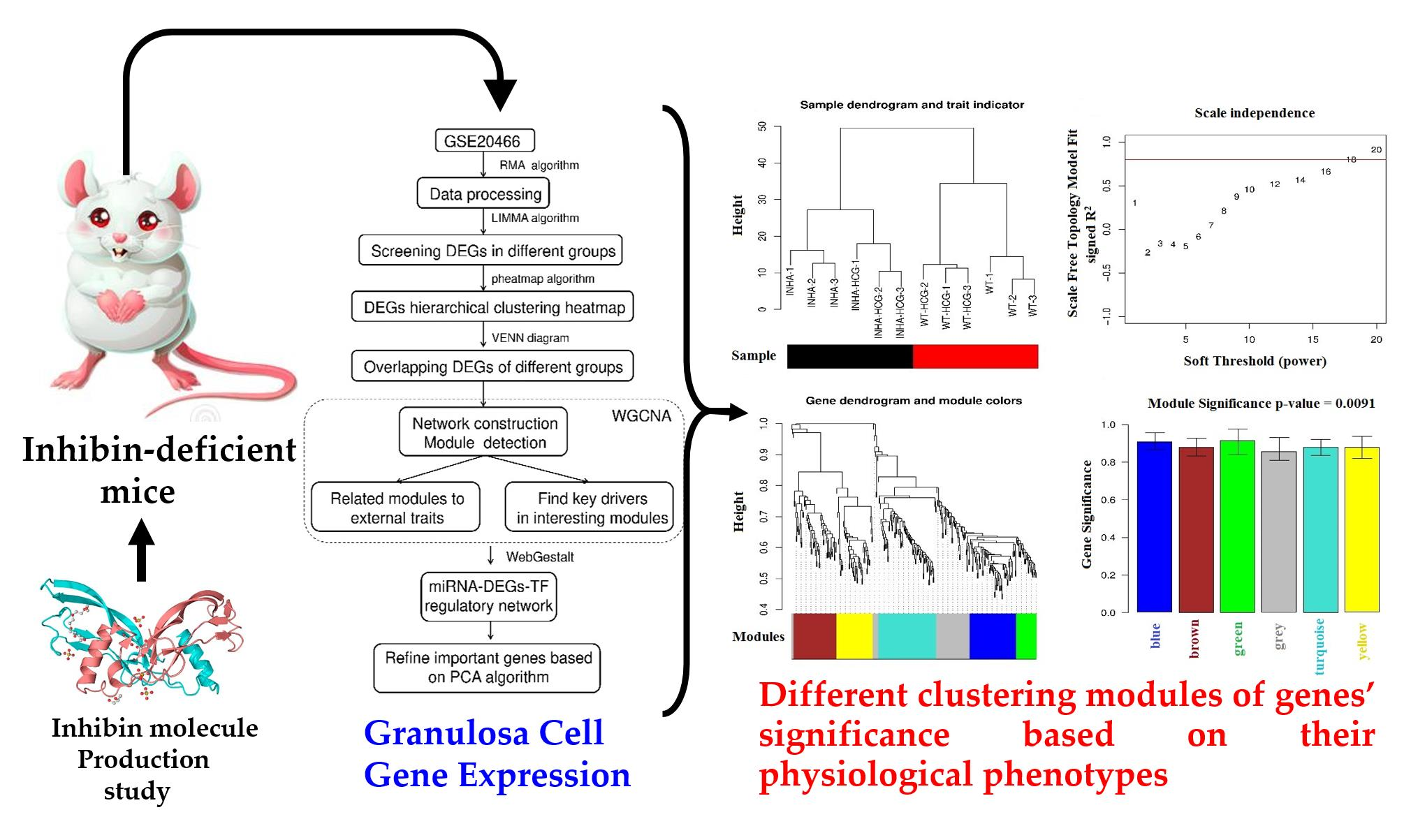
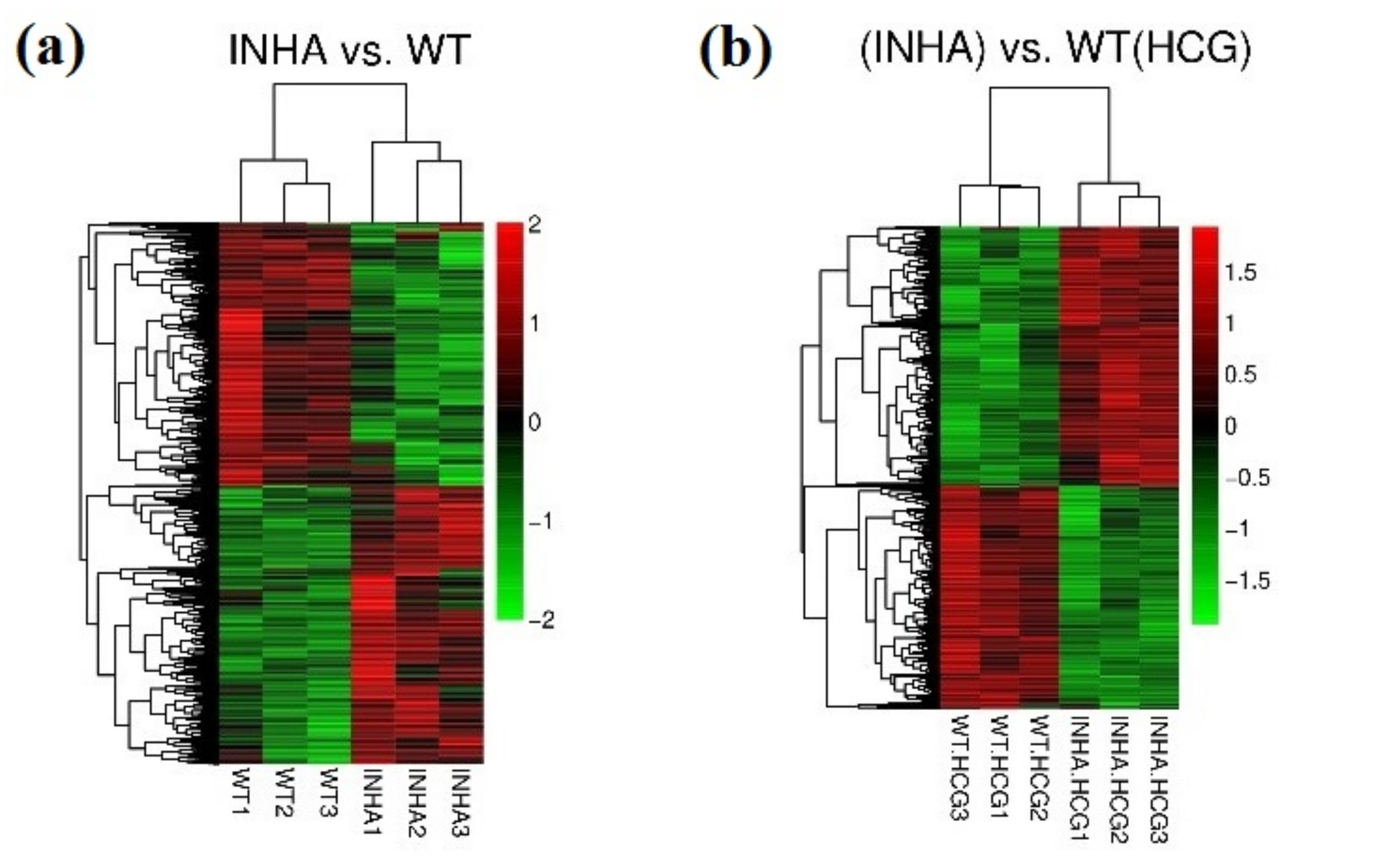
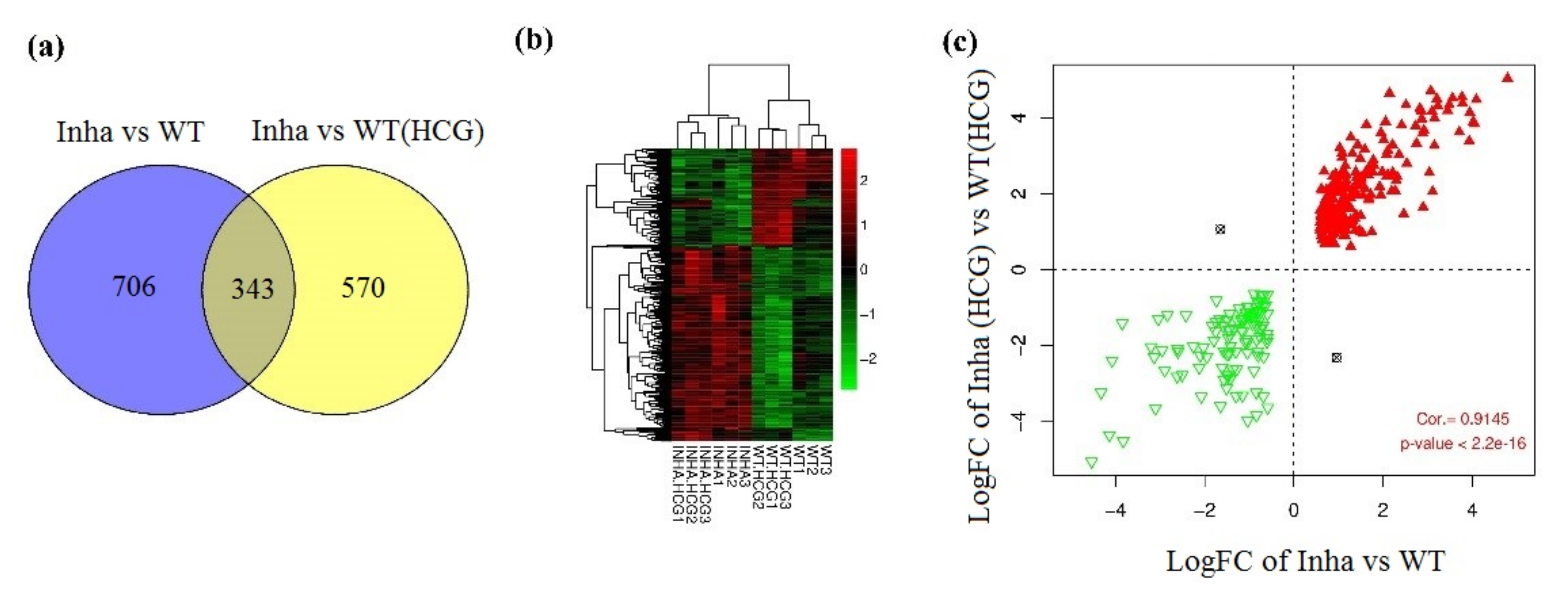
2.1. Hierarchical Clustering and Comparison Analysis of Selected DEGs in Different Groups
2.2. GO and KEGG Pathway Enrichment Analysis for the ODEGs
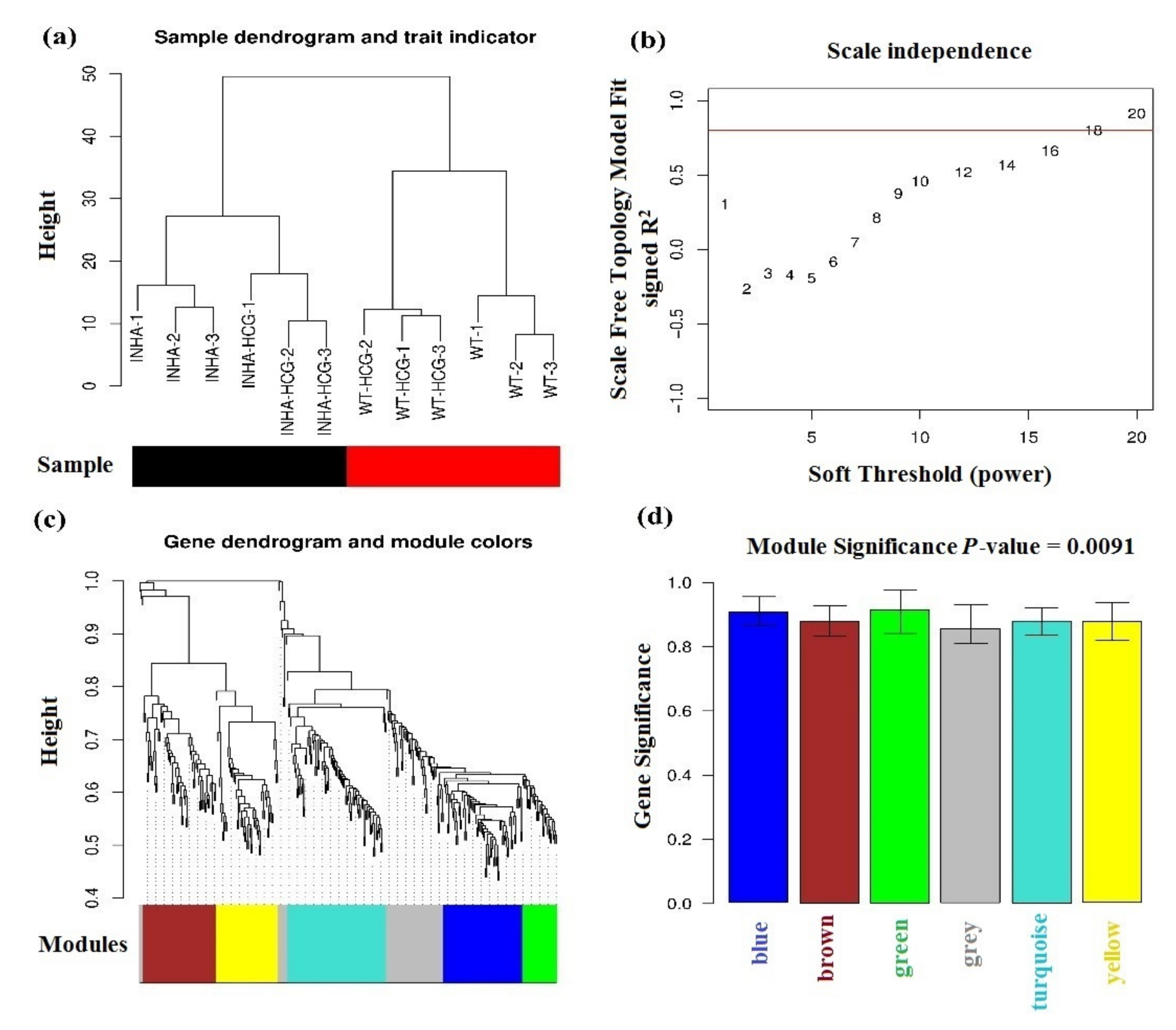
2.3. Physiological Phenotypes R Modules and Genes Identification Based on WGCNA
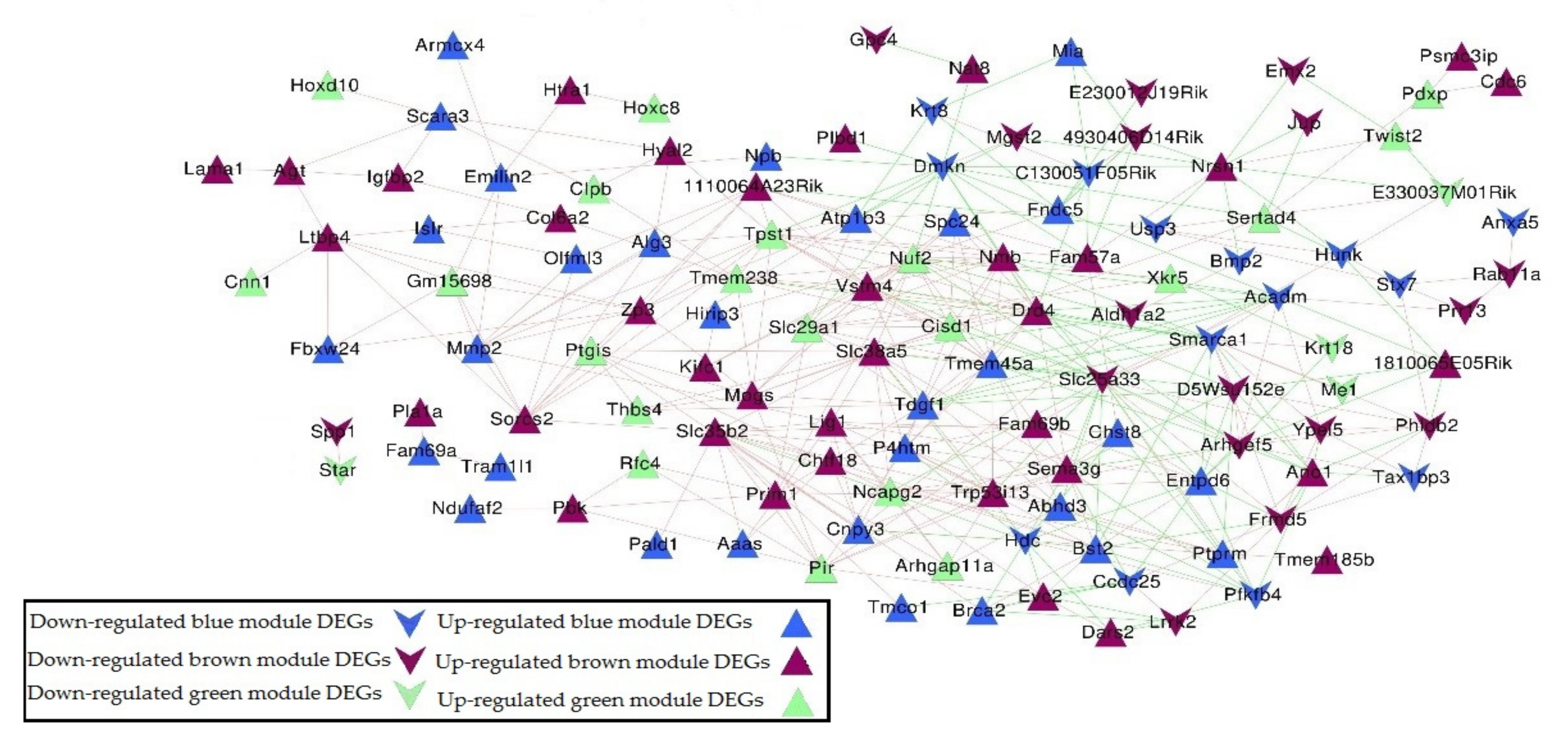
2.4. Co-Expression Network Construction
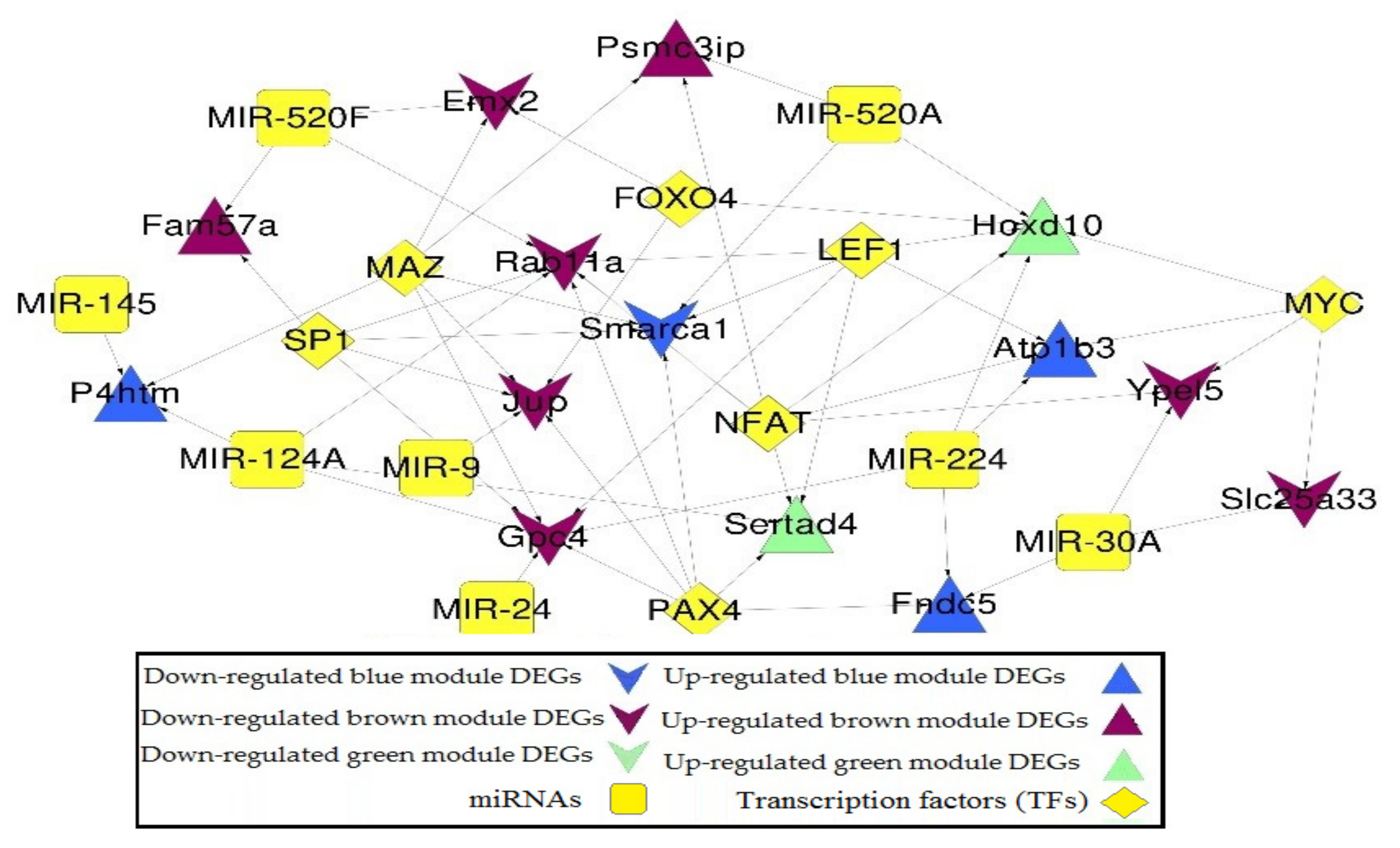
2.5. miRNA-DEGs-TF Regulatory Network Construction
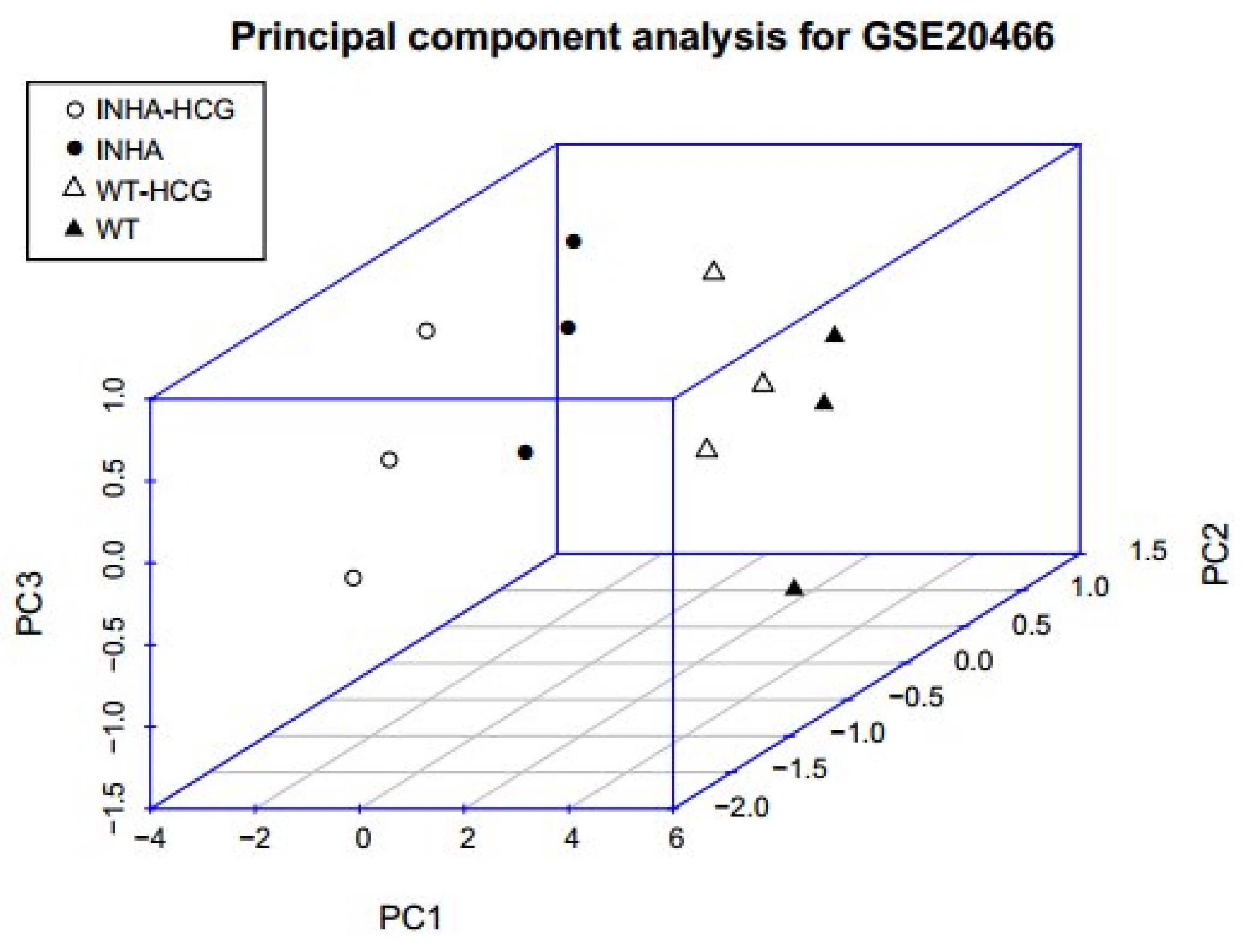
2.6. PCA for Genes in Regulatory Network
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Data and Experimental Design
4.3. Data Reprocessing and Differentially Expressed Genes (DEGs) Screening
4.4. Hierarchical Clustering and Comparison Analysis of Selected DEGs in Different Groups
4.5. Enrichment Analysis for the Overlapping DEGs
4.6. Physiological Phenotypes-Related Modules and Genes Identification Based on WGCNA
4.7. Co-Expression Network Construction
4.8. miRNA-DEGs-TF Target Regulatory Network Analysis
4.9. Principal Component Analysis (PCA) for Genes in the Regulatory Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dufour, S.; Quérat, B.; Tostivint, H.; Pasqualini, C.; Vaudry, H.; Rousseau, K. Origin and Evolution of the Neuroendocrine Control of Reproduction in Vertebrates, With Special Focus on Genome and Gene Duplications. Physiol. Rev. 2020, 100, 869–943. [Google Scholar] [CrossRef] [PubMed]
- Filatov, M.; Khramova, Y.; Parshina, E.; Bagaeva, T.; Semenova, M. Influence of gonadotropins on ovarian follicle growth and development in vivo and in vitro. Zygote 2017, 25, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Kandaraki, E.A.; Chatzigeorgiou, A.; Papageorgiou, E.; Piperi, C.; Adamopoulos, C.; Papavassiliou, A.G.; Koutsilieris, M.; Diamanti-Kandarakis, E. Advanced glycation end products interfere in luteinizing hormone and follicle stimulating hormone signaling in human granulosa KGN cells. Exp. Biol. Med. 2018, 243, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Bassil, R.; Meriano, J.; Samara, N.; Barzilay, E.; Gonen, N.; Casper, R.F. Does daily co-administration of letrozole and gonadotropins during ovarian stimulation improve IVF outcome? Reprod. Biol. Endocrinol. 2017, 15, 70. [Google Scholar] [CrossRef]
- Han, L.; Wu, C.; Riaz, H.; Bai, L.; Chen, J.; Zhen, Y.; Guo, A.; Yang, L. Characterization of the mechanism of inhibin alpha-subunit gene in mouse anterior pituitary cells by RNA interference. PLoS ONE 2013, 8, e74596. [Google Scholar]
- Calcaterra, V.; Cena, H.; Regalbuto, C.; Vinci, F.; Porri, D.; Verduci, E.; Chiara, M.; Zuccotti, G.V. The Role of Fetal, Infant, and Childhood Nutrition in the Timing of Sexual Maturation. Nutrients 2021, 13, 419. [Google Scholar] [CrossRef]
- Robertson, D.M.; Pruysers, E.; Jobling, T. Inhibin as a diagnostic marker for ovarian cancer. Cancer Lett. 2007, 249, 14–17. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Finegold, M.J.; Su, J.G.; Hsueh, A.J.; Bradley, A. Alpha-inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature 1992, 360, 313–319. [Google Scholar] [CrossRef]
- Rathore, R.; Arora, D.; Agarwal, S.; Sharma, S. Correlation of foxl2 with inhibin and calretinin in the diagnosis of ovarian sex cord stromal tumors. Turk. J. Pathol. 2017, 33, 121–128. [Google Scholar] [CrossRef]
- Doroszko, M.; Chrusciel, M.; Belling, K.; Vuorenoja, S.; Dalgaard, M.; Leffers, H.; Nielsen, H.B.; Huhtaniemi, I.; Toppari, J.; Rahman, N.A. Novel genes involved in pathophysiology of gonadotropin-dependent adrenal tumors in mice. Mol. Cell. Endocrinol. 2017, 444, 9–18. [Google Scholar] [CrossRef]
- Haverfield, J.T.; Stanton, P.G.; Loveland, K.L.; Zahid, H.; Nicholls, P.K.; Olcorn, J.S.; Makanji, Y.; Itman, C.M.; Simpson, E.R.; Meachem, S.J. Suppression of Sertoli cell tumour development during the first wave of spermatogenesis in inhibin α-deficient mice. Reprod. Fertil. Dev. 2017, 29, 609–620. [Google Scholar] [CrossRef]
- Hetzler, K.L.; Hardee, J.P.; LaVoie, H.A.; Murphy, E.A.; Carson, J.A. Ovarian function’s role during cancer cachexia progression in the female mouse. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E447–E459. [Google Scholar] [CrossRef]
- Nagaraja, A.K.; Agno, J.E.; Kumar, T.R.; Matzuk, M.M. Luteinizing hormone promotes gonadal tumorigenesis in inhibin-deficient mice. Mol. Cell. Endocrinol. 2008, 294, 19–28. [Google Scholar] [CrossRef]
- Kumar, T.R.; Palapattu, G.; Wang, P.; Woodruff, T.K.; Boime, I.; Byrne, M.C.; Matzuk, M.M. Transgenic models to study gonadotropin function: The role of follicle-stimulating hormone in gonadal growth and tumorigenesis. Mol. Endocrinol. 1999, 13, 851–865. [Google Scholar] [CrossRef]
- Chermuła, B.; Brązert, M.; Iżycki, D.; Ciesiółka, S.; Kranc, W.; Celichowski, P.; Ożegowska, K.; Nawrocki, M.J. New Gene Markers of Angiogenesis and Blood Vessels Development in Porcine Ovarian Granulosa Cells during Short-Term Primary Culture In Vitro. BioMed Res. Intl. 2019, 2019, 6545210. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Zhao, Y. scRNA-seq of ovarian follicle granulosa cells from different fertility goats reveals distinct expression patterns. Reprod. Domest. Anim. 2021, 56, 801–811. [Google Scholar] [CrossRef]
- Nia, A.M.; Chen, T.; Barnette, B.L.; Khanipov, K.; Ullrich, R.L.; Bhavnani, S.K.; Emmett, M.R. Efficient identification of multiple pathways: RNA-Seq analysis of livers from (56)Fe ion irradiated mice. BMC Bioinform. 2020, 21, 118. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Gouda, M.; Huang, Z.; Liu, Y.; He, Y.; Li, X. Physicochemical impact of bioactive terpenes on the microalgae biomass structural characteristics. Bioresour. Technol. 2021, 334, 125232. [Google Scholar] [CrossRef]
- Gouda, M.; Chen, K.; Li, X.; Liu, Y.; He, Y. Detection of microalgae single-cell antioxidant and electrochemical potentials by gold microelectrode and Raman micro-spectroscopy combined with chemometrics. Sens. Actuators B Chem. 2021, 329, 129229. [Google Scholar] [CrossRef]
- Shankar, V.; Gouda, M.; Moncivaiz, J.; Gordon, A.; Reo, N.V.; Hussein, L.; Paliy, O. Differences in Gut Metabolites and Microbial Composition and Functions between Egyptian and U.S. Children Are Consistent with Their Diets. mSystems 2017, 2, e00169-16. [Google Scholar] [CrossRef] [Green Version]
- FarmanUllah; Liang, X.; Khan, F.; Salim, M.; Rehman, Z.; Khan, M.; Talpur, H.; Schreurs, N.; Gouda, M.; Khan, S.; et al. Transcriptomic in silico analysis of bovine Escherichia coli mastitis highlights its immune-related expressed genes as an effective biomarker. J. Genet. Eng. Biotechnol. 2021, 19, 00290–00291. [Google Scholar] [CrossRef]
- Ahmed, F.E.; Gouda, M.M.; Hussein, L.A.; Ahmed, N.C.; Vos, P.W.; Mohammad, M.A. Role of Melt Curve Analysis in Interpretation of Nutrigenomics’ MicroRNA Expression Data. Cancer Genom. Proteom. 2017, 14, 469–481. [Google Scholar]
- Fong, M.Y.; Kakar, S.S. Ovarian cancer mouse models: A summary of current models and their limitations. J. Ovarian Res. 2009, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Kadariya, I.; Wang, J.; ur Rehman, Z.; Ali, H.; Riaz, H.; He, J.; Bhattarai, D.; Liu, J.J.; Zhang, S.J. RNAi-mediated knockdown of inhibin α subunit increased apoptosis in granulosa cells and decreased fertility in mice. J. Steroid Biochem. Mol. Biol. 2015, 152, 161–170. [Google Scholar] [CrossRef]
- Vasilache, A.M.; Kugelberg, U.; Blomqvist, A.; Nilsberth, C. Minor changes in gene expression in the mouse preoptic hypothalamic region by inflammation-induced prostaglandin E2. J. Neuroendocrinol. 2013, 25, 635–643. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, X.; Li, A.; Xie, L.; Miao, X. Genome-Wide Analysis of mRNAs and lncRNAs of Intramuscular Fat Related to Lipid Metabolism in Two Pig Breeds. Cell. Physiol. Biochem. 2018, 50, 2406–2422. [Google Scholar] [CrossRef]
- Chen, C.; Ahmad, M.J.; Ye, T.; Du, C.; Zhang, X.; Liang, A.; Yang, L. Cathepsin B Regulates Mice Granulosa Cells’ Apoptosis and Proliferation In Vitro. Int. J. Mol. Sci. 2021, 22, 11827. [Google Scholar] [CrossRef]
- Jing, R.; Gu, L.; Li, J.; Gong, Y. A transcriptomic comparison of theca and granulosa cells in chicken and cattle follicles reveals ESR2 as a potential regulator of CYP19A1 expression in the theca cells of chicken follicles. Comp. Biochem. Physiol. Part D Genom. Proteom. 2018, 27, 40–53. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, L.; Qin, Y.; Liu, S.; Qiao, Y.; Wan, X.; Zeng, H.; Tang, X.; Liu, M.; Hou, Y. Effects of differential distributed-JUP on the malignancy of gastric cancer. J. Adv. Res. 2021, 28, 195–208. [Google Scholar] [CrossRef]
- Peng, M.; Zhang, H.; Jaafar, L.; Risinger, J.I.; Huang, S.; Mivechi, N.F.; Ko, L. Human ovarian cancer stroma contains luteinized theca cells harboring tumor suppressor gene GT198 mutations. J. Biol. Chem. 2013, 288, 33387–33397. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. All in the CCN family: Essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 2006, 119, 4803–4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Xiao, L. Junction plakoglobin, a potential prognostic marker of oral squamous cell carcinoma, promotes proliferation, migration and invasion. J. Oral Pathol. Med. 2020, 49, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Zangen, D.; Kaufman, Y.; Zeligson, S.; Perlberg, S.; Fridman, H.; Kanaan, M.; Abdulhadi-Atwan, M.; Abu Libdeh, A.; Gussow, A.; Kisslov, I.; et al. XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes coactivation of estrogen-driven transcription. Am. J. Hum. Genet. 2011, 89, 572–579. [Google Scholar] [CrossRef]
- Yin, T.; Getsios, S.; Caldelari, R.; Kowalczyk, A.P.; Müller, E.J.; Jones, J.C.R.; Green, K.J. Plakoglobin suppresses keratinocyte motility through both cell–cell adhesion-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2005, 102, 5420–5425. [Google Scholar] [CrossRef]
- Hofland, J.; Steenbergen, J.; Voorsluijs, J.M.; Verbiest, M.M.; de Krijger, R.R.; Hofland, L.J.; de Herder, W.W.; Uitterlinden, A.G.; Feelders, R.A.; de Jong, F.H. Inhibin alpha-subunit (INHA) expression in adrenocortical cancer is linked to genetic and epigenetic INHA promoter variation. PLoS ONE 2014, 9, e104944. [Google Scholar] [CrossRef]
- Nagaraja, A.K.; Middlebrook, B.S.; Rajanahally, S.; Myers, M.; Li, Q.; Matzuk, M.M.; Pangas, S.A. Defective gonadotropin-dependent ovarian folliculogenesis and granulosa cell gene expression in inhibin-deficient mice. Endocrinology 2010, 151, 4994–5006. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, W.; Huang, R.; Miao, X.; Li, J.; Yu, D.; Li, Y.; Hsu, W.; Qiu, M.; Zhang, Z.; et al. The function of Wls in ovarian development. Mol. Cell. Endocrinol. 2021, 522, 111142. [Google Scholar] [CrossRef]
- Wang, L.; Cao, C.; Ma, Q.; Zeng, Q.; Wang, H.; Cheng, Z.; Zhu, G.; Qi, J.; Ma, H.; Nian, H.; et al. RNA-seq analyses of multiple meristems of soybean: Novel and alternative transcripts, evolutionary and functional implications. BMC Plant Biol. 2014, 14, 169. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Diao, C.; Xi, Y.; Xiao, T. Identification and analysis of key genes in osteosarcoma using bioinformatics. Oncol. Lett. 2018, 15, 2789–2794. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
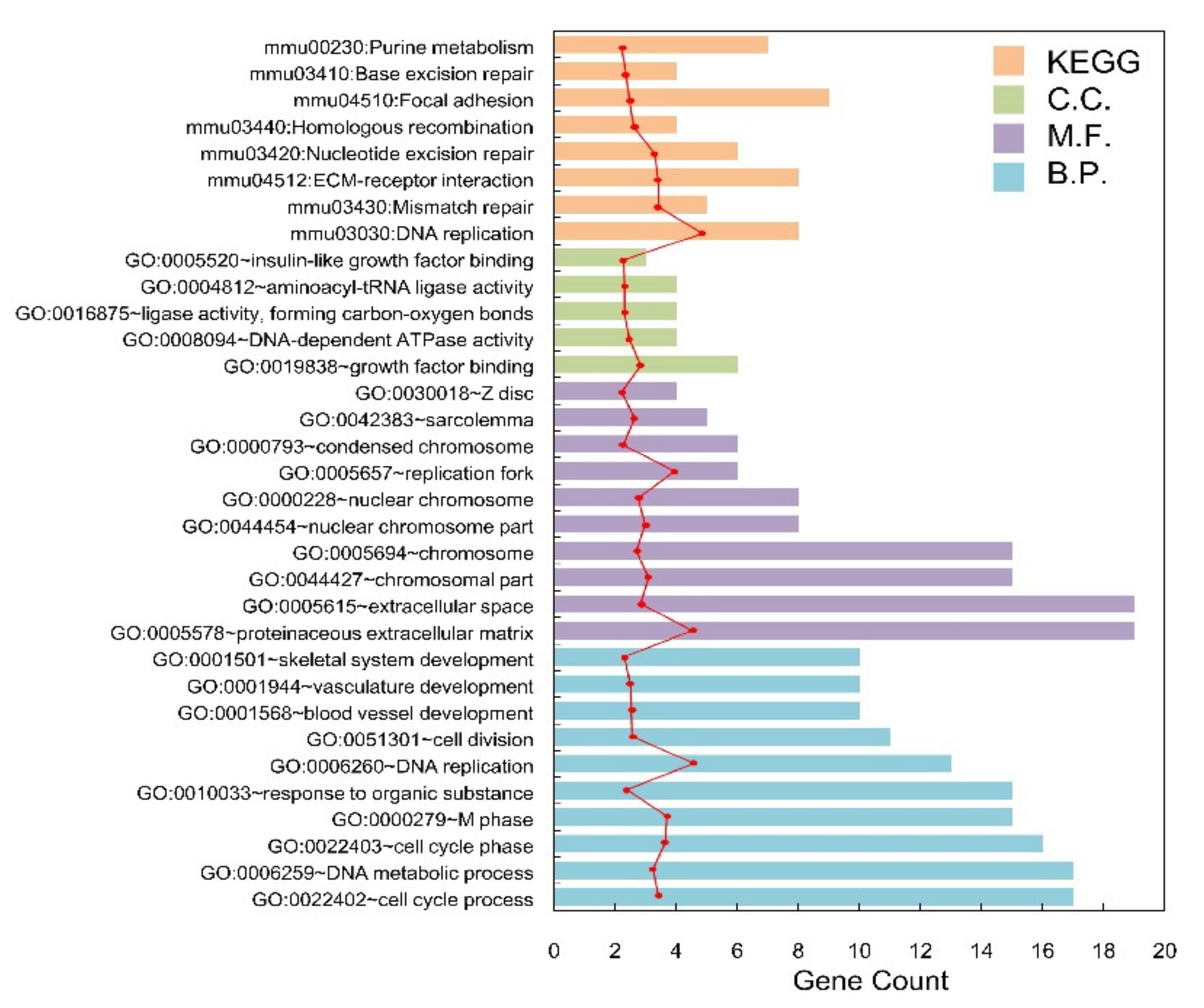
| Category | Term | Count | p-Value |
|---|---|---|---|
| Biology Process | GO:0022402~cell cycle process | 17 | 4.05 × 10−6 |
| GO:0006259~DNA metabolic process | 17 | 8.56 × 10−4 | |
| GO:0022403~cell cycle phase | 16 | 1.79 × 10−4 | |
| GO:0000279~M phase | 15 | 1.31 × 10−4 | |
| GO:0010033~response to organic substance | 15 | 2.53 × 10−2 | |
| GO:0006260~DNA replication | 13 | 4.14 × 10−6 | |
| GO:0051301~cell division | 11 | 1.21 × 10−2 | |
| GO:0001568~blood vessel development | 10 | 1.37 × 10−2 | |
| GO:0001944~vasculature development | 10 | 1.58 × 10−2 | |
| GO:0001501~skeletal system development | 10 | 3.33 × 10−2 | |
| Cellular Component | GO:0005578~proteinaceous extracellular matrix | 19 | 5.11 × 10−6 |
| GO:0005615~extracellular space | 19 | 4.03 × 10−3 | |
| GO:0044427~chromosomal part | 15 | 1.49 × 10−3 | |
| GO:0005694~chromosome | 15 | 7.06 × 10−3 | |
| GO:0044454~nuclear chromosome part | 8 | 2.25 × 10−3 | |
| GO:0000228~nuclear chromosome | 8 | 5.51 × 10−3 | |
| GO:0005657~replication fork | 6 | 5.21 × 10−5 | |
| GO:0000793~condensed chromosome | 6 | 4.05 × 10−2 | |
| GO:0042383~sarcolemma | 5 | 9.21 × 10−3 | |
| GO:0030018~Z disc | 4 | 4.69 × 10−2 | |
| Molecular Function | GO:0019838~growth factor binding | 6 | 4.09 × 10−3 |
| GO:0008094~DNA-dependent ATPase activity | 4 | 1.80 × 10−2 | |
| GO:0016875~ligase activity, forming carbon-oxygen bonds | 4 | 3.32 × 10−2 | |
| GO:0004812~aminoacyl-tRNA ligase activity | 4 | 3.32 × 10−2 | |
| GO:0005520~insulin-like growth factor binding | 3 | 4.08 × 10−2 | |
| KEGG Pathway | mmu03030:DNA replication | 8 | 1.35 × 10−6 |
| mmu03430:Mismatch repair | 5 | 4.27 × 10−4 | |
| mmu04512:ECM-receptor interaction | 8 | 4.34 × 10−4 | |
| mmu03420:Nucleotide excision repair | 6 | 6.87 × 10−4 | |
| mmu03440:Homologous recombination | 4 | 9.92 × 10−3 | |
| mmu04510:Focal adhesion | 9 | 1.73 × 10−3 | |
| mmu03410:Base excision repair | 4 | 2.87 × 10−2 | |
| mmu00230:Purine metabolism | 7 | 4.71 × 10−2 |
| Color | Gene Count | Correlation Coefficient (R2) |
|---|---|---|
| blue | 65 | 0.9218561 |
| green | 28 | 0.9203381 |
| brown | 60 | 0.8912573 |
| turquoise | 81 | 0.8894315 |
| yellow | 51 | 0.889348 |
| grey | 58 | 0.8815949 |
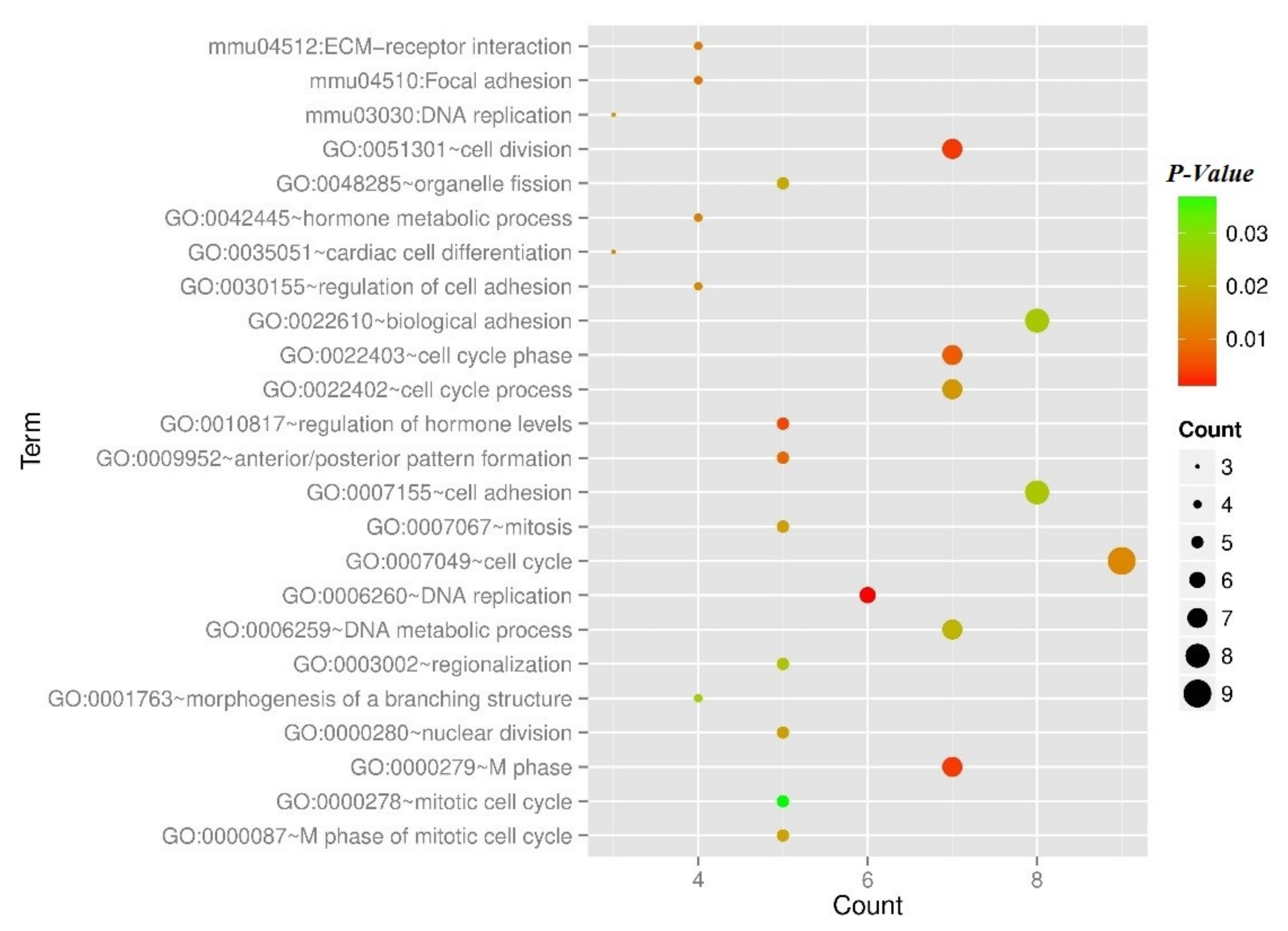
| Parameter | Term | Count | p-Value |
|---|---|---|---|
| Biology Process | GO:0007049~cell cycle | 9 | 0.012955 |
| GO:0007155~cell adhesion | 8 | 0.02503 | |
| GO:0022610~biological adhesion | 8 | 0.025243 | |
| GO:0051301~cell division | 7 | 0.003211 | |
| GO:0000279~M phase | 7 | 0.003325 | |
| GO:0022403~cell cycle phase | 7 | 0.00678 | |
| GO:0022402~cell cycle process | 7 | 0.015593 | |
| GO:0006259~DNA metabolic process | 7 | 0.021132 | |
| GO:0006260~DNA replication | 6 | 0.001106 | |
| GO:0010817~regulation of hormone levels | 5 | 0.004264 | |
| GO:0009952~anterior/posterior pattern formation | 5 | 0.007972 | |
| GO:0000280~nuclear division | 5 | 0.016588 | |
| GO:0007067~mitosis | 5 | 0.016588 | |
| GO:0000087~M phase of mitotic cell cycle | 5 | 0.017769 | |
| GO:0048285~organelle fission | 5 | 0.018688 | |
| GO:0003002~regionalization | 5 | 0.024455 | |
| GO:0000278~mitotic cell cycle | 5 | 0.03702 | |
| GO:0042445~hormone metabolic process | 4 | 0.011174 | |
| GO:0030155~regulation of cell adhesion | 4 | 0.012565 | |
| GO:0001763~morphogenesis of a branching structure | 4 | 0.026619 | |
| GO:0035051~cardiac cell differentiation | 3 | 0.012461 | |
| KEGG pathway | mmu04512:ECM-receptor interaction | 4 | 0.010701 |
| mmu04510:Focal adhesion | 4 | 0.009668 | |
| mmu03030:DNA replication | 3 | 0.015987 |
| miRNA | ID | p-Value | FDR |
|---|---|---|---|
| mmu_TGCCTTA,MIR-124A | DB_ID:590 | 9.65 × 10−3 | 0.0014 |
| mmu_GTGACTT,MIR-224 | DB_ID:524 | 0.0002 | 0.0014 |
| mmu_CTCTGGA,MIR-520A | DB_ID:484 | 0.0036 | 0.0126 |
| mmu_ACCAAAG,MIR-9 | DB_ID:588 | 0.0029 | 0.0126 |
| mmu_ACTGAAA,MIR-30A | DB_ID:464 | 0.0065 | 0.0182 |
| mmu_CTGAGCC,MIR-24 | DB_ID:539 | 0.0107 | 0.0194 |
| mmu_AACTGGA,MIR-145 | DB_ID:614 | 0.0101 | 0.0194 |
| mmu_AAGCACT,MIR-520F | DB_ID:615 | 0.0111 | 0.0194 |
| TF | ID | p-Value | FDR |
|---|---|---|---|
| PAX4 | DB_ID:1830 | 8.59 × 10−6 | 2.58 × 10−5 |
| MAZ | DB_ID:1815 | 7.72 × 10−6 | 2.58 × 10−5 |
| MYC | DB_ID:1819 | 4.99 × 10−6 | 2.58 × 10−5 |
| NFAT | DB_ID:1822 | 1.40 × 10−5 | 3.15 × 10−5 |
| FOXO4 | DB_ID:1801 | 3.72 × 10−5 | 6.70 × 10−5 |
| SP1 | DB_ID:1837 | 2.00 × 10−4 | 3.00 × 10−4 |
| LEF1 | DB_ID:1813 | 4.00 × 10−4 | 4.00 × 10−4 |
| Gene | Contribution to PC1-3 |
|---|---|
| Fndc5 | 0.97 |
| Sertad4 | 0.97 |
| Atp1b3 | 0.96 |
| Fam57a | 0.95 |
| P4htm | 0.89 |
| Hoxd10 | 0.85 |
| Psmc3ip | 0.85 |
| Rab11a | −0.85 |
| Ypel5 | −0.9 |
| Emx2 | −0.91 |
| Jup | −0.92 |
| Gpc4 | −0.93 |
| Slc25a33 | −0.94 |
| Smarca1 | −0.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talpur, H.S.; Rehman, Z.u.; Gouda, M.; Liang, A.; Bano, I.; Hussain, M.S.; FarmanUllah, F.; Yang, L. Molecular Genomic Study of Inhibin Molecule Production through Granulosa Cell Gene Expression in Inhibin-Deficient Mice. Molecules 2022, 27, 5595. https://doi.org/10.3390/molecules27175595
Talpur HS, Rehman Zu, Gouda M, Liang A, Bano I, Hussain MS, FarmanUllah F, Yang L. Molecular Genomic Study of Inhibin Molecule Production through Granulosa Cell Gene Expression in Inhibin-Deficient Mice. Molecules. 2022; 27(17):5595. https://doi.org/10.3390/molecules27175595
Chicago/Turabian StyleTalpur, Hira Sajjad, Zia ur Rehman, Mostafa Gouda, Aixing Liang, Iqra Bano, Mir Sajjad Hussain, FarmanUllah FarmanUllah, and Liguo Yang. 2022. "Molecular Genomic Study of Inhibin Molecule Production through Granulosa Cell Gene Expression in Inhibin-Deficient Mice" Molecules 27, no. 17: 5595. https://doi.org/10.3390/molecules27175595
APA StyleTalpur, H. S., Rehman, Z. u., Gouda, M., Liang, A., Bano, I., Hussain, M. S., FarmanUllah, F., & Yang, L. (2022). Molecular Genomic Study of Inhibin Molecule Production through Granulosa Cell Gene Expression in Inhibin-Deficient Mice. Molecules, 27(17), 5595. https://doi.org/10.3390/molecules27175595