

Copper-catalyzed S-arylation of Furanose-Fused Oxazolidine-2-thiones

 , , ,
, , ,
Abstract
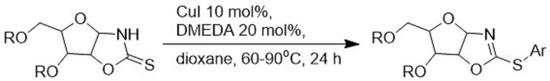
:
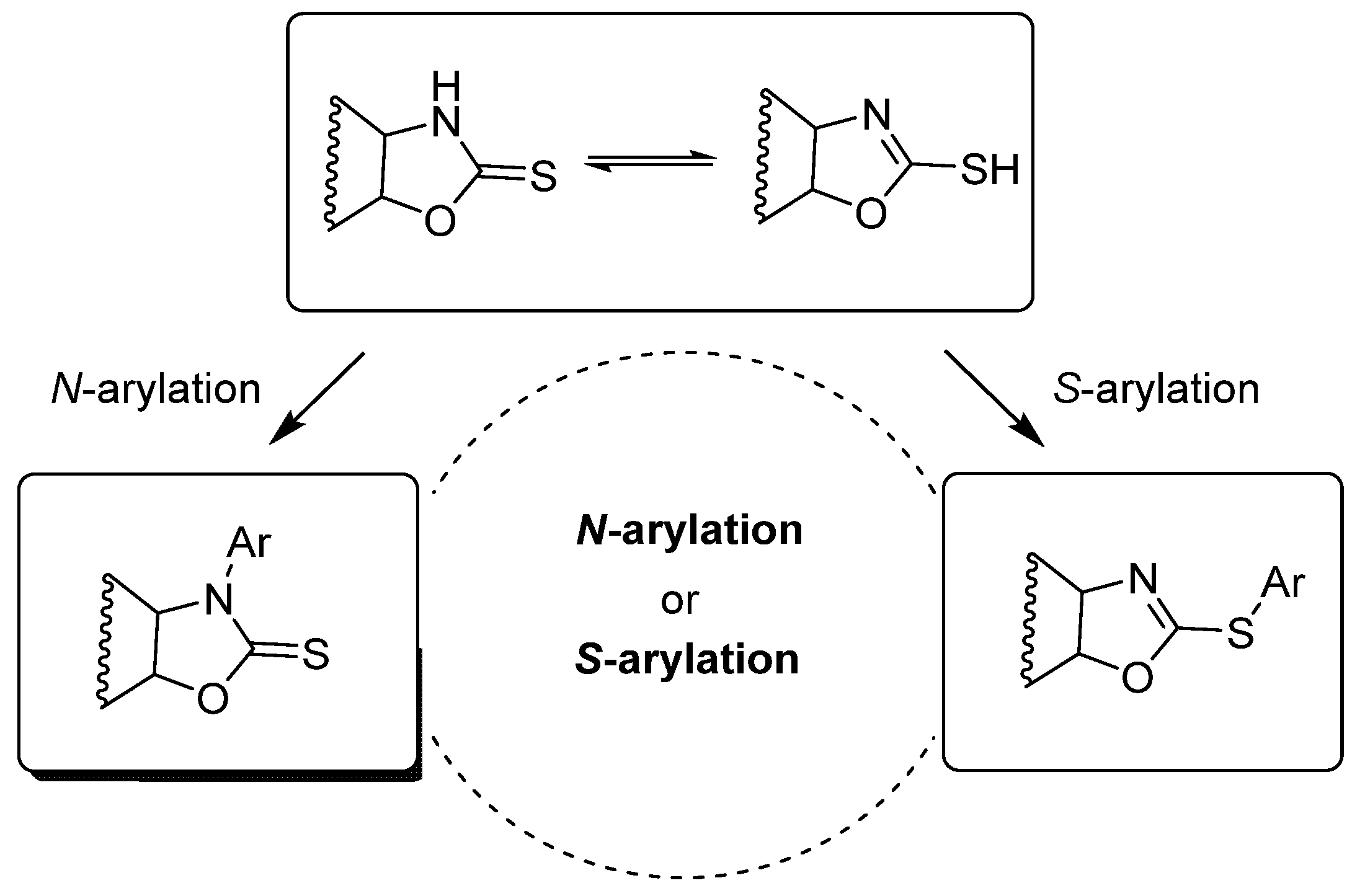
1. Introduction
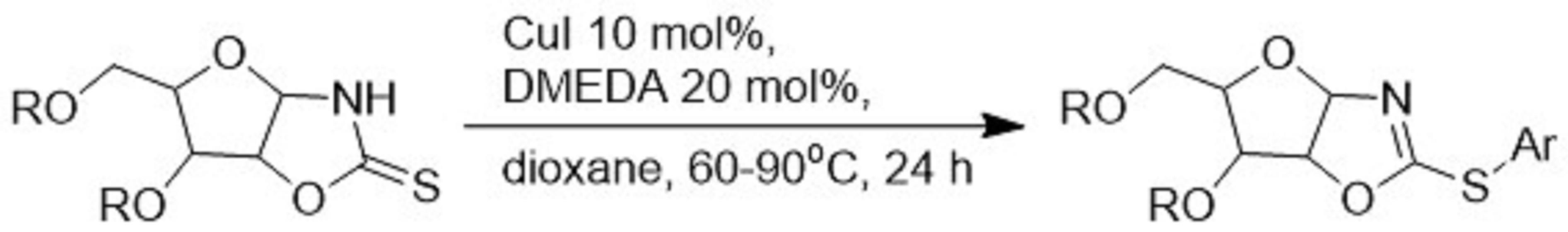
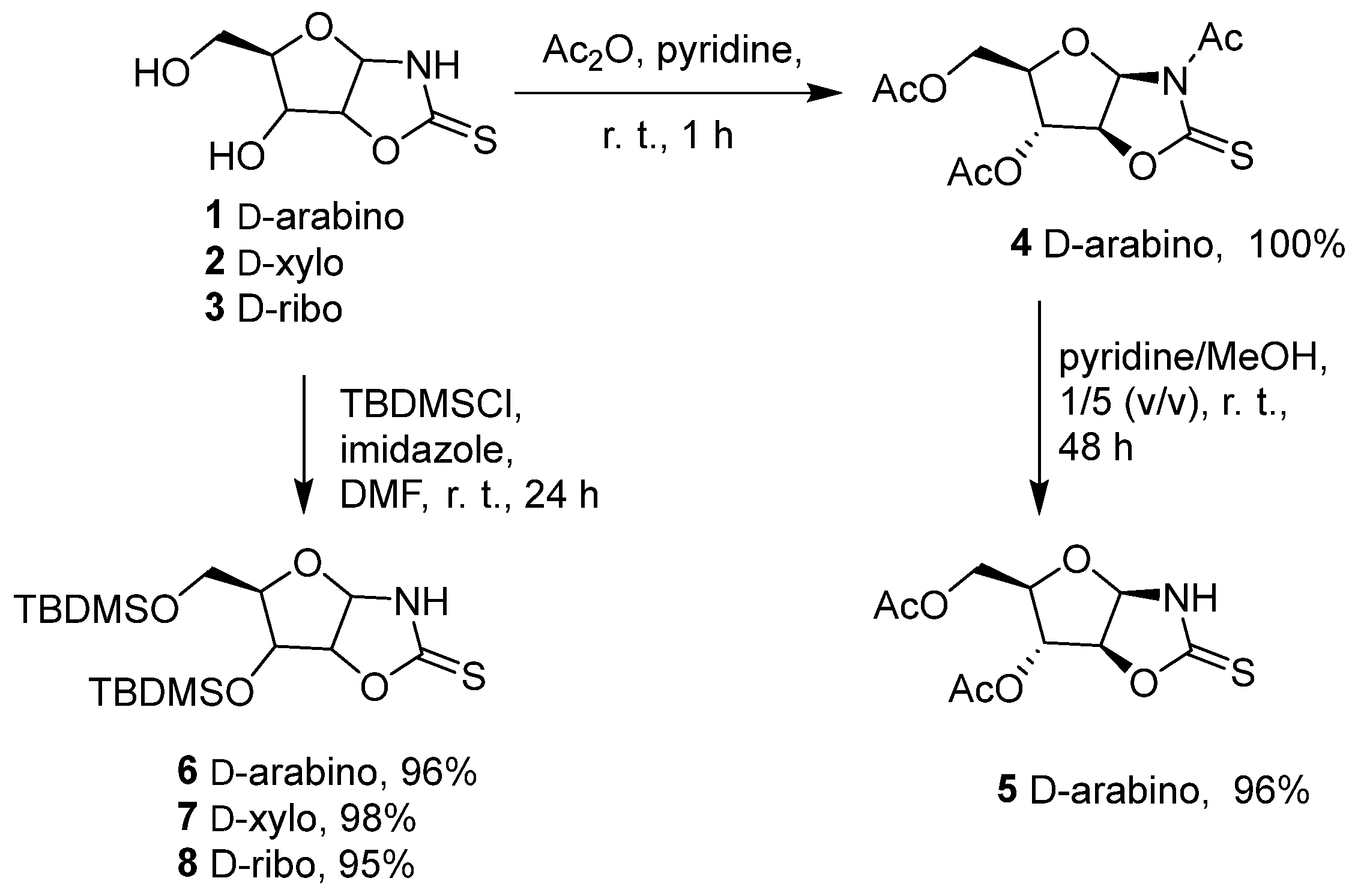
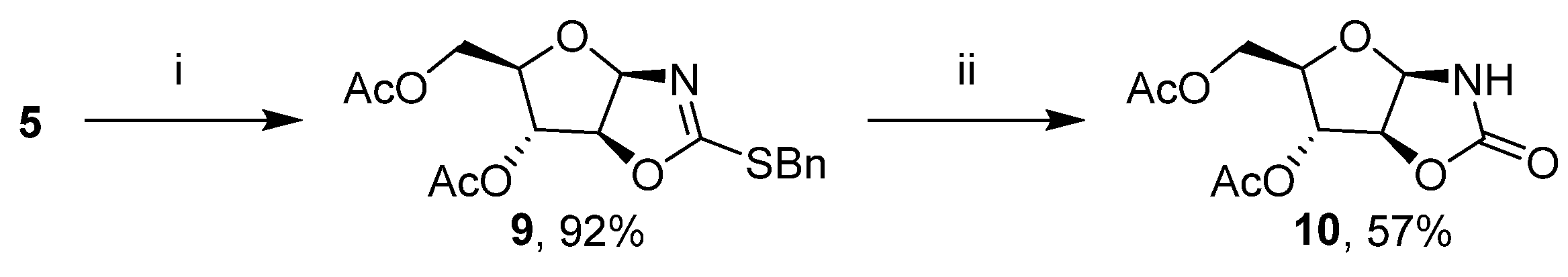
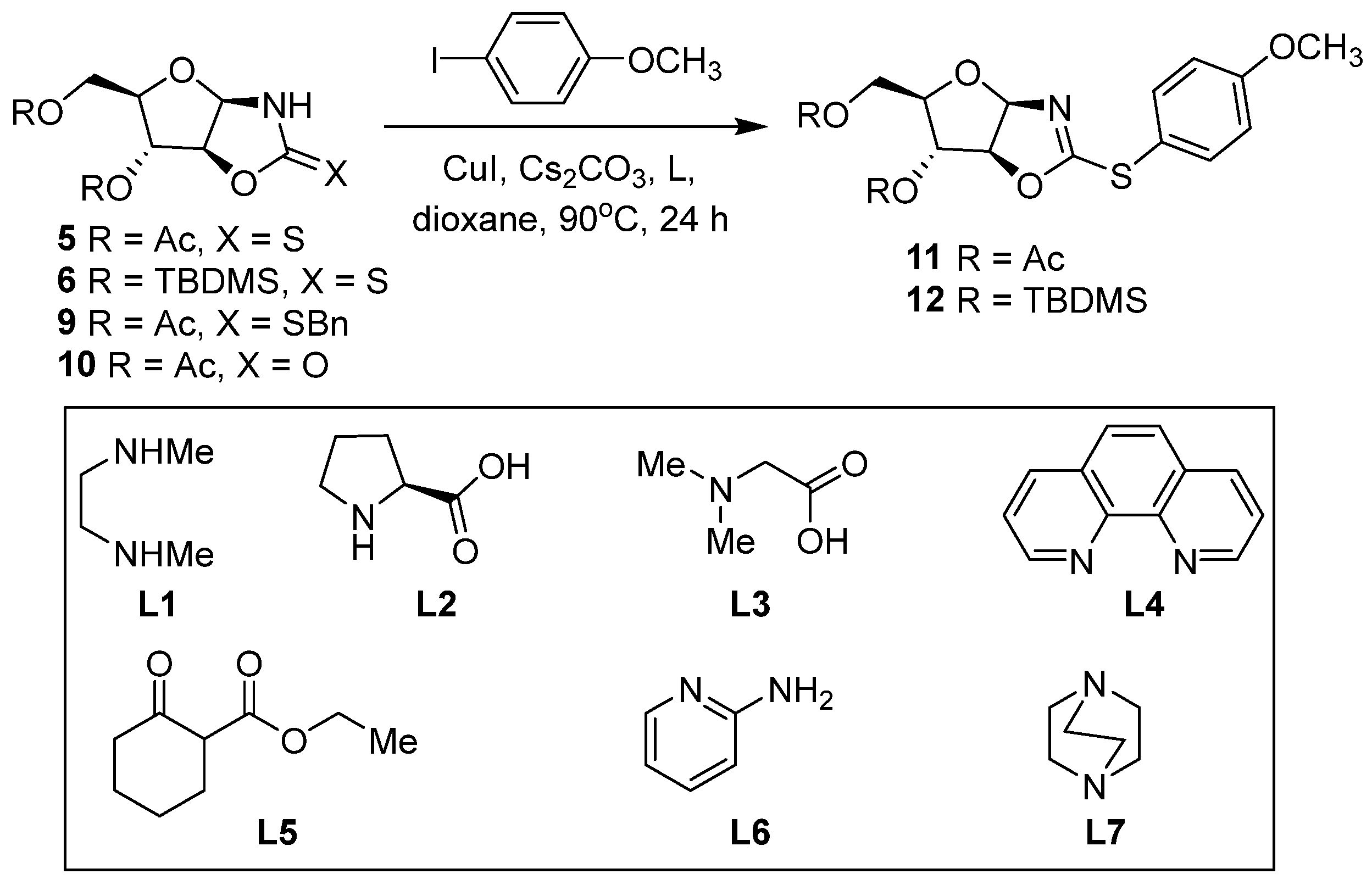
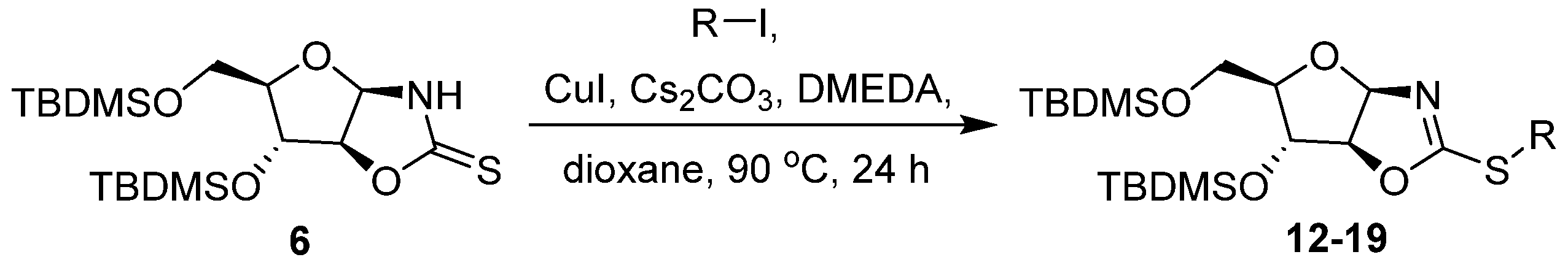
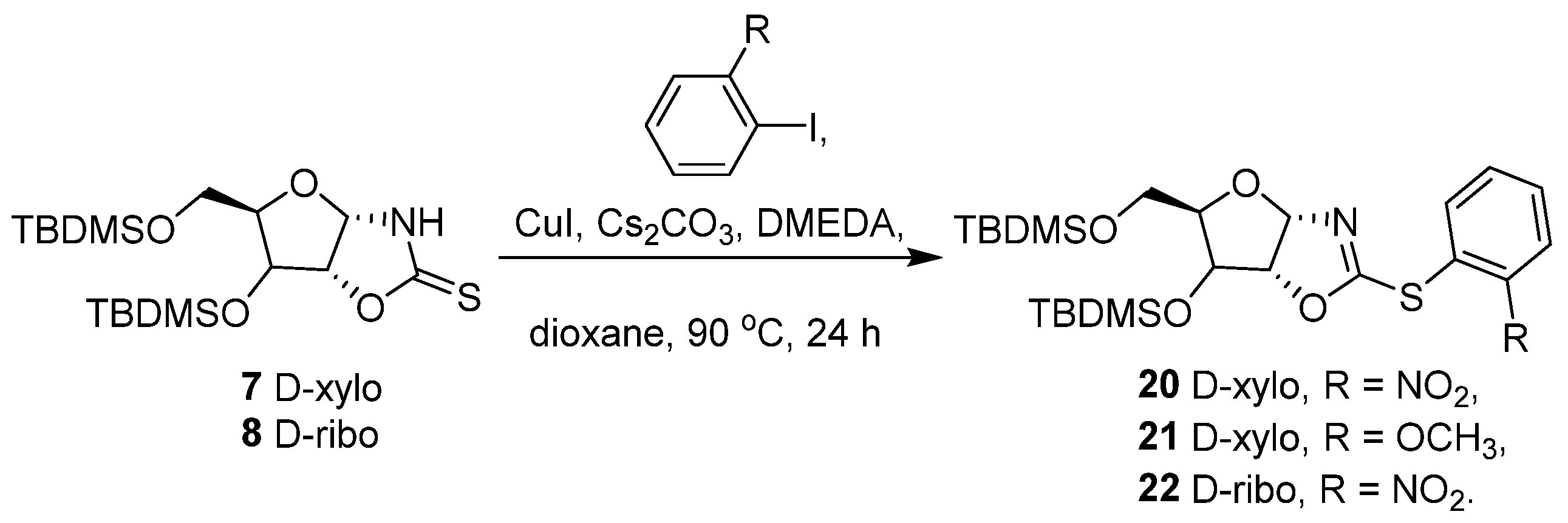
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of N-acetyl-4,5-dihydro(3′,5′-di-O-acetyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazolidine-2-thione (4)
3.3. Synthesis of 4,5-dihydro(3′,5′-di-O-acetyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazolidine-2-thione (5)
3.4. Synthesis of 2-benzylsulfanyl-4,5-dihydro(3′,5′-di-O-acetyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazole (9)
3.5. Synthesis of 4,5-dihydro(3′,5′-di-O-acetyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazolidine-2-one (10)
3.6. Synthesis of 4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazolidine-2-thione (6)
3.7. Synthesis of 4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-α-D-xylofuranoso)-[1,2-d]-oxazolidine-2-thione (7)
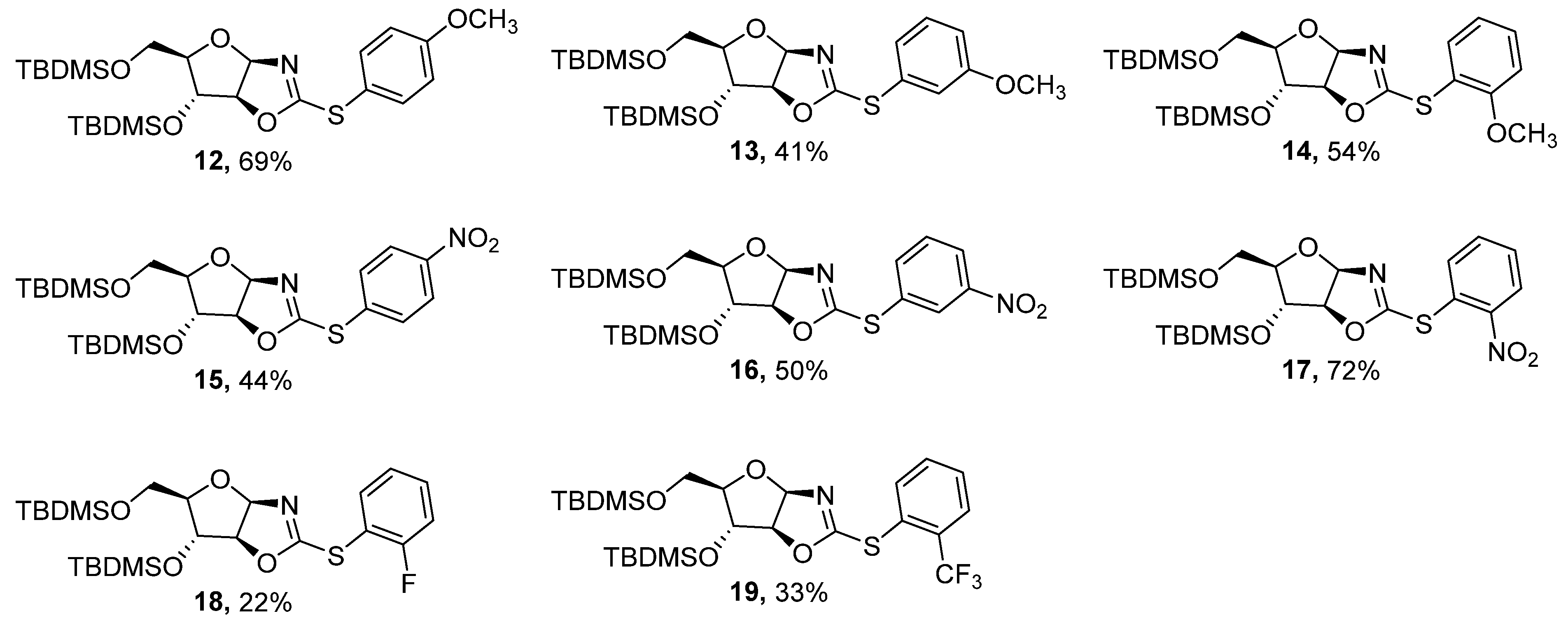
3.8. General Procedure: Copper-catalyzed S-arylation of Compounds 11–21
3.8.1. 2-[(4-Methoxyphenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-acetyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazole (11)
3.8.2. 2-[(4-Methoxyphenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazole (12)
3.8.3. 2-[(3-Methoxyphenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazole (13)
3.8.4. 2-[(2-Methoxyphenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazole (14)
3.8.5. 2-[(4-Nitrophenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazole (15)
3.8.6. 2-[(3-Nitrophenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazole (16)
3.8.7. 2-[(2-Nitrophenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazole (17)
3.8.8. 2-[(2-Fluorophenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-di-deoxy-β-D-arabinofuranoso)-[1,2-d]-oxazole (18)
3.8.9. 2-[(2-Trifluoromethylphenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethyl-silyl-1′,2′-dideoxy-β-D-arabinofuranoso)-[1,2-d]-oxazole (19)
3.8.10. 2-[(2-Nitrophenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-α-D-xylofuranoso)-[1,2-d]-oxazole (20)
3.8.11. 2-[(2-Methoxyphenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-α-D-xylofuranoso)-[1,2-d]-oxazole (21)
3.8.12. 2-[(2-Nitrophenyl)sulfanyl]-4,5-dihydro(3′,5′-di-O-tert-butyldimethylsilyl-1′,2′-dideoxy-β-D-ribofuranoso)-[1,2-d]-oxazole (22)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Awan, S.I.; Werz, D.B. Syntheses of 1,2-annulated and 1-spiroannulated carbohydrate derivatives: Recent developments. Bioorg. Med. Chem. 2012, 20, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Girniene, J.; Tatibouet, A.; Sackus, A.; Yang, J.; Holman, G.D.; Rollin, P. Inhibition of the D-Fructose Transporter Protein GLUT5 by Fused-Ring Glyco-1,3-Oxazolidin-2-Thiones and -Oxazolidin-2-Ones. Carbohydr. Res. 2003, 338, 711–719. [Google Scholar] [CrossRef]
- Igual, M.O.; Nunes, P.S.G.; da Costa, R.M.; Mantoani, S.P.; Tostes, R.C.; Carvalho, I. Novel glucopyranoside C2-derived 1,2,3-triazoles displaying selective inhibition of O-GlcNAcase (OGA). Carbohydr. Res. 2019, 471, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Velueta-Viveros, M.; Martínez-Bailén, M.; Puerta, A.; Romero-Hernández, L.L.; Křen, V.; Merino-Montiel, P.; Montiel-Smith, S.; Fernandes, M.X.; Moreno-Vargas, A.J.; Padrón, J.M.; et al. Carbohydrate-derived bicyclic selenazolines as new dual inhibitors (cholinesterases/OGA) against Alzheimer’s disease. Bioorganic Chem. 2022, 127, 105983–105990. [Google Scholar] [CrossRef] [PubMed]
- Chennaiah, A.; Dubbu, S.; Parasuraman, K.; Vankar, Y.D. Stereoselective Synthesis of 1,2-Annulated Sugars Having Substituted Tetrahydropyran/(-furan) Scaffolds Using the PrinsReaction. Eur. J. Org. Chem. 2018, 2018, 6706–6713. [Google Scholar] [CrossRef]
- Verma, A.K.; Chennaiah, A.; Dubbu, S.; Vankar, Y.D. Palladium catalyzed synthesis of sugar-fused indolines via C(sp2)-H/N-H activation. Carbohydr. Res. 2019, 473, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Leibeling, M.; Milde, B.; Kratzert, D.; Stalke, D.; Werz, D.B. Intermolecular Twofold Carbopalladation/Cyclization Sequence to Access Chromans and Isochromans from Carbohydrates. Chem.—A Eur. J. 2011, 17, 9888–9892. [Google Scholar] [CrossRef]
- Xiong, W.; Zhang, S.; Li, H.; Zhang, Z.; Xu, T. Pd-Catalyzed Decarboxylative Cycloaddition of Vinylethylene Carbonates with Isothiocyanates. J. Org. Chem. 2020, 85, 8773–8779. [Google Scholar] [CrossRef]
- Vessally, E.; Mohammadi, R.; Hosseinian, A.; Didehban, K.; Edjlali, L. S-arylation of 2-mercaptobenzazoles: A comprehensive review. J. Sulfur Chem. 2018, 39, 443–463. [Google Scholar] [CrossRef]
- Crimmins, M.T.; King, B.W.; Tabet, E.A. Pav. Asymmetric Aldol Additions with Titanium Enolates of Acyloxazolidinethiones: Dependence of Selectivity on Amine Base and Lewis Acid Stoichiometry. J. Am. Chem. Soc. 1997, 119, 7883–7884. [Google Scholar] [CrossRef]
- Crimmins, M.T.; King, B.W.; Tabet, E.A.; Chaudhary, K. Asymmetric Aldol Additions: Use of Titanium Tetrachloride and (−)-Sparteine for the Soft Enolization of N-Acyl Oxazolidinones, Oxazolidinethiones, and Thiazolidinethione. J. Org. Chem. 2001, 66, 894–902. [Google Scholar] [CrossRef]
- Crimmins, M.T.; McDougall, P.J. Anti-Selective Aldol Reactions with Titanium Enolates of N-Glycolyloxazolidinethiones. Org. Lett. 2003, 5, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Carter, R.G.; Yokochi, A.F.T. Unified Synthesis of C19-C26 Subunits of Amphidinolides B1, B2, and B3 by Exploiting Unexpected Stereochemical Differences in Crimmins’ and Evans’ Aldol Reactions. J. Org. Chem. 2004, 69, 2569–2572. [Google Scholar] [CrossRef] [PubMed]
- Crimmins, M.T.; Shamszad, M. Highly Selective Acetate Aldol Additions Using Mesityl-Substituted Chiral Auxiliaries. Org. Lett. 2007, 9, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Agerbirk, N.; Olsen, C.E. Glucosinolate Structures in Evolution. Phytochemistry 2012, 77, 16–45. [Google Scholar] [CrossRef]
- Rollin, P.; Tatibouët, A. Glucosinolates: The Synthetic Approach. C. R. Chim. 2011, 14, 194–210. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Mishra, A. Prospects and Problems of Dietary Glucosinolates in Animal Feeding. Adv. Dairy Res. 2017, 5, 1000180. [Google Scholar] [CrossRef]
- Rollin, P.; Tatibouet, A. Sulfur Metabolites in Brassicales: From Daily Vegetables to Thiofunctional Chemistry. Phosphorus Sulfur Silicon Relat. Elem. 2011, 186, 1130–1136. [Google Scholar] [CrossRef]
- Xie, Z.; Shi, Y.; Wang, Z.; Wang, R.; Li, Y. Biotransformation of Glucosinolates Epiprogoitrin and Progoitrin to (R)- and (S)-Goitrin in Radix Isatidis. J. Agric. Food Chem. 2011, 59, 12467–12472. [Google Scholar] [CrossRef]
- Ishikawa, S.; Maruyama, A.; Yamamoto, Y.; Hara, S. Extraction and Characterization of Glucosinolates and Isothiocyanates from Rape Seed Meal. J. Oleo Sci. 2014, 63, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Agerbirk, N.; Olsen, C.E.; Cipollini, D.; Orgaard, M.; Linde-Laursen, I.; Chew, F.S. Specific Glucosinolate Analysis Reveals Variable Levels of Epimeric Glucobarbarins, Dietary Precursors of 5-Phenyloxazolidine-2-Thiones, in Watercress Types with Contrasting Chromosome Numbers. J. Agric. Food Chem. 2014, 62, 9586–9596. [Google Scholar] [CrossRef]
- Seo, B.; Yun, J.; Lee, S.; Kim, M.; Hwang, K.; Kim, J.; Min, K.R.; Kim, Y.; Moon, D. Barbarin as a New Tyrosinase Inhibitor from Barbarea Orthocerus. Planta Med. 1999, 65, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Mithofer, A.; Boland, W. Plant Defense against Herbivores: Chemical Aspects. Annu Rev. Plant. Biol 2012, 63, 431–450. [Google Scholar] [CrossRef] [PubMed]
- Mishra, K.B.; Agrahari, A.K.; Tiwari, V.K. One-pot synthesis of oxazolidine-2-thione and thiazolidine-2-thione from sugar azido-alcohols. Carbohydr. Res. 2017, 450, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Radulović, N.S.; Todorovska, M.M.; Zlatković, D.B.; Stojanović, N.M.; Randjelović, P.J. Two goitrogenic 1,3-oxazolidine-2-thione derivatives from Brassicales taxa: Challenging identification, occurrence and immunomodulatory effects. Food Chem. Toxicol. 2017, 110, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Sansinenea, E. The synthetic versatility of oxazolidinethiones. J. Sulfur Chem. 2007, 28, 109–147. [Google Scholar] [CrossRef]
- Li, G.; Qian, X.; Cui, J.; Huang, Q.; Zhang, R.; Guan, H. Synthesis and Herbicidal Activity of Novel 3-Aminocarbonyl-2-oxazolidinethione Derivatives Containing a Substituted Pyridine Ring. J. Agric. Food Chem. 2006, 54, 125–129. [Google Scholar] [CrossRef]
- Morales-Nava, R.; Fernandez-Zertuche, M.; Ordonez, M. Microwave-Assisted Improved Synthesis of Oxazolidin-2-Ones, Oxazolidine-2-Thiones and Thiazolidine-2-Thione Chiral Auxiliaries. Molecules 2011, 16, 8803–8814. [Google Scholar] [CrossRef]
- Cano, I.; Gomez-Bengoa, E.; Landa, A.; Maestro, M.; Mielgo, A.; Olaizola, I.; Oiarbide, M.; Palomo, C. N-(Diazoacetyl)oxazolidin-2-thiones as Sulfur-Donor Reagents: Asymmetric Synthesis of Thiiranes from Aldehydes. Angew. Chem. Int. Ed. 2012, 51, 10856–10860. [Google Scholar] [CrossRef]
- Han, Y.-Y.; Chen, W.-B.; Han, W.-Y.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. Highly Efficient and Stereoselective Construction of Dispiro-[Oxazolidine-2-Thione]Bisoxindoles and Dispiro[Imidazolidine-2-Thione]Bisoxindoles. Org. Lett. 2012, 14, 490–493. [Google Scholar] [CrossRef]
- Munive, L.; Rivas, V.M.; Ortiz, A.; Olivo, H.F. Oxazolidine-2-Thiones and Thiazolidine-2-Thiones as Nucleophiles in Intermolecular Michael Additions. Org. Lett. 2012, 14, 3514–3517. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Qiao, C. Synthesis of Epigoitrin from (R)-(+)-4-Hydroxy-γ-Butyrolactone. J. Heterocycl. Chem. 2013, 50, 1290–1293. [Google Scholar] [CrossRef]
- Saygili, N.; Ozalp, M.; Yildirim, L.T. Synthesis, X-Ray Analysis, and Biological Activities of Novel Oxazolidinethiones. J. Heterocycl. Chem. 2014, 51, 1264–1269. [Google Scholar] [CrossRef]
- Simao, A.C.; Rousseau, J.; Silva, S.; Rauter, A.P.; Tatibouet, A.; Rollin, P. Thionocarbamates on Carbohydrate Scaffolds–from Synthesis to Bioactivity. Carbohydr. Chem. 2009, 35, 127–172. [Google Scholar] [CrossRef]
- Leconte, N.; Pellegatti, L.; Tatibouet, A.; Suzenet, F.; Rollin, P.; Guillaumet, G. Benzylsulfanyloxazolines in Palladium-Catalyzed Cross-Coupling Reactions: A Novel Approach to Chiral Oxazolines. Synthesis 2007, 857–864. [Google Scholar] [CrossRef]
- Silva, S.; Sylla, B.; Suzenet, F.; Tatibouet, A.; Rauter, A.P.; Rollin, P. Oxazolinethiones and Oxazolidinethiones for the First Copper-Catalyzed Desulfurative Cross-Coupling Reaction and First Sonogashira Applications. Org. Lett. 2008, 10, 853–856. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Tardy, S.; Routier, S.; Suzenet, F.; Tatibouet, A.; Rauter, A.P.; Rollin, P. 1,3-Oxazoline- and 1,3-Oxazolidine-2-Thiones as Substrates in Direct Modified Stille and Suzuki Cross-Coupling. Tetrahedron Lett. 2008, 49, 5583–5586. [Google Scholar] [CrossRef]
- Domingues, M.; Jaszczyk, J.; Ismael, M.I.; Figueiredo, J.A.; Daniellou, R.; Lafite, P.; Schuler, M.; Tatibouet, A. Conformationally Restricted Oxazolidin-2-One Fused Bicyclic Iminosugars as Potential Glycosidase Inhibitors. Eur. J. Org. Chem. 2020, 2020, 6109–6126. [Google Scholar] [CrossRef]
- Shen, C.; Xia, H.; Yan, H.; Chen, X.; Ranjit, S.; Xie, X.; Tan, D.; Lee, R.; Yang, Y.; Xing, B.; et al. Concise, Efficient Synthesis of Sugar-Based Benzothiazoles through Chemoselective Intramolecular C-S Coupling. Chem. Sci. 2012, 3, 2388–2393. [Google Scholar] [CrossRef]
- Brachet, E.; Brion, J.-D.; Alami, M.; Messaoudi, S. Nickel-Catalyzed Arylation, Alkenylation, and Alkynylation of Unprotected Thioglycosides at Room Temperature. ChemEur. J. 2013, 19, 15276–15280. [Google Scholar] [CrossRef]
- Brachet, E.; Brion, J.-D.; Alami, M.; Messaoudi, S. Stereoselective Palladium-Catalyzed Alkenylation and Alkynylation of Thioglycosides. Adv. Synth. Catal. 2013, 355, 2627–2636. [Google Scholar] [CrossRef]
- Soeta, T.; Matsumoto, A.; Sakata, Y.; Ukaji, Y. Development of a One-Pot Synthetic Method for Multifunctional Oxazole Derivatives Using Isocyanide Dichloride. J. Org. Chem. 2017, 82, 4930–4935. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-H.; Lu, Y.; Zhang, Q.; Lu, R.; Bao, L.-Q.; Shen, M.-H.; Xu, H.-D. Selective S-Arylation of 2-Oxazolidinethiones and Selective N-Arylation of 2-Benzoxazolinones/2-Benzimidazolinones. Org. Biomol. Chem. 2017, 15, 4058–4063. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Kriščiūnienė, V.; Matulevičiūtė, G.; Paliulis, O.; Rollin, P.; Šačkus, A. Conversion of 2-thioxo-2,3-dihydroquinazolin-4(1H)-ones to N(3)-unsubstituted 2-(het)arylquinazolin-4(3H)-ones by copper-mediated Pd-catalysed cross-coupling reactions. Heterocycles 2016, 93, 150–163. [Google Scholar] [CrossRef]
- Jha, M.; Enaohwo, O.; Guy, S. Yttrium Triflate-Catalyzed Efficient Chemoselective S-Benzylation of Indoline-2-Thiones Using Benzyl Alcohols. Tetrahedron Lett. 2011, 52, 684–687. [Google Scholar] [CrossRef]
- Gosselin, G.; Bergogne, M.C.; De Rudder, J.; De Clercq, E.; Imbach, J.L. Systematic Synthesis and Biological Evaluation of α- and β-D-Xylofuranosyl Nucleosides of the Five Naturally Occurring Bases in Nucleic Acids and Related Analogs. J. Med. Chem. 1986, 29, 203–213. [Google Scholar] [CrossRef]
- Girniene, J.; Gueyrard, D.; Tatibouet, A.; Sackus, A.; Rollin, P. Base-Modified Nucleosides from Carbohydrate Derived Oxazolidinethiones: A Five-Step Process. Tetrahedron Lett. 2001, 42, 2977–2980. [Google Scholar] [CrossRef]
- Tatibouet, A.; Lawrence, S.; Rollin, P.; Holman, G.D. Selective Formation of 1,3-Oxazolidine-2-Thiones on Ketohexose Templates. Synlett 2004, 11, 1945–1948. [Google Scholar] [CrossRef]
- Girniene, J.; Apremont, G.; Tatibouet, A.; Sackus, A.; Rollin, P. Small Libraries of Fused Quinazolinone-Sugars. Access to Quinazolinedione Nucleosides. Tetrahedron 2004, 60, 2609–2619. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, W.; Ma, D. L -Proline-Promoted CuI-Catalyzed C-S Bond Formation between Aryl Iodides and Thiols. Synthetic Commun. 2007, 37, 25–35. [Google Scholar] [CrossRef]
- Kwong, F.Y.; Klapars, A.; Buchwald, S.L. Copper-Catalyzed Coupling of Alkylamines and Aryl Iodides: An Efficient System Even in an Air Atmosphere. Org. Lett. 2002, 4, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Kederienė, V.; Jaglinskaitė, I.; Voznikaitė, P.; Rousseau, J.; Rollin, P.; Šačkus, A.; Tatibouët, A. Mild copper-catalyzed, l-proline-promoted cross-coupling of methyl 3-amino-1-benzothiophene-2-carboxylate. Molecules 2021, 26, 6822. [Google Scholar] [CrossRef] [PubMed]
- Surry, D.S.; Buchwald, S.L. Diamine Ligands in Copper-Catalyzed Reactions. Chem. Sci. 2010, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Klapars, A.; Huang, X.; Buchwald, S.L. A General and Efficient Copper Catalyst for the Amidation of Aryl Halides. J. Am. Chem. Soc. 2002, 124, 7421–7428. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.-F.; Correa, A.; Carril, M.; Norrby, P.-O.; Bolm, C. Copper-Catalyzed Cross-Couplings with Part-per-Million Catalyst Loadings. Angew. Chem. Int. Ed. 2009, 48, 5691–5693. [Google Scholar] [CrossRef]
- Larsson, P.-F.; Bolm, C.; Norrby, P.-O. Kinetic Investigation of a Ligand-Accelerated Sub-Mol % Copper-Catalyzed C-N Cross-Coupling Reaction. Chem. Eur. J. 2010, 16, 13613–13616. [Google Scholar] [CrossRef]
- Sekar, R.; Srinivasan, M.; Marcelis, A.T.M.; Sambandam, A. S-arylation of mercaptobenzimidazoles using Cu(I) catalysts—experimental and theoretical observations. Tet. Lett. 2011, 52, 3347–3352. [Google Scholar] [CrossRef]
- Strieter, E.R.; Bhayana, B.; Buchwald, S.L. Mechanistic studies on the Copper-catalyzed N-arylation of amides. JACS. 2009, 131, 78–88. [Google Scholar] [CrossRef]
- Jones, G.O.; Liu, P.; Houk, K.N.; Buchwald, S.L. Computational Explorations of Mechanisms and Ligand-Directed Selectivities of Copper-Catalyzed Ullmann-Type Reactions. J. Am. Chem. Soc. 2010, 132, 6205–6213. [Google Scholar] [CrossRef]
- Tye, J.W.; Weng, Z.; Giri, R.; Hartwig, J.F. Copper(I) phenoxide complexes in the etherification of aryl halides. Angew. Chem. Int. Ed. 2010, 49, 2185–2189. [Google Scholar] [CrossRef] [PubMed]
- Tye, J.W.; Weng, Z.; Jons, A.M.; Incarvito, C.D.; Hartwig, J.F. Copper complexes of anionic nitrogen ligands in the amidation and imidation of aryl halides. J. Am. Chem. Soc. 2008, 130, 9971–9983. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-L.; Liu, L.; Fu, Y.; Guo, Q.-X. Theoretical study on Copper(I)-catalyzed cross-coupling between aryl halides and amides. Organometallics 2007, 26, 4546–4554. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Molecule | CuI:Ligand (Ratio) [b] | Ligand (L) | Product, Yield (%) [c] |
|---|---|---|---|---|
| 1 | 5 | 1:0 | - | 11, traces |
| 2 | 5 | 1:1 | L1 | 11, 15 |
| 3 | 5 | 1:2 | L1 | 11, 20 |
| 4 | 5 | 1:3 | L1 | 11, 13 |
| 5 | 5 | 1:2 | L1 | 11, 35 [d] |
| 6 | 6 | 1:2 | L1 | 12, 22 [d] |
| 7 | 6 | 1:2 | L1 | 12, 69 |
| 8 | 6 | 1:2 | L2 | 12, traces |
| 9 | 6 | 1:2 | L3 | 12, traces |
| 10 | 6 | 1:2 | L4 | 12, 8 |
| 11 | 6 | 1:2 | L5 | 12, -[e] |
| 12 | 6 | 1:2 | L6 | 12, traces |
| 13 | 6 | 1:2 | L7 | 12, -[e] |
| 14 | 9 | 1:2 | L1 | -[e] |
| 15 | 10 | 1:2 | L1 | -[e] |
| Entry | Substituent (R) | Product | Yield (%) [b] |
|---|---|---|---|
| 1 | 4-OCH3C6H4 | 12 | 69 |
| 2 | 3-OCH3C6H4 | 13 | 41 |
| 3 | 2-OCH3C6H4 | 14 | 54 |
| 4 | 4-NO2C6H4 | 15 | 44 |
| 5 | 3-NO2C6H4 | 16 | 50 |
| 6 | 2-NO2C6H4 | 17 | 72 |
| 7 | 2-FC6H4 | 18 | 22 |
| 8 | 2-CF3C6H4 | 19 | 33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kederienė, V.; Rousseau, J.; Schuler, M.; Šačkus, A.; Tatibouët, A. Copper-catalyzed S-arylation of Furanose-Fused Oxazolidine-2-thiones. Molecules 2022, 27, 5597. https://doi.org/10.3390/molecules27175597
Kederienė V, Rousseau J, Schuler M, Šačkus A, Tatibouët A. Copper-catalyzed S-arylation of Furanose-Fused Oxazolidine-2-thiones. Molecules. 2022; 27(17):5597. https://doi.org/10.3390/molecules27175597
Chicago/Turabian StyleKederienė, Vilija, Jolanta Rousseau, Marie Schuler, Algirdas Šačkus, and Arnaud Tatibouët. 2022. "Copper-catalyzed S-arylation of Furanose-Fused Oxazolidine-2-thiones" Molecules 27, no. 17: 5597. https://doi.org/10.3390/molecules27175597
APA StyleKederienė, V., Rousseau, J., Schuler, M., Šačkus, A., & Tatibouët, A. (2022). Copper-catalyzed S-arylation of Furanose-Fused Oxazolidine-2-thiones. Molecules, 27(17), 5597. https://doi.org/10.3390/molecules27175597