



Structural Basis of Artemisinin Binding Sites in Serum Albumin with the Combined Use of NMR and Docking Calculations

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
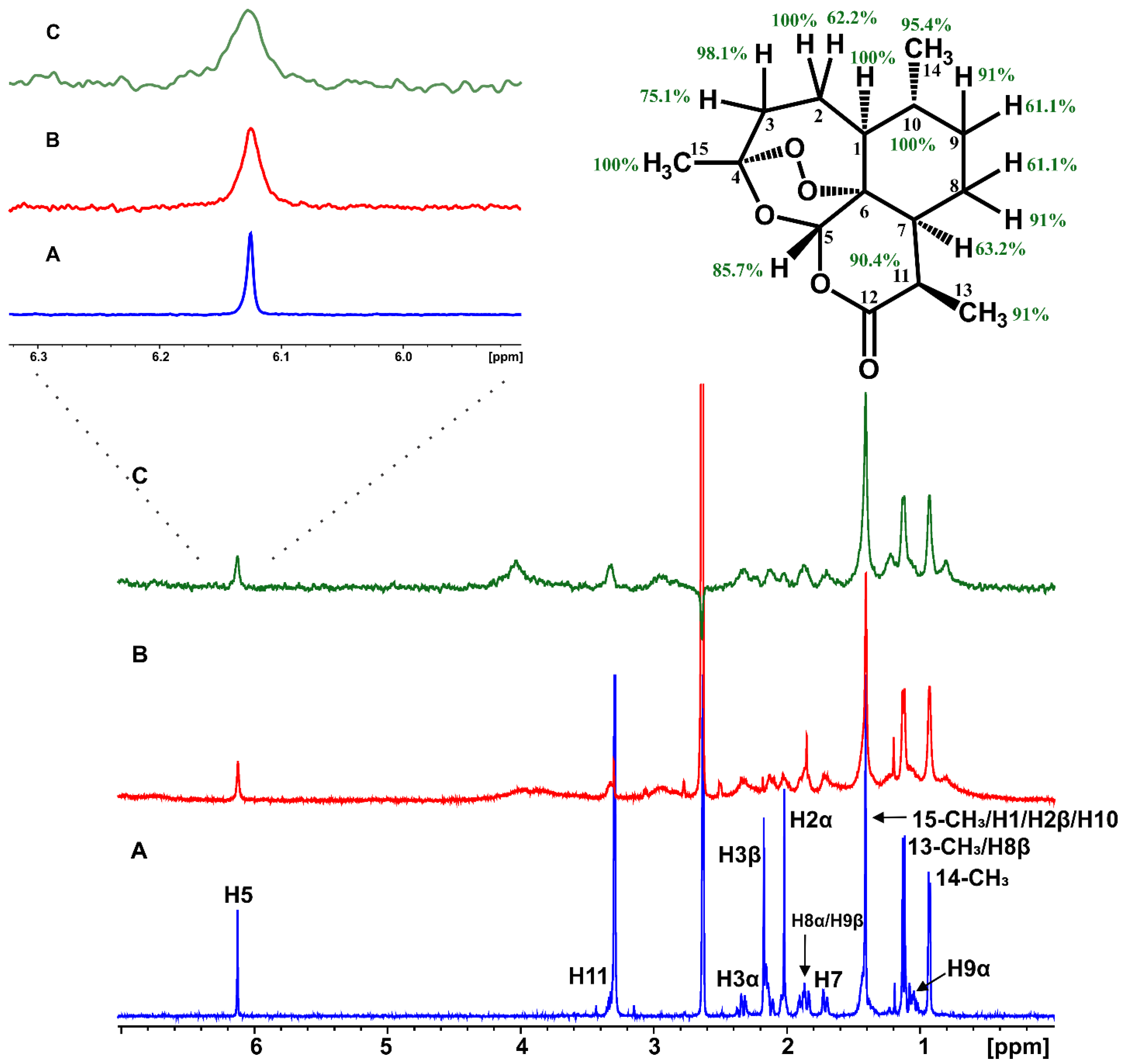
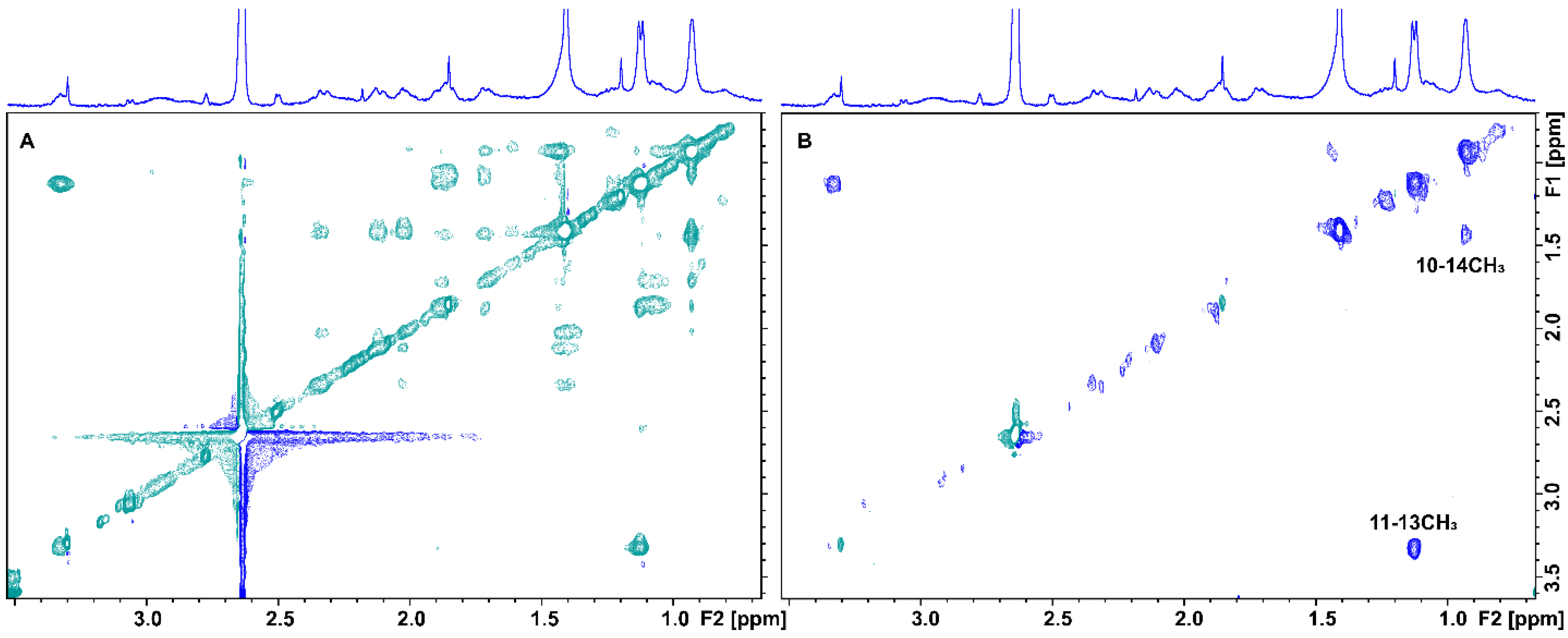
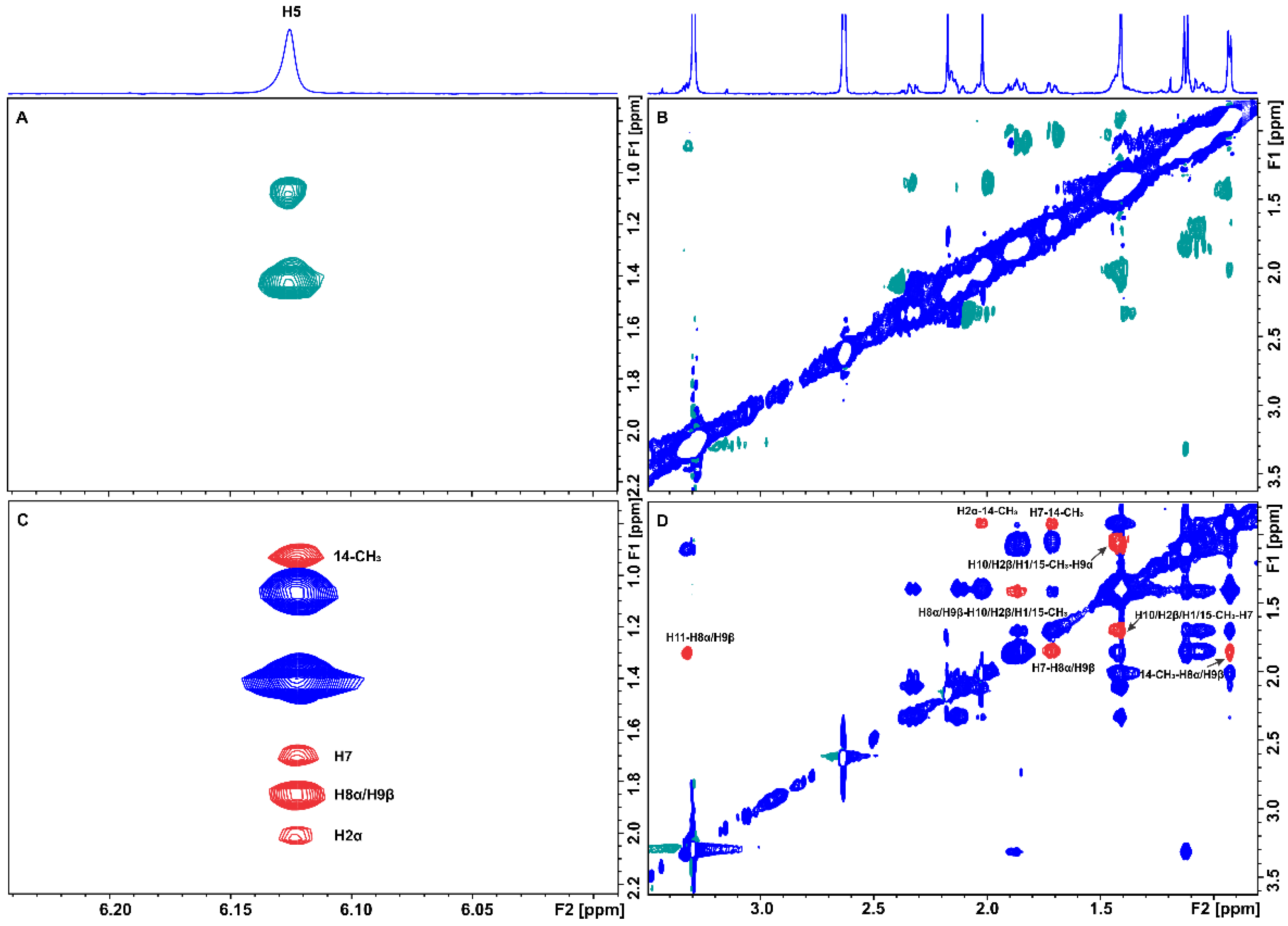
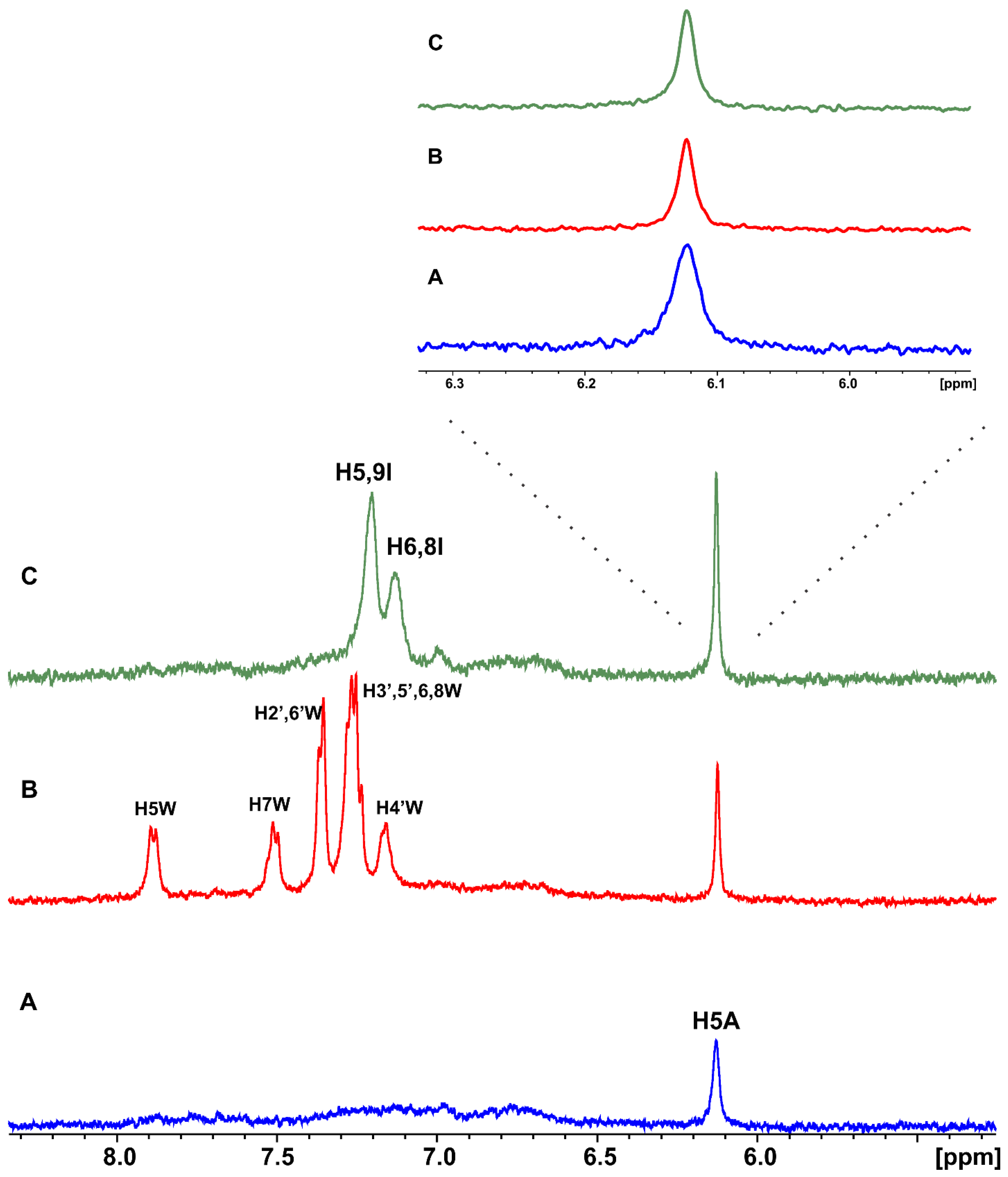
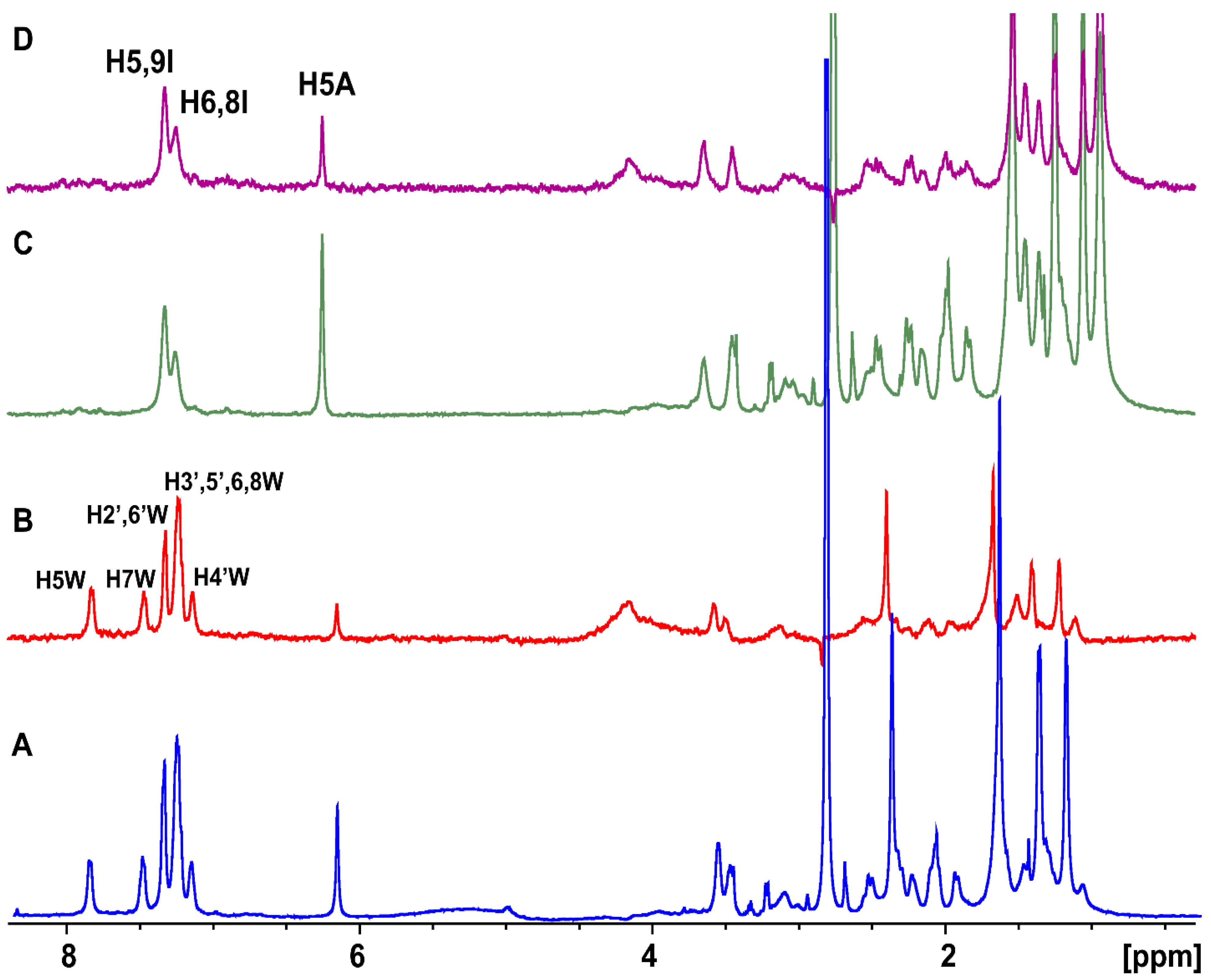
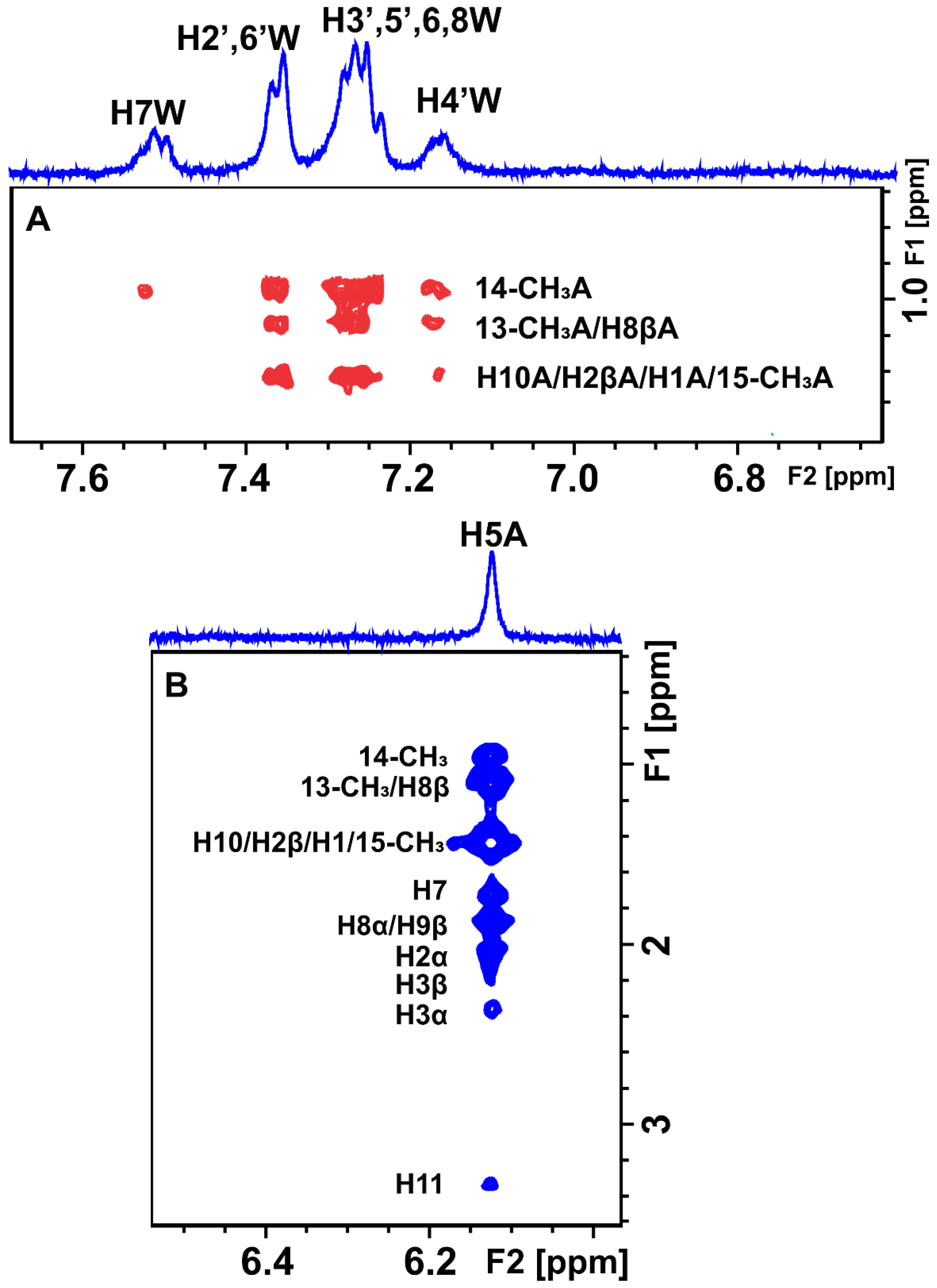
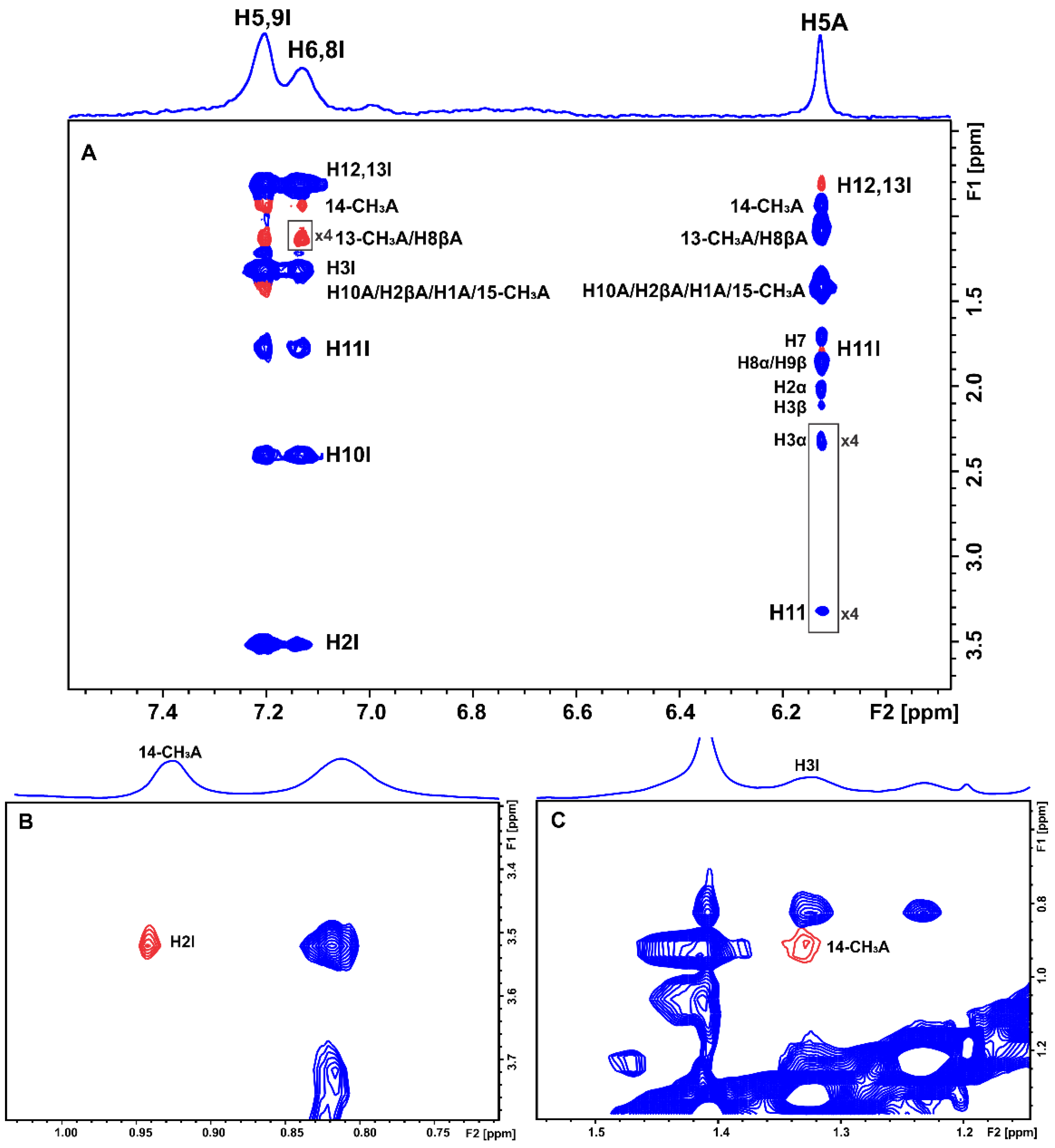
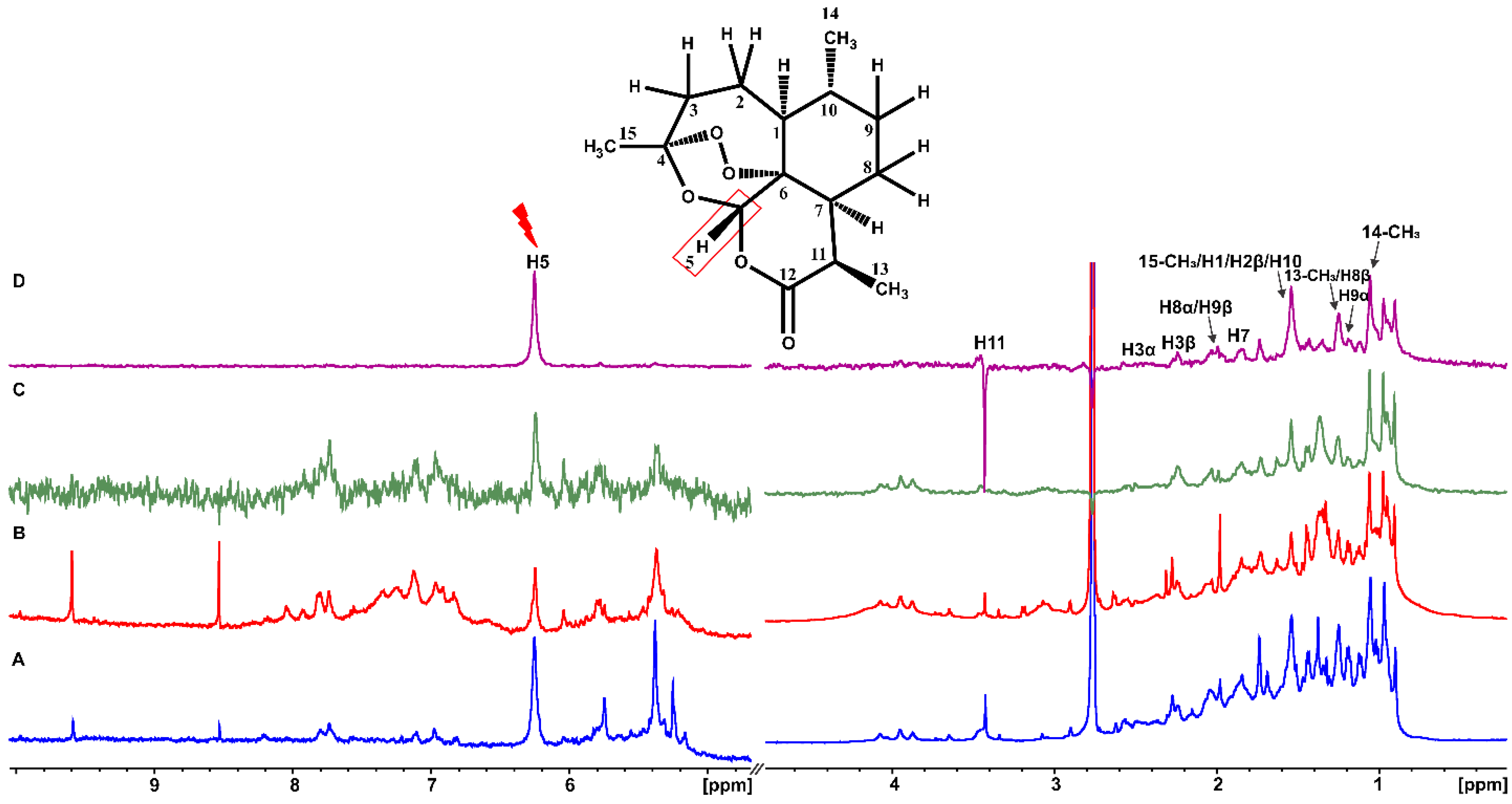
2.1. Epitope Mapping of Bound Artemisinin with the Combined Use of STD, STD-TOCSY, and Tr-NOESY NMR
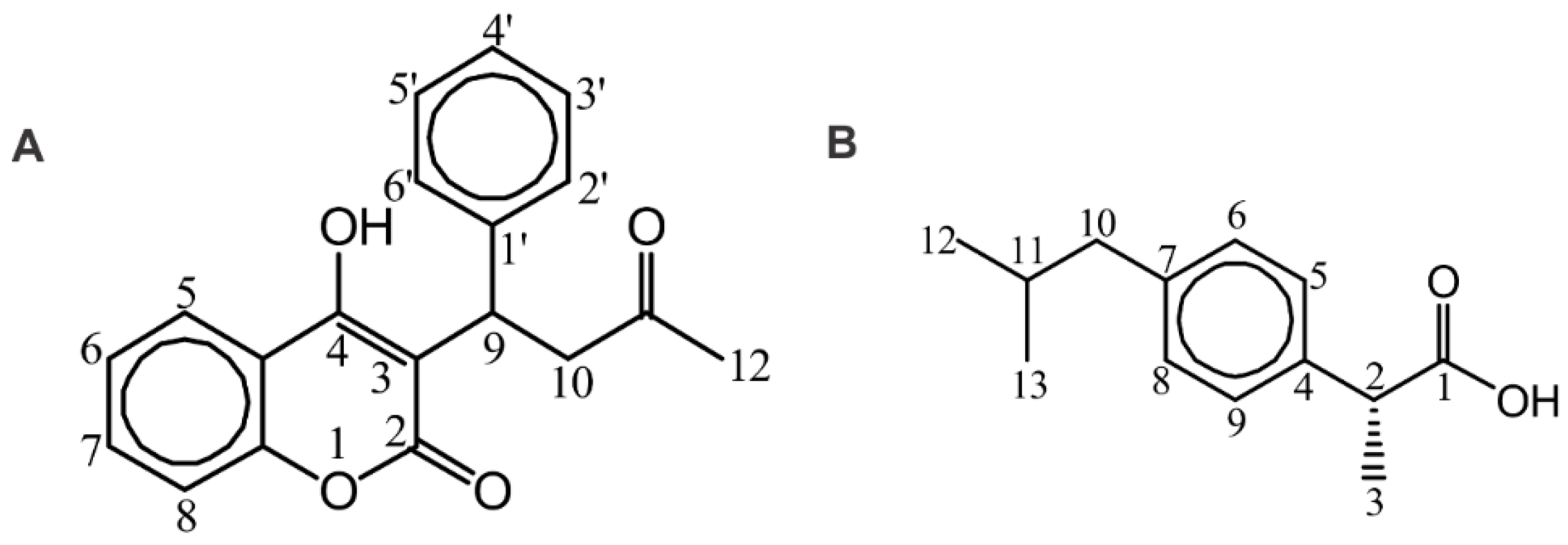
2.2. STD and INPHARMA NMR Competition Experiments of Artemisinin with Warfarin and Ibuprofen
2.3. Docking Calculations
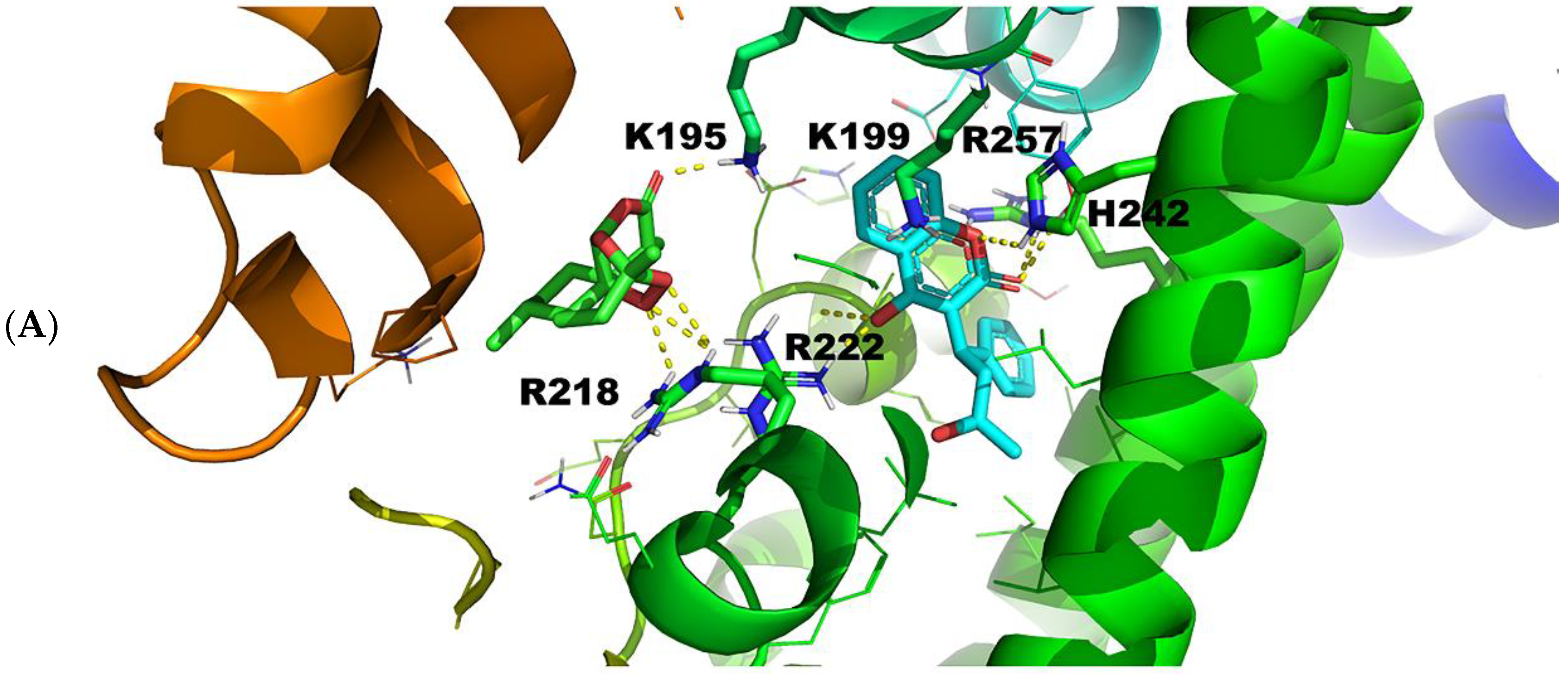
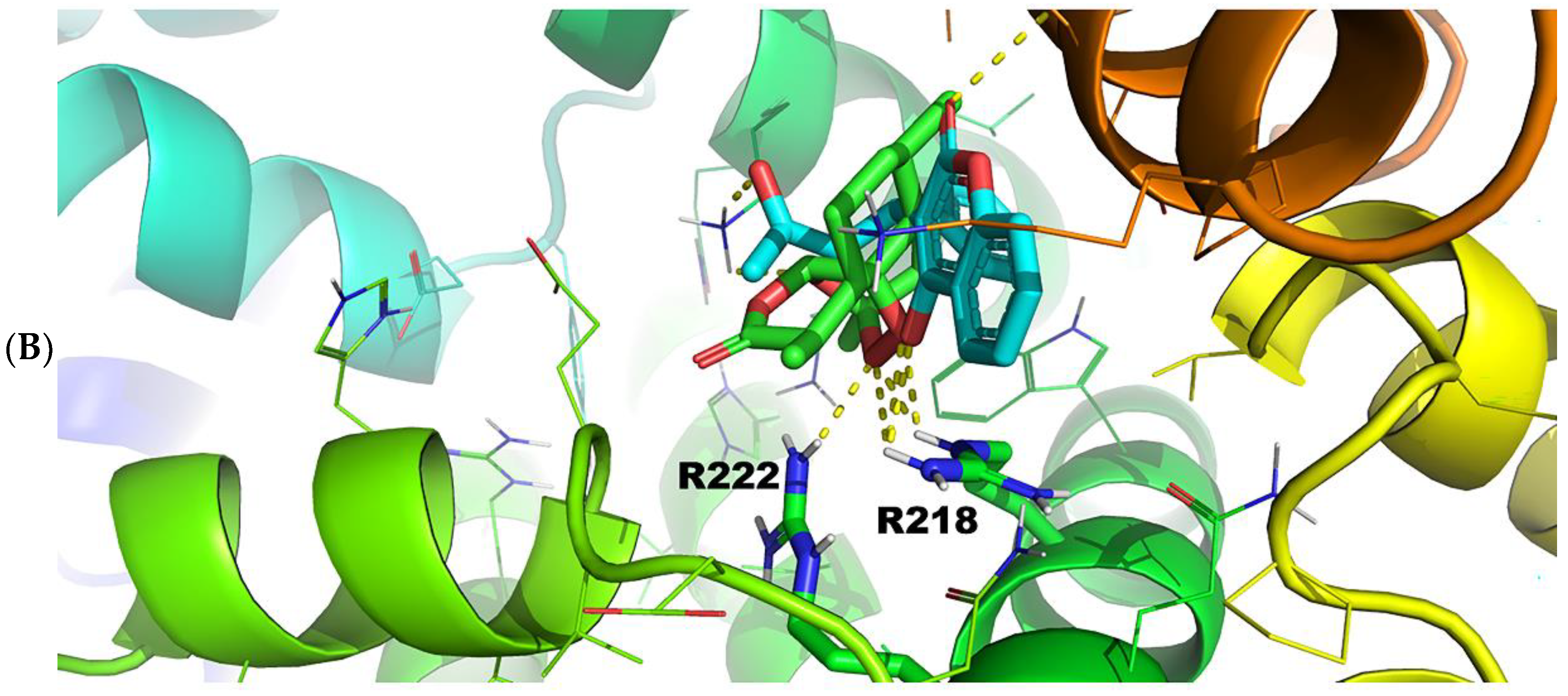
2.3.1. Warfarin Binding Site FA7
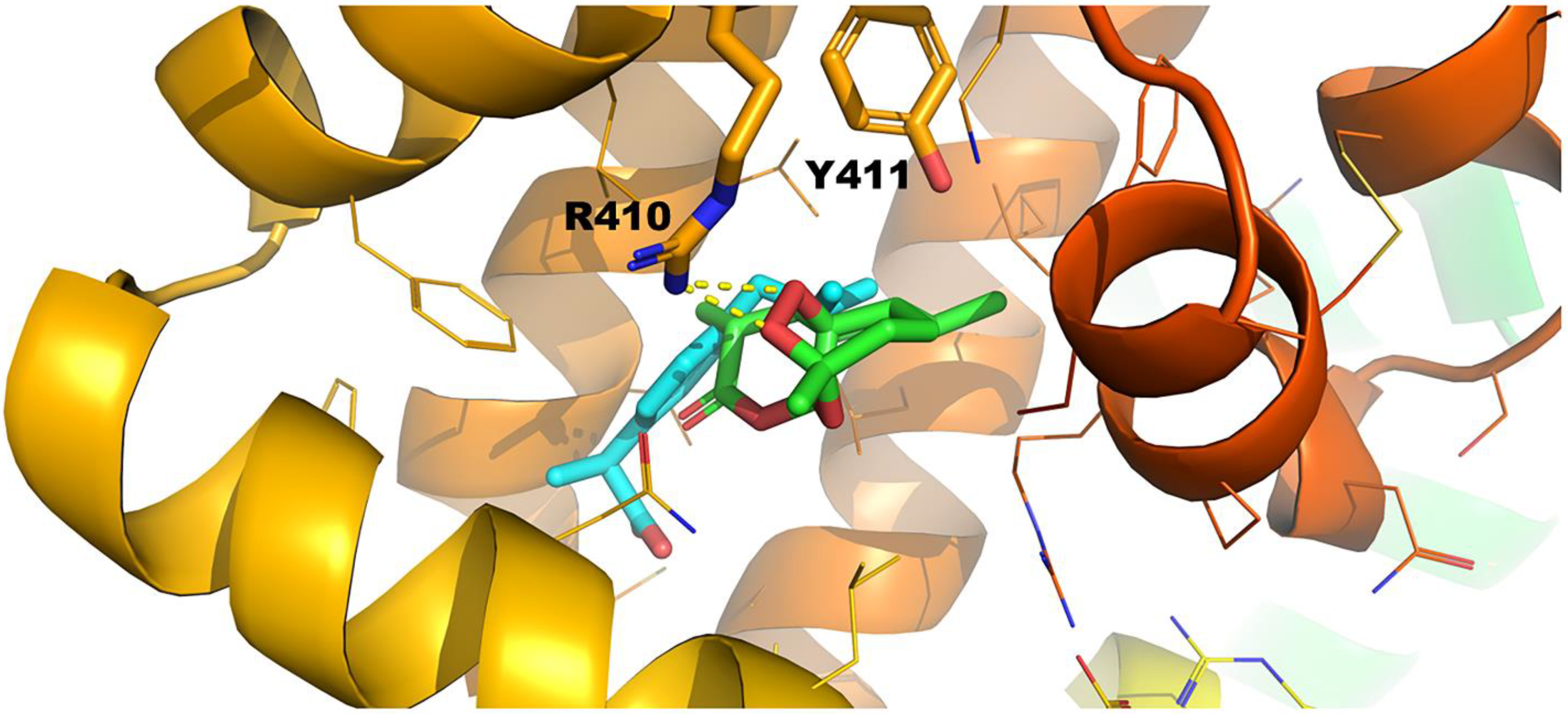
2.3.2. Ibuprofen Binding Site (FA4)
3. Investigation of the Interaction of Artemisia annua Extract with BSA
4. Materials and Methods
4.1. Chemicals and Standards
4.2. Artemisia Annua Extraction
4.3. NMR Methodologies
4.4. Computational Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Callaway, E.; Cyranoski, D. China celebrates first Nobel. Nature 2015, 526, 174–175. [Google Scholar] [CrossRef]
- White, N.J. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Microb. Agents Chemother. 1997, 41, 1413–1422. [Google Scholar] [CrossRef]
- Wong, Y.K.; Xu, C.; Kalesh, K.A.; He, Y.; Lin, Q.; Wong, W.S.F.; Shen, H.-M.; Wang, J. Artemisinin as an anticancer drug: Recent advances in target profiling and mechanisms of action. Med. Res. Rev. 2017, 37, 1492–1517. [Google Scholar] [CrossRef]
- Lu, B.W.; Baum, L.; So, K.F.; Chiu, K.; Xie, L.K. More than anti-malarial agents: Therapeutic potential of artemisinins in neurodegeneration. Neural. Regen. Res. 2019, 14, 1494–1498. [Google Scholar] [CrossRef]
- Zhou, Y.; Gilmore, K.; Ramirez, S.; Settels, E.; Gammeltoft, K.A.; Pham, L.V.; Fahnøe, U.; Feng, S.; Offersgaard, A.; Trimpert, J.; et al. In vitro efficacy of artemisinin-based treatments against SARS-CoV-2. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Cao, R.; Hu, H.; Li, Y.; Wang, X.; Xu, M.; Liu, J.; Zhang, H.; Yan, Y.; Zhao, L.; Li, W.; et al. Anti-SARS-CoV-2 potential of artemisinins in vitro. ACS Infect. Dis. 2020, 6, 2524–2531. [Google Scholar] [CrossRef]
- Ginosyan, S.; Grabski, H.; Tiratsuyan, S. In vitro and in silico identification of the mechanism of interaction of antimalarial drug—Artemisinin with human serum albumin and genomic DNA. Jpn. J. Food Eng. 2019, 20, 18. [Google Scholar] [CrossRef]
- Bhattacharya, A.A.; Grüne, T.; Curry, S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 2000, 303, 721–732. [Google Scholar] [CrossRef]
- Krenzel, E.S.; Chen, Z.; Hamilton, J.A. Correspondence of fatty acid and drug binding sites on human serum albumin: A two-dimensional nuclear magnetic resonance study. Biochemistry 2013, 52, 1559–1567, Erratum on Biochemistry 2013, 52, 2382.. [Google Scholar] [CrossRef]
- Chen, R.; Jiang, H.; Pu, H. Interaction of artemisinin and its derivatives with human serum albumin studied using spectroscopies and molecular modeling methods. Mol. Biol. Rep. 2013, 40, 4791–4804. [Google Scholar] [CrossRef]
- Liu, R.; Cheng, Z.; Jiang, X. Comparative studies on the interactions of dihydroartemisinin and artemisinin with bovine serum albumin using spectroscopic methods. Luminescence 2014, 29, 1033–1046. [Google Scholar] [CrossRef]
- Bian, H.; Li, M.; Yu, Q.; Chen, Z.; Tian, J.; Liang, H. Study of the interaction of artemisinin with bovine serum albumin. Int. J. Biol. Macromol. 2006, 39, 291–297. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Barton, V.E.; Ward, S.A. The molecular mechanism of acti–n of artemisinin—The debate continues. Molecules 2010, 15, 1705–1721. [Google Scholar] [CrossRef]
- Viegas, A.; Nobrega, F.L.; Cabrita, E.J. Saturation-Transfer Difference (STD) NMR: A Simple and Fast Method for Ligand Screening and Characterization of Protein Binding. J. Chem. Educ. 2011, 88, 990–994. [Google Scholar] [CrossRef]
- Primikyri, A.; Sayyad, N.; Quilici, G.; Vrettos, E.I.; Lim, K.; Chi, S.-W.; Musco, G.; Gerothanassis, I.P.; Tzakos, A.G. Probing the interaction of a quercetin bioconjugate with Bcl-2 in living human cancer cells with in-cell NMR spectroscopy. FEBS Lett. 2018, 592, 3367–3379. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef]
- Tanoli, S.A.K.; Tanoli, N.U.; Bondancia, T.M.; Usmani, S.; Ul-Haq, Z.; Fernandes, J.B.; Thomasia, S.S.; Ferreiraa, A.G. Human serum albumin-specific recognition of the natural herbal extract of Stryphnodendron polyphyllum through STD NMR, hyphenations and docking simulation studies. RSC Adv. 2015, 5, 23431–23442. [Google Scholar] [CrossRef]
- Tanoli, S.A.K.; Tanoli, N.U.; Bondancia, T.M.; Usmani, S.; Kerssebaum, R.; Ferreira, A.G.; Fernandesb, J.B.; Ul-Haq, Z. Crude to leads: A triple-pronged direct NMR approach in coordination with docking simulation. Analyst 2013, 138, 5137–5145. [Google Scholar] [CrossRef]
- Becker, W.; Bhattiprolu, K.C.; Gubensäk, N.; Zangger, K. Investigating Protein–Ligand Interactions by solution nuclear magnetic resonance spectroscopy. Chem. Phys. Chem. 2018, 19, 895–906. [Google Scholar] [CrossRef]
- Petitpas, I.; Bhattacharya, A.A.; Twine, S.; East, M.; Curry, S. Crystal structure analysis of warfarin binding to human serum albumin. Anatomy of drug site I. J. Biol. Chem. 2001, 276, 22804–22809. [Google Scholar] [CrossRef] [Green Version]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef]
- Simard, J.R.; Zunszain, P.A.; Ha, C.E.; Hamilton, J.A. Locating high-affinity fatty acid-binding sites on albumin by x-ray crystallography and NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2005, 102, 17958–17963. [Google Scholar] [CrossRef]
- Fehske, K.J.; Schläfer, U.; Wollert, U.M.W. Characterization of an important drug binding area on human serum albumin including the high-affinity binding sites of warfarin and azapropazone. Mol. Pharmacol. 1982, 21, 387–393. [Google Scholar]
- Larsen, F.G.; Larsen, C.G.; Jakobsen, P.; Brodersen, R. Interaction of warfarin with human serum albumin. A stoichiometric description. Mol. Pharmacol. 1985, 27, 263–270. [Google Scholar]
- Kragh-Hansen, U. Evidence for a large and flexible region of human serum albumin possessing high affinity binding sites for salicylate, warfarin, and other ligands. Mol. Pharmacol. 1988, 34, 106–171. [Google Scholar]
- Kragh-Hansen, U. Relations between high-affinity binding sites of markers for binding regions on human serum albumin. Biochem. J. 1985, 225, 629–638. [Google Scholar] [CrossRef]
- Ràfols, C.; Amézqueta, S.; Fuguet, E.; Bosch, E. Molecular interactions between waIrin and human (HSA) or bovine (BSA) serum albumin evaluated by isothermal titration calorimetry (ITC), fluorescence spectrometry (FS) and frontal analysis capillary electrophoresis (FA/CE). J. Pharm. Biomed. Anal. 2018, 150, 452–459. [Google Scholar] [CrossRef]
- Rizzuti, B.; Bartucci, R.; Pey, A.L.; Guzzi, R. Warfarin increases thermal resistance of albumin through stabilization of the protein lobe that includes its binding site. Arch. Biochem. Biophys. 2019, 676, 108–123. [Google Scholar] [CrossRef]
- Carlomagno, T. NMR in natural products: Understanding conformation, configuration and receptor interactions. Nat. Prod. Rep. 2012, 29, 536–554. [Google Scholar] [CrossRef]
- Orts, J.; Tuma, J.; Reese, M.; Grimm, K.; Monecke, P.; Bartoschek, S.; Schiffer, A.; Wendt, K.U.; Griesinger, C.; Carlomagno, T. Crystallography-independent determination of ligand binding modes. Angew. Chem. Int. Ed. 2008, 47, 7736–7740. [Google Scholar] [CrossRef]
- Sánchez-Pedregal, V.M.; Reese, M.; Meiler, J.; Blommers, M.J.J.; Griesinger, C.; Carlomagno, T. The INPHARMA method: Protein-mediated interligand NOEs for pharmacophore mapping. Angew. Chem. Int. Ed. 2005, 44, 4172–4175. [Google Scholar] [CrossRef]
- Alexandri, E.; Primikyri, A.; Papamokos, G.; Venianakis, T.; Gkalpinos, V.K.; Tzakos, A.G.; Karydis-Messinis, A.; Moschovas, D.; Avgeropoulos, A.; Gerothanassis, I.P. NMR and computational studies reveal novel aspects in molecular recognition of unsaturated fatty acids with non-labeled serum albumin. FEBS J. 2022. [Google Scholar] [CrossRef]
- Hernychova, L.; Alexandri, E.; Tzakos, A.G.; Zatloukalová, M.; Primikyri, A.; Gerothanassis, I.P.; Uhrik, L.; Šebela, M.; Kopečný, D.; Jedinák, L.; et al. Serum albumin as a primary non-covalent binding protein for nitro-oleic acid. Int. J. Biol. Macromol. 2022, 203, 116–129. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Notari, S.; Fanali, G.; Fesce, R.; Fasano, M. Allosteric modulation of drug binding to human serum albumin. Mini Rev. Med. Chem. 2006, 6, 483–489. [Google Scholar] [CrossRef]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. Des. Sel. 1999, 12, 439–446. [Google Scholar] [CrossRef]
- Kontogianni, V.G.; Primikyri, A.; Sakka, M.; Gerothanassis, I.P. Simultaneous determination of artemisinin and its analogs and flavonoids in Artemisia annua crude extracts with the use of NMR spectroscopy. Magn. Reson. Chem. 2020, 58, 232–244. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 6.0.16; Semichem Inc.: Shawnee Mission, KS, USA, 2019.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model. 2018, 58, 1697–1706. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comp. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Wenskowsky, L.; Schreuder, H.; Derdau, V.; Matter, H.; Volkmar, J.; Nazare, M.; Opatz, T.; Petry, S. Identification and characterization of a single high-affinity fatty acid binding site in human serum albumin. Angew. Chem. Int. Ed. 2018, 57, 1044–1048. [Google Scholar] [CrossRef]
- Wallaart, T.E.; Pras, N.; Wim, J.; Quax, W.J. Isolation and Identification of dihydroartemisinic acid hydroperoxide from artemisia annua: A novel biosynthetic precursor of artemisinin. J. Nat. Prod. 1999, 62, 1160–1162. [Google Scholar] [CrossRef]
- Kontogianni, V.G.; Gerothanassis, I.P. Analytical and structural tools of lipid hydroperoxides: Present state and future perspectives. Molecules 2022, 27, 2139. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Primikyri, A.; Papamokos, G.; Venianakis, T.; Sakka, M.; Kontogianni, V.G.; Gerothanassis, I.P. Structural Basis of Artemisinin Binding Sites in Serum Albumin with the Combined Use of NMR and Docking Calculations. Molecules 2022, 27, 5912. https://doi.org/10.3390/molecules27185912
Primikyri A, Papamokos G, Venianakis T, Sakka M, Kontogianni VG, Gerothanassis IP. Structural Basis of Artemisinin Binding Sites in Serum Albumin with the Combined Use of NMR and Docking Calculations. Molecules. 2022; 27(18):5912. https://doi.org/10.3390/molecules27185912
Chicago/Turabian StylePrimikyri, Alexandra, Georgios Papamokos, Themistoklis Venianakis, Marianna Sakka, Vassiliki G. Kontogianni, and Ioannis P. Gerothanassis. 2022. "Structural Basis of Artemisinin Binding Sites in Serum Albumin with the Combined Use of NMR and Docking Calculations" Molecules 27, no. 18: 5912. https://doi.org/10.3390/molecules27185912
APA StylePrimikyri, A., Papamokos, G., Venianakis, T., Sakka, M., Kontogianni, V. G., & Gerothanassis, I. P. (2022). Structural Basis of Artemisinin Binding Sites in Serum Albumin with the Combined Use of NMR and Docking Calculations. Molecules, 27(18), 5912. https://doi.org/10.3390/molecules27185912