

Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies

 ,
,  , , , , ,
, , , , ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Testing
2.2.1. In Vitro Anti-proliferative Activities against HepG-2 and HCT-116
2.2.2. In Vitro VEGFR-2 Enzyme Assay
2.2.3. Effect on Cell Cycle Phases
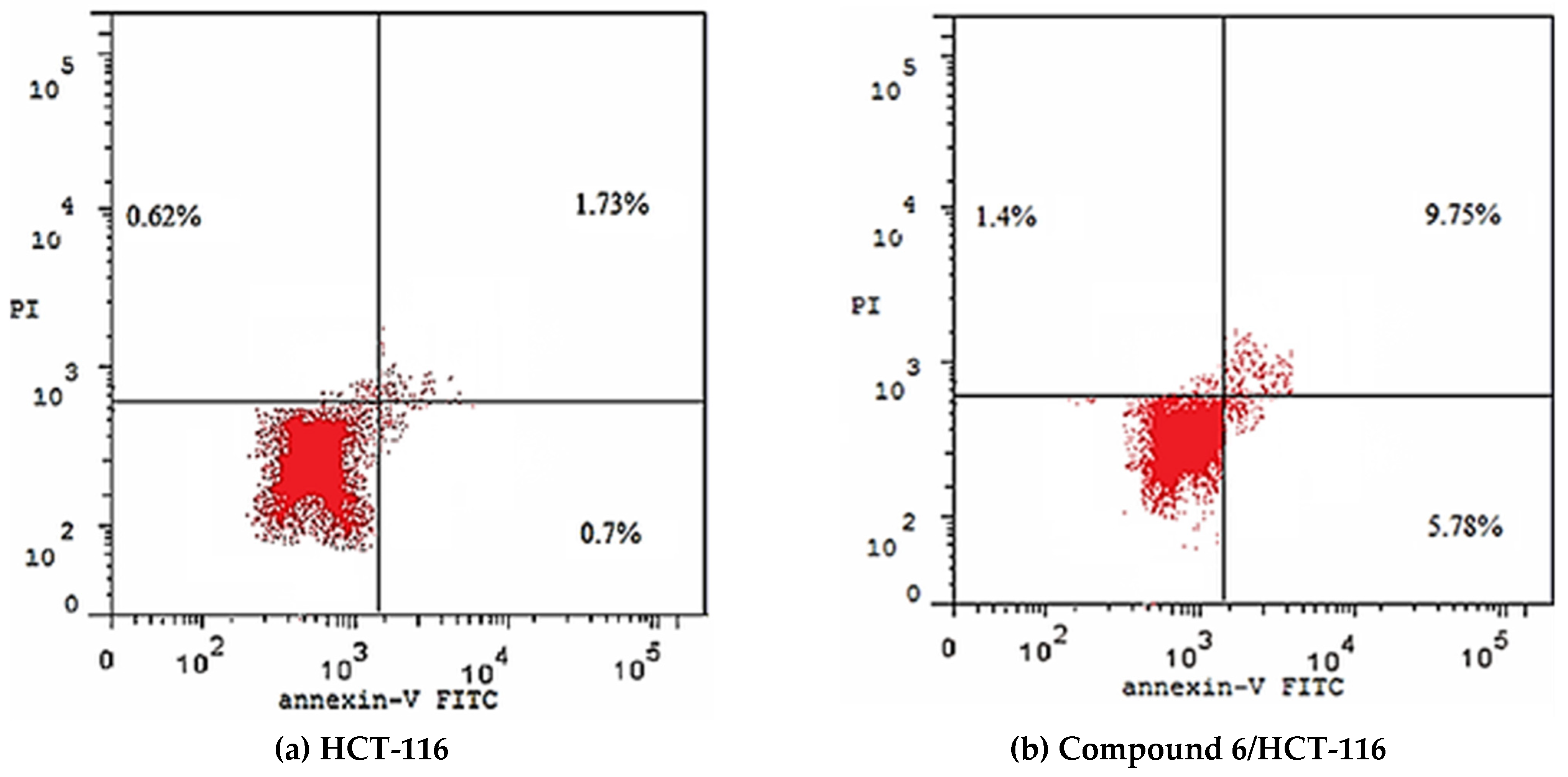
2.2.4. Detection of Apoptosis
2.2.5. In Vitro Immunomodulatory Assay
2.3. In Silico Studies
2.3.1. Molecular Docking
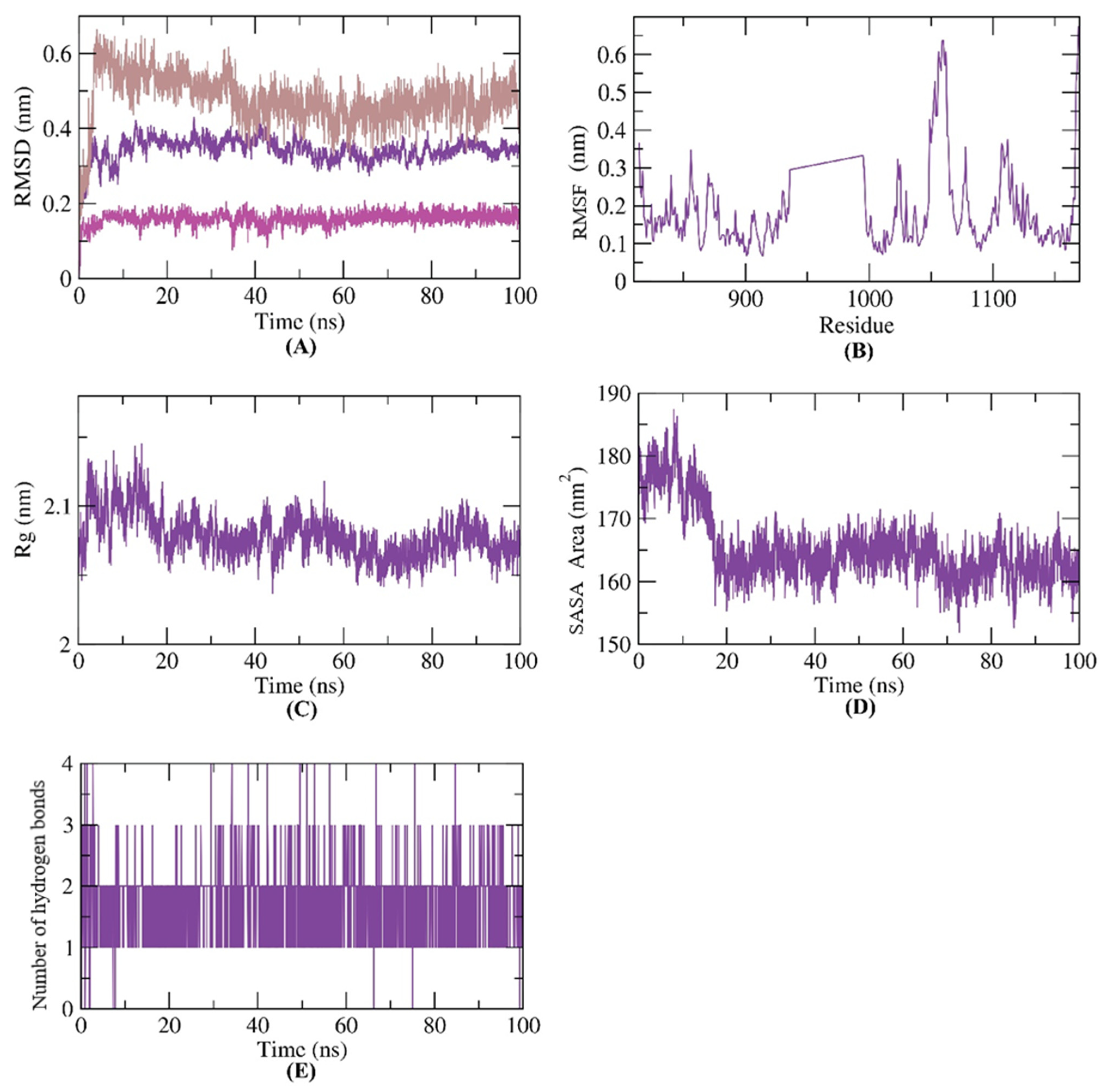
2.3.2. MD Simulations
2.3.3. MM-PBSA Studies
2.3.4. In Silico ADMET Analysis
2.3.5. Toxicity Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Synthesis of Congeners 6, 7, and 8
N-{4-[2-(4-Chlorobenzylidene)hydrazine-1-carbonyl]phenyl}nicotinamide 6
N-{4-[2-(2-Hydroxybenzylidene)hydrazine-1-carbonyl]phenyl}nicotinamide 7
N-{4-[2-((1H-Indol-3-yl)methylene)hydrazine-1-carbonyl]phenyl}nicotinamide 8
N-{4-[1-(2-Carbamoylhydrazineylidene)ethyl]phenyl}nicotinamide 10
3.2. Biological Testing
3.2.1. In Vitro Anti-proliferative Activities against HepG-2 and HCT-116
3.2.2. In Vitro VEGFR-2 Enzyme Inhibition Assay
3.2.3. Flow Cytometry Analysis for Cell Cycle
3.2.4. Flow Cytometry Analysis for Apoptosis
3.3. In Silico Studies
3.3.1. Docking Studies
3.3.2. ADMET Studies
3.3.3. Toxicity Studies
3.3.4. Molecular Dynamics Simulation & MM/PBSA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; Dahab, M.A.; Ibrahim, M.K.; Alsaif, N.A.; Alanazi, A.; Eissa, S.I.; Mehany, A.B.; Beauchemin, A.M. Design and discovery of new antiproliferative 1, 2, 4-triazin-3 (2H)-ones as tubulin polymerization inhibitors targeting colchicine binding site. Bioorganic Chem. 2021, 112, 104965. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.J.; Kulkarni, V.M. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: Medicinal chemistry perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Modi, S.J.; Kulkarni, V.M. Discovery of VEGFR-2 inhibitors exerting significant anticancer activity against CD44+ and CD133+ cancer stem cells (CSCs): Reversal of TGF-β induced epithelial-mesenchymal transition (EMT) in hepatocellular carcinoma. Eur. J. Med. Chem. 2020, 207, 112851. [Google Scholar] [CrossRef]
- Sana, S.; Reddy, V.G.; Bhandari, S.; Reddy, T.S.; Tokala, R.; Sakla, A.P.; Bhargava, S.K.; Shankaraiah, N. Exploration of carbamide derived pyrimidine-thioindole conjugates as potential VEGFR-2 inhibitors with anti-angiogenesis effect. Eur. J. Med. Chem. 2020, 200, 112457. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef]
- Suleimen, Y.M.; Metwaly, A.M.; Mostafa, A.E.; Elkaeed, E.B.; Liu, H.-W.; Basnet, B.B.; Suleimen, R.N.; Ishmuratova, M.Y.; Turdybekov, K.M.; van Heсke, K. Isolation Crystal Structure, and In Silico Aromatase Inhibition Activity of Ergosta-5, 22-dien-3β-ol from the Fungus Gyromitra esculenta. J. Chem. 2021, 2021, 5529786. [Google Scholar] [CrossRef]
- El-Adl, K.; Sakr, H.M.; Yousef, R.G.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; Eissa, I.H. Discovery of new quinoxaline-2 (1h)-one-based anticancer agents targeting vegfr-2 as inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorganic Chem. 2021, 114, 105105. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Mahdy, H.A.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Dahab, M.A.; Eissa, I.H. New bis ([1, 2, 4] triazolo)[4, 3-a: 3′, 4′-c] quinoxaline derivatives as vegfr-2 inhibitors and apoptosis inducers: Design, synthesis, in silico studies, and anticancer evaluation. Bioorganic Chem. 2021, 112, 104949. [Google Scholar] [CrossRef]
- Oguro, Y.; Cary, D.R.; Miyamoto, N.; Tawada, M.; Iwata, H.; Miki, H.; Hori, A.; Imamura, S. Design, synthesis, and evaluation of novel VEGFR2 kinase inhibitors: Discovery of [1, 2, 4] triazolo [1, 5-a] pyridine derivatives with slow dissociation kinetics. Biorg. Med. Chem. 2013, 21, 4714–4729. [Google Scholar] [CrossRef]
- AbdelHaleem, A.; Mansour, A.O.; AbdelKader, M.; Arafa, R.K. Selective VEGFR-2 inhibitors: Synthesis of pyridine derivatives, cytotoxicity and apoptosis induction profiling. Bioorganic Chem. 2020, 103, 104222. [Google Scholar] [CrossRef]
- Gu, W.; Dai, Y.; Qiang, H.; Shi, W.; Liao, C.; Zhao, F.; Huang, W.; Qian, H. Discovery of novel 2-substituted-4-(2-fluorophenoxy) pyridine derivatives possessing pyrazolone and triazole moieties as dual c-Met/VEGFR-2 receptor tyrosine kinase inhibitors. Bioorganic Chem. 2017, 72, 116–122. [Google Scholar] [CrossRef]
- Yousef, R.G.; Ibrahim, A.; Khalifa, M.M.; Eldehna, W.M.; Gobaara, I.M.; Mehany, A.B.; Elkaeed, E.B.; Alsfouk, A.A.; Metwaly, A.M.; Eissa, I.H. Discovery of new nicotinamides as apoptotic VEGFR-2 inhibitors: Virtual screening, synthesis, anti-proliferative, immunomodulatory, ADMET, toxicity, and molecular dynamic simulation studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1389–1403. [Google Scholar] [CrossRef] [PubMed]
- El-Metwally, S.A.; Abou-El-Regal, M.M.; Eissa, I.H.; Mehany, A.B.; Mahdy, H.A.; Elkady, H.; Elwan, A.; Elkaeed, E.B. Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorganic Chem. 2021, 112, 104947. [Google Scholar] [CrossRef] [PubMed]
- Elkady, H.; Elwan, A.; El-Mahdy, H.A.; Doghish, A.S.; Ismail, A.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Dahab, M.A.; Mahdy, H.A. New benzoxazole derivatives as potential VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, anti-proliferative evaluation, flowcytometric analysis, and in silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 397–410. [Google Scholar] [CrossRef] [PubMed]
- El-Helby, A.G.A.; Sakr, H.; Eissa, I.H.; Abulkhair, H.; Al-Karmalawy, A.A.; El-Adl, K. Design, synthesis, molecular docking, and anticancer activity of benzoxazole derivatives as VEGFR-2 inhibitors. Arch. Pharm. 2019, 352, 1900113. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.M.; Abdel-Rahman, A.A.H.; Omar, A.M.; Khalifa, M.M.; El-Adl, K. Pyridine-derived VEGFR-2 inhibitors: Rational design, synthesis, anticancer evaluations, in silico ADMET profile, and molecular docking. Arch. Pharm. 2021, 354, 2100085. [Google Scholar] [CrossRef]
- Eissa, I.H.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.; Abdelhady, A.A.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Mahdy, H.A. Design, molecular docking, in vitro, and in vivo studies of new quinazolin-4 (3H)-ones as VEGFR-2 inhibitors with potential activity against hepatocellular carcinoma. Bioorganic Chem. 2021, 107, 104532. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Ibrahim, M.K.; Eissa, I.H.; El-Adl, K. Design, synthesis, in silico admet, docking, and antiproliferative evaluations of [1, 2, 4] triazolo [4, 3-c] quinazolines as classical DNA intercalators. Arch. Pharm. 2022, 355, 2100412. [Google Scholar] [CrossRef]
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G.; Talukdar, S.N.; Eissa, I.H.; Metwaly, A.M.; Khalil, A. Discovery of new anticancer thiourea-azetidine hybrids: Design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorganic Chem. 2021, 115, 105206. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; El-Adl, K.; El-Hddad, S.S.; Elhady, M.M.; Saleh, N.M.; Khalifa, M.M.; Khedr, F.; Alswah, M.; Nayl, A.A.; Ghoneim, M.M.; et al. Design, Molecular Docking, Synthesis, Anticancer and Anti-Hyperglycemic Assessments of Thiazolidine-2, 4-diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPARγ Agonists. Pharmaceuticals 2022, 15, 226. [Google Scholar] [CrossRef] [PubMed]
- Yousef, R.G.; Sakr, H.M.; Eissa, I.H.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; El-Adl, K. New quinoxaline-2 (1 H)-ones as potential VEGFR-2 inhibitors: Design, synthesis, molecular docking, ADMET profile and anti-proliferative evaluations. New J. Chem. 2021, 45, 16949–16964. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Eissa, I.H.; Alsaif, N.A.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.F.; Albassam, H.; Elkady, H.; Elwan, A. Design, synthesis, docking, admet studies, and anticancer evaluation of new 3-methylquinoxaline derivatives as vegfr-2 inhibitors and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1760–1782. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.; Li, W.; Qin, Y.; Yu, T.; Liu, Z.; Zhou, M.; Liu, C.; Qiao, T.; Li, X.; Yousef, R.G. Inhibition of vascular smooth muscle and cancer cell proliferation by new VEGFR inhibitors and their immunomodulator effect: Design, synthesis, and biological evaluation. Oxidative Med. Cell. Longev. 2021, 2021, 8321400. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein–ligand docking: Current status and future challenges. Proteins Struct. Funct. Bioinform. 2006, 65, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Oostenbrink, C.; van Gunsteren, W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef]
- Liu, P.; Lu, J.; Yu, H.; Ren, N.; Lockwood, F.E.; Wang, Q.J. Lubricant shear thinning behavior correlated with variation of radius of gyration via molecular dynamics simulations. J. Chem. Phys. 2017, 147, 084904. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Anbarasu, A.; Ramaiah, S. Molecular docking and molecular dynamics studies on β-lactamases and penicillin binding proteins. Mol. BioSystems 2014, 10, 891–900. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef]
- Xia, X.; Maliski, E.G.; Gallant, P.; Rogers, D. Classification of kinase inhibitors using a Bayesian model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA QSAR, ADMET and Predictive Toxicology. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html (accessed on 1 May 2020).
- Venkatapathy, R.; Wang, N.C.Y.; Martin, T.M.; Harten, P.F.; Young, D. Structure–Activity Relationships for Carcinogenic Potential. Gen. Appl. Syst. Toxicol. 2009. [Google Scholar] [CrossRef]
- Goodrnan, G.; Wilson, R. Comparison of the dependence of the TD50 on maximum tolerated dose for mutagens and nonmutagens. Risk Anal. 1992, 12, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Council, N.R. Correlation Between Carcinogenic Potency and the Maximum Tolerated Dose: Implications for Risk Assessment. In Issues in Risk Assessment; National Academies Press (US): Washington, DC, USA, 1993. [Google Scholar]
- Diaza, R.G.; Manganelli, S.; Esposito, A.; Roncaglioni, A.; Manganaro, A.; Benfenati, E. Comparison of in silico tools for evaluating rat oral acute toxicity. SAR QSAR Environ. Res. 2015, 26, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, F.; Benfenati, E. In silico models for repeated-dose toxicity (RDT): Prediction of the no observed adverse effect level (NOAEL) and lowest observed adverse effect level (LOAEL) for drugs. In In Silico Methods for Predicting Drug Toxicity; Springer: Berlin/Heidelberg, Germany, 2016; pp. 163–176. [Google Scholar]
- Venkatapathy, R.; Moudgal, C.J.; Bruce, R.M. Assessment of the oral rat chronic lowest observed adverse effect level model in TOPKAT, a QSAR software package for toxicity prediction. J. Chem. Inf. Comput. Sci. 2004, 44, 1623–1629. [Google Scholar] [CrossRef]
- Chaitra, G.; Rohini, R. Synthesis and biological activities of [1, 3]-oxazine derivatives. Der Pharma Chem. 2018, 10, 96–101. [Google Scholar]
- Eldehna, W.M.; Al-Rashood, S.T.; Al-Warhi, T.; Eskandrani, R.O.; Alharbi, A.; el Kerdawy, A.M. Novel oxindole/benzofuran hybrids as potential dual CDK2/GSK-3β inhibitors targeting breast cancer: Design, synthesis, biological evaluation, and in silico studies. J. Enzym. Inhib. Med. Chem. 2021, 36, 270–285. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Al-Ansary, G.H.; Elsayed, Z.M.; Maklad, R.M.; Elkaeed, E.B.; Abdelgawad, M.A.; Bukhari, S.N.A.; Abdel-Aziz, M.M.; Suliman, H.; Eldehna, W.M. Development of 3-methyl/3-(morpholinomethyl) benzofuran derivatives as novel antitumor agents towards non-small cell lung cancer cells. J. Enzym. Inhib. Med. Chem. 2021, 36, 987–999. [Google Scholar] [CrossRef]
- Abou-Seri, S.M.; Eldehna, W.M.; Ali, M.M.; el Ella, D.A.A. 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: Synthesis and in vitro biological evaluation. Eur. J. Med. Chem. 2016, 107, 165–179. [Google Scholar] [CrossRef]
- Wang, J.; Lenardo, M.J. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J. Cell Sci. 2000, 113, 753–757. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Hassan, G.S.; Al-Rashood, S.T.; Al-Warhi, T.; Altyar, A.E.; Alkahtani, H.M.; Almehizia, A.A.; Abdel-Aziz, H.A. Synthesis and in vitro anticancer activity of certain novel 1-(2-methyl-6-arylpyridin-3-yl)-3-phenylureas as apoptosis-inducing agents. J. Enzym. Inhib. Med. Chem. 2019, 34, 322–332. [Google Scholar] [CrossRef]
- Lo, K.K.-W.; Lee, T.K.-M.; Lau, J.S.-Y.; Poon, W.-L.; Cheng, S.-H. Luminescent biological probes derived from ruthenium (II) estradiol polypyridine complexes. Inorg. Chem. 2008, 47, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Sabt, A.; Abdelhafez, O.M.; El-Haggar, R.S.; Madkour, H.M.; Eldehna, W.M.; El-Khrisy, E.E.-D.A.; Abdel-Rahman, M.A.; Rashed, L.A. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: Synthesis, in vitro biological evaluation, and QSAR studies. J. Enzym. Inhib. Med. Chem. 2018, 33, 1095–1107. [Google Scholar] [CrossRef]
- Suleimen, Y.M.; Jose, R.A.; Suleimen, R.N.; Arenz, C.; Ishmuratova, M.; Toppet, S.; Dehaen, W.; Alsfouk, A.A.; Elkaeed, E.B.; Eissa, I.H. Isolation and In Silico Anti-SARS-CoV-2 Papain-Like Protease Potentialities of Two Rare 2-Phenoxychromone Derivatives from Artemisia spp. Molecules 2022, 27, 1216. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Elwan, A.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Taghour, M.S.; Eissa, I.H. Discovery of new 3-methylquinoxalines as potential anti-cancer agents and apoptosis inducers targeting VEGFR-2: Design, synthesis, and in silico studies. J. Enzym. Inhib. Med. Chem. 2021, 36, 1732–1750. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Yousef, R.G.; Elkady, H.; Alsfouk, A.A.; Husein, D.Z.; Ibrahim, I.M.; Metwaly, A.M.; Eissa, I.H. New anticancer theobromine derivative targeting egfrwt and egfrt790m: Design, semi-synthesis, in silico, and in vitro anticancer studies. Molecules 2022, 27, 5859. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | In Vitro Cytotoxicity IC50 (µM) a | VEGFR-2 IC50 (nM) a | |
|---|---|---|---|
| HepG-2 | HCT-116 | ||
| 6 | 7.80 ± 0.025 | 9.30 ± 0.02 | 60.83 ± 3.81 |
| 7 | 10.20 ± 0.035 | 15.90 ± 0.041 | 129.30 ± 8.32 |
| 8 | 16.03 ± 0.051 | 20.50 ± 0.052 | 229.50 ± 10.22 |
| 10 | 17.60 ± 0.048 | 24.20 ± 0.06 | 63.61 ± 3.31 |
| Sorafenib | 5.28 ± 0.21 | 7.28 ± 0.58 | 53.65 ± 2.55 |
| Sample | % G0-G1 | % S | % G2-M | % Pre-G1 |
|---|---|---|---|---|
| Compound 6/HCT-116 | 36.71 | 27.45 | 35.84 | 16.93 |
| HCT-116 | 55.62 | 31.47 | 12.91 | 3.05 |
| Sample | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| Compound 6/HCT-116 | 16.93 | 5.78 | 9.75 | 1.40 |
| HCT-116 | 3.05 | 0.70 | 1.73 | 0.62 |
| Comp. | % Inhibition of TNF-α pg/mL | % Inhibition of IL6 pg/mL |
|---|---|---|
| 6 | 66.42 | 57.34 |
| 7 | 60.54 | 52.75 |
| Dexamethasone | 82.47 | 93.15 |
| Comp. | ∆G | Residues (H-bond) |
|---|---|---|
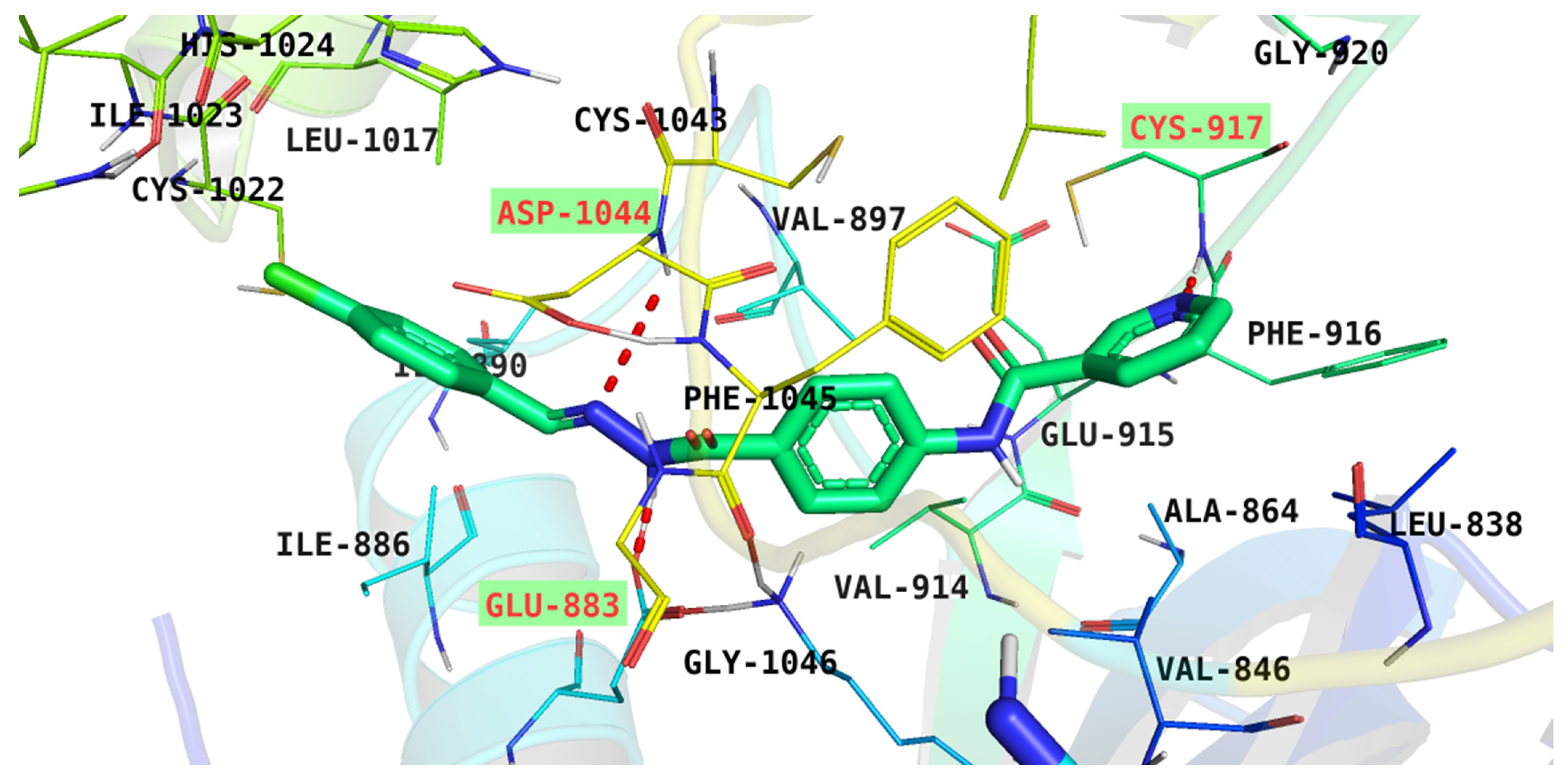
| 6 | −28.52 | Cys917, Asp1044, and Glu883 |
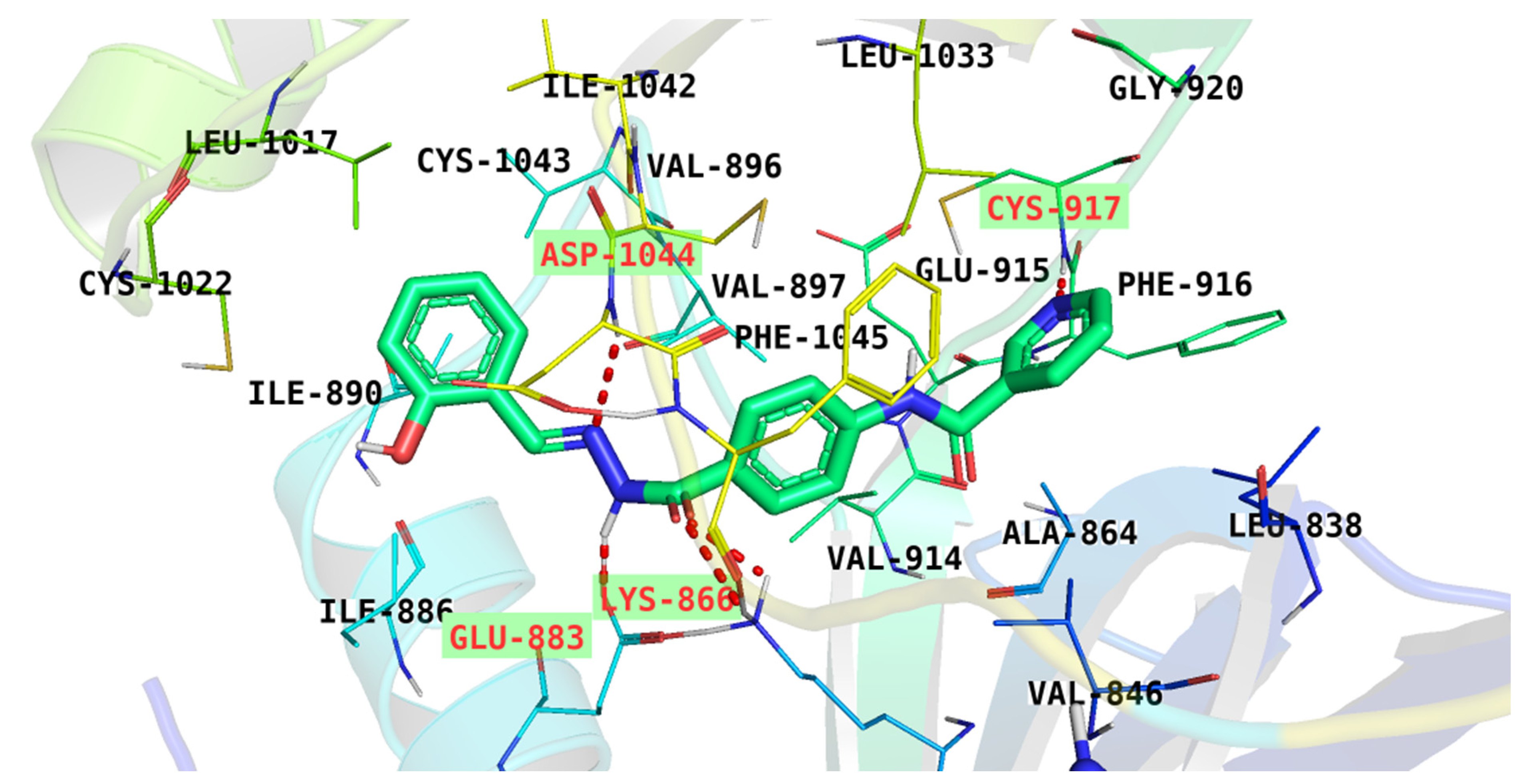
| 7 | −26.77 | Cys917, Asp1044, Glu883, and Lys886 |
| 8 | −23.09 | Cys917, Asp1044, and Glu883 |
| 10 | −16.46 | Cys917, Asp1044, and Glu883 |
| Co-crystallized ligand | −28.02 | Cys917, Asp1044, and Glu833 |
| Comp. | BBB-P a | Sol-L a | Int-A a | CYP2D6-Inh b | PP-Bind c |
|---|---|---|---|---|---|
| 6 | -- | -- | ---- | Non-inh | Mr |
| 7 | -- | --- | ---- | Non-inh | Mr |
| 8 | -- | -- | ---- | Non-inh | Ls |
| 10 | -- | ---- | ---- | Non-inh | Ls |
| Sorafenib | - | - | ---- | Non-inh | Mr |
| Comp. | R TD50 mg/kg | R MTD, g/kg | R LD50, g/kg | R LOAEL, g/kg | Skin Irritancy | Ocular Irritancy |
|---|---|---|---|---|---|---|
| 6 | 12.294 | 0.154 | 1.320 | 0.096 | None | Mild |
| 7 | 276.321 | 0.414 | 1.531 | 0.481 | None | Mild |
| 8 | 14.966 | 0.181 | 1.860 | 0.316 | None | Mild |
| 10 | 54.777 | 0.144 | 0.744 | 0.230 | None | Mild |
| Sorafenib | 14.244 | 0.089 | 0.823 | 0.005 | None | Mild |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkaeed, E.B.; Yousef, R.G.; Khalifa, M.M.; Ibrahim, A.; Mehany, A.B.M.; Gobaara, I.M.M.; Alsfouk, B.A.; Eldehna, W.M.; Metwaly, A.M.; Eissa, I.H.; et al. Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies. Molecules 2022, 27, 6203. https://doi.org/10.3390/molecules27196203
Elkaeed EB, Yousef RG, Khalifa MM, Ibrahim A, Mehany ABM, Gobaara IMM, Alsfouk BA, Eldehna WM, Metwaly AM, Eissa IH, et al. Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies. Molecules. 2022; 27(19):6203. https://doi.org/10.3390/molecules27196203
Chicago/Turabian StyleElkaeed, Eslam B., Reda G. Yousef, Mohamed M. Khalifa, Albaraa Ibrahim, Ahmed B. M. Mehany, Ibraheem M. M. Gobaara, Bshra A. Alsfouk, Wagdy M. Eldehna, Ahmed M. Metwaly, Ibrahim H. Eissa, and et al. 2022. "Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies" Molecules 27, no. 19: 6203. https://doi.org/10.3390/molecules27196203
APA StyleElkaeed, E. B., Yousef, R. G., Khalifa, M. M., Ibrahim, A., Mehany, A. B. M., Gobaara, I. M. M., Alsfouk, B. A., Eldehna, W. M., Metwaly, A. M., Eissa, I. H., & El-Zahabi, M. A. (2022). Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies. Molecules, 27(19), 6203. https://doi.org/10.3390/molecules27196203