
Phyto-Computational Intervention of Diabetes Mellitus at Multiple Stages Using Isoeugenol from Ocimum tenuiflorum: A Combination of Pharmacokinetics and Molecular Modelling Approaches
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Data Retrieval and ADMET Profiling
2.2. Molecular Docking Simulation
2.3. Molecular Dynamics Simulation
2.4. Binding Free Energy Calculation
2.5. Druglikeness, Pharmacokinetics, and PASS Analysis of the Representative Compounds
3. Results and Discussion
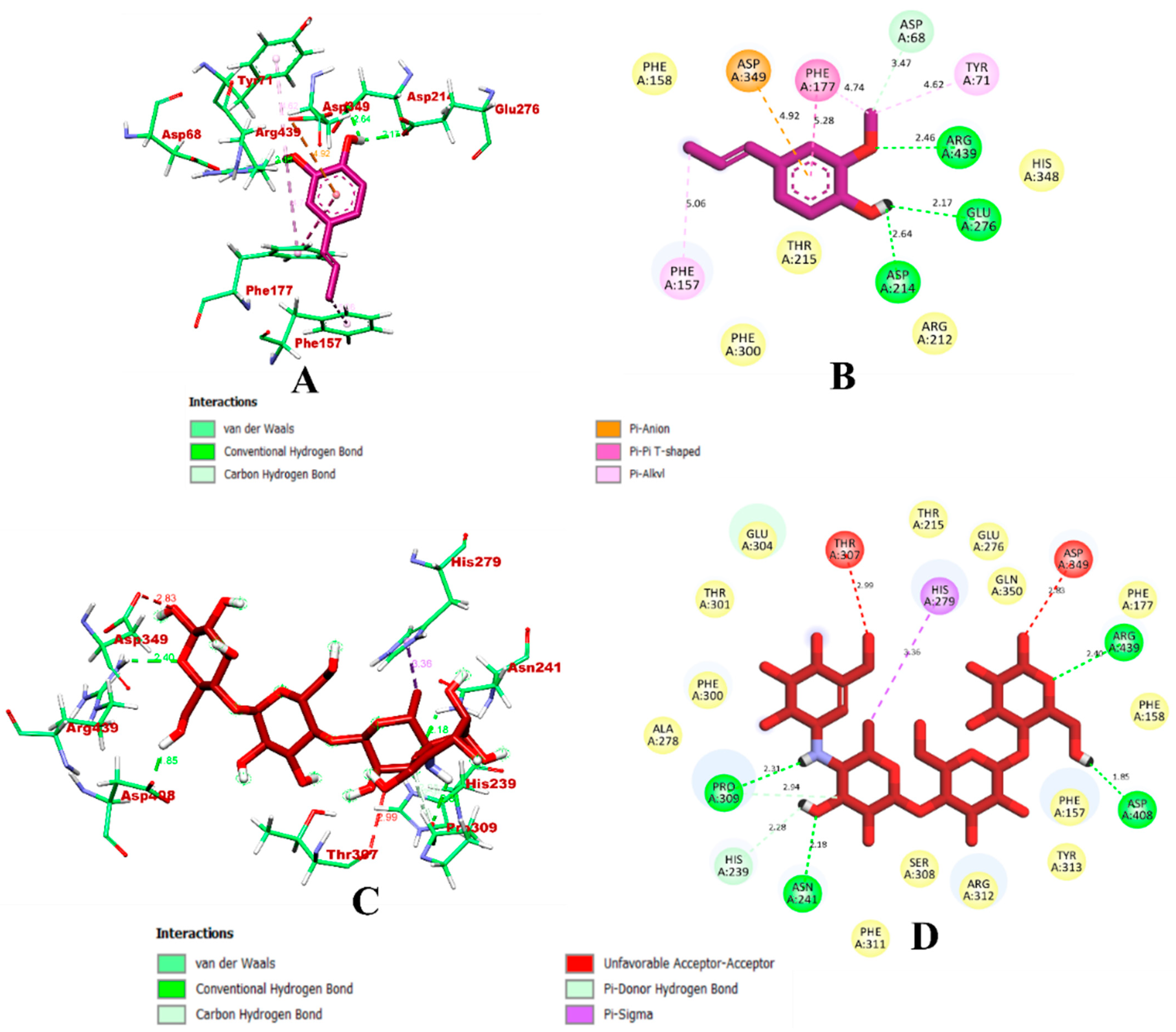
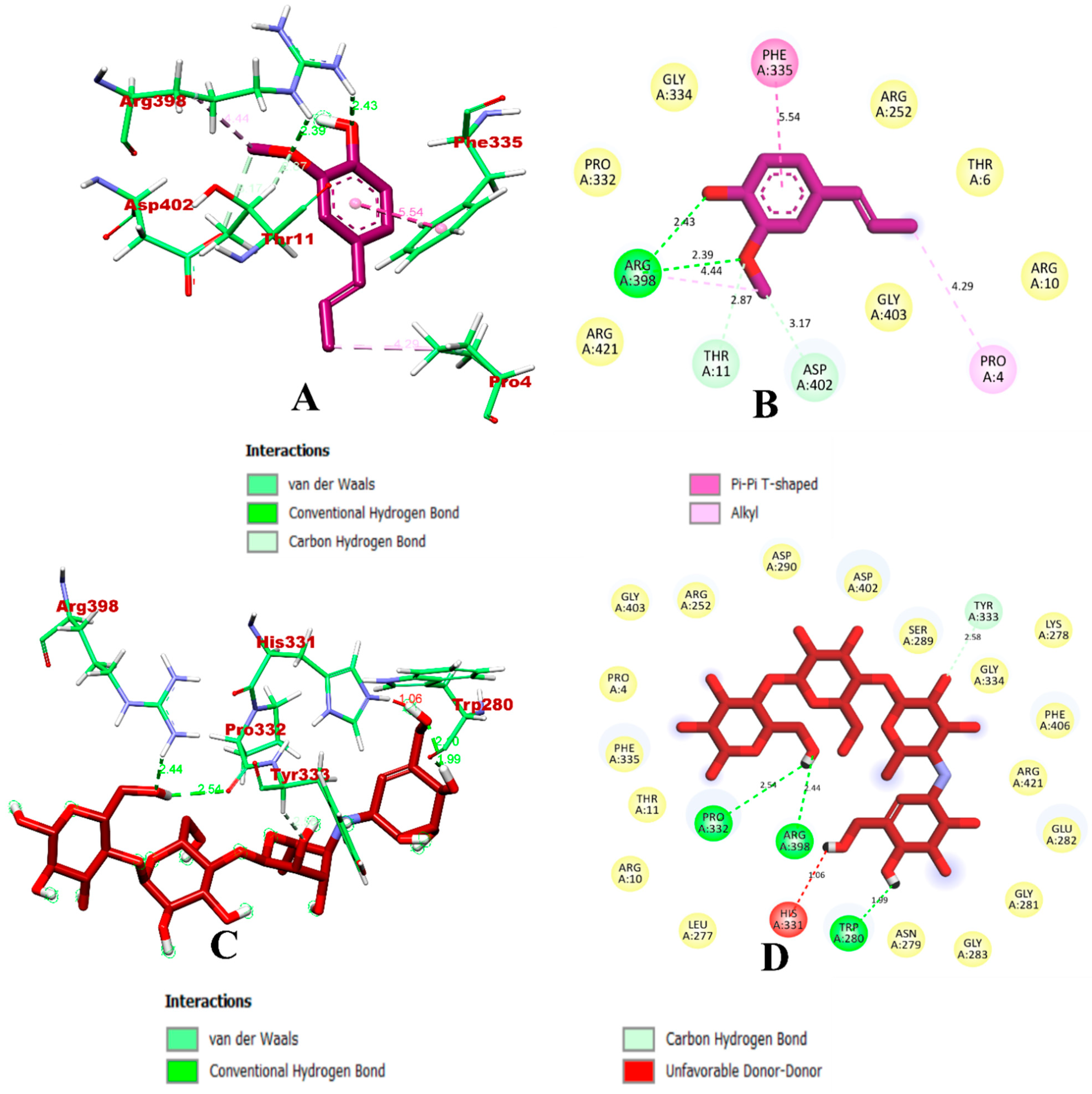
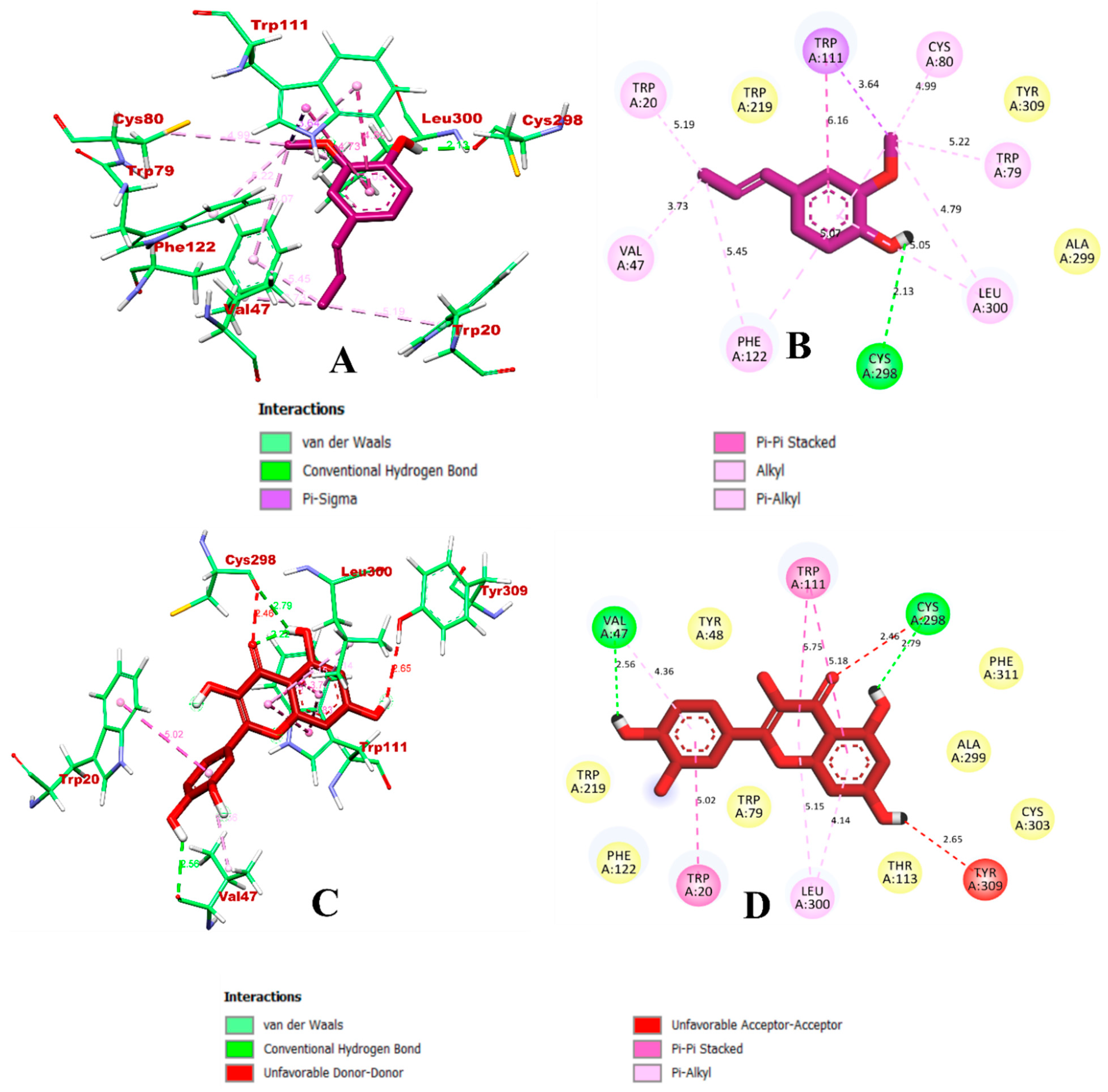
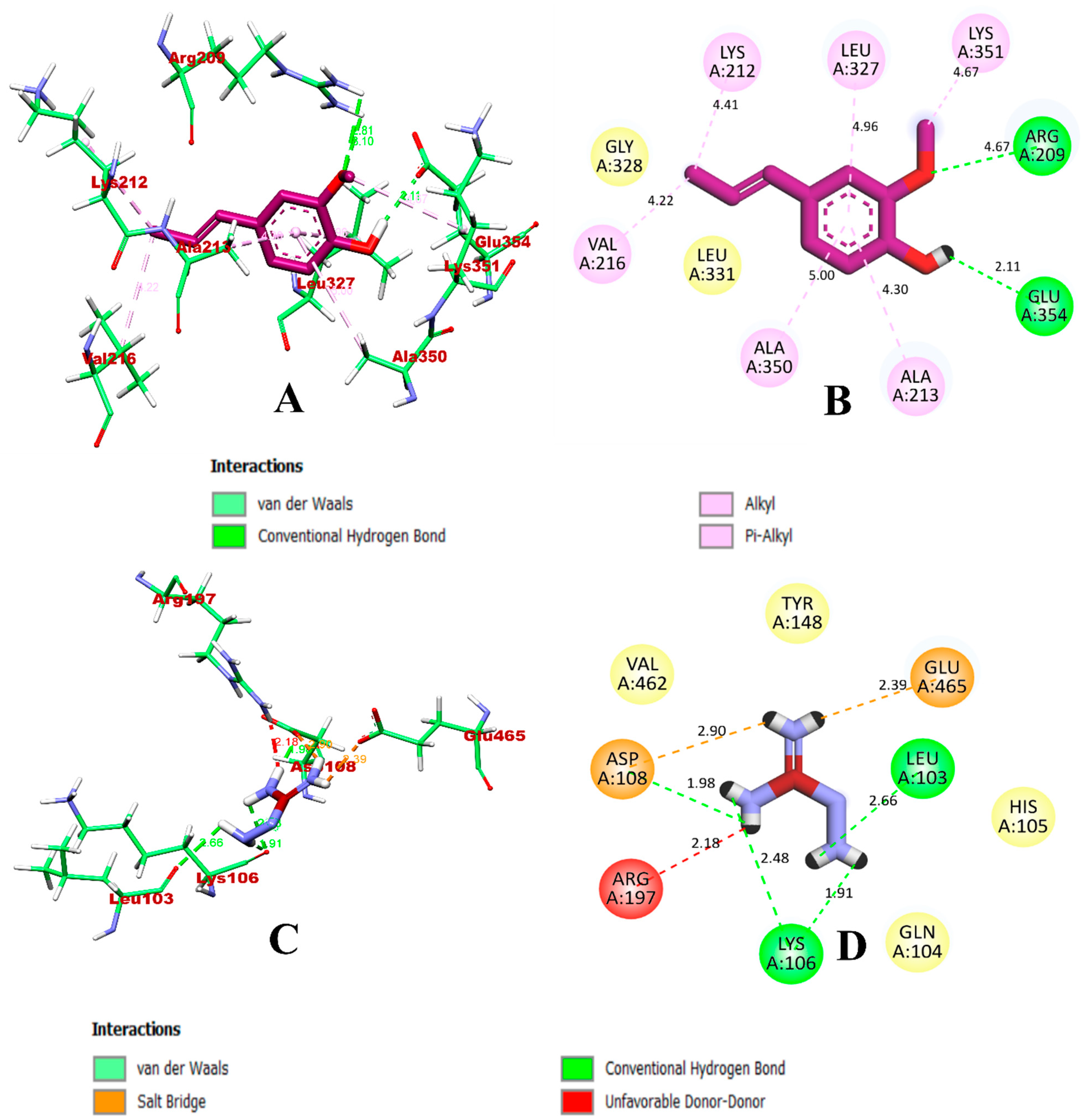
3.1. Virtual Screening through ADMET and Molecular Docking Simulation
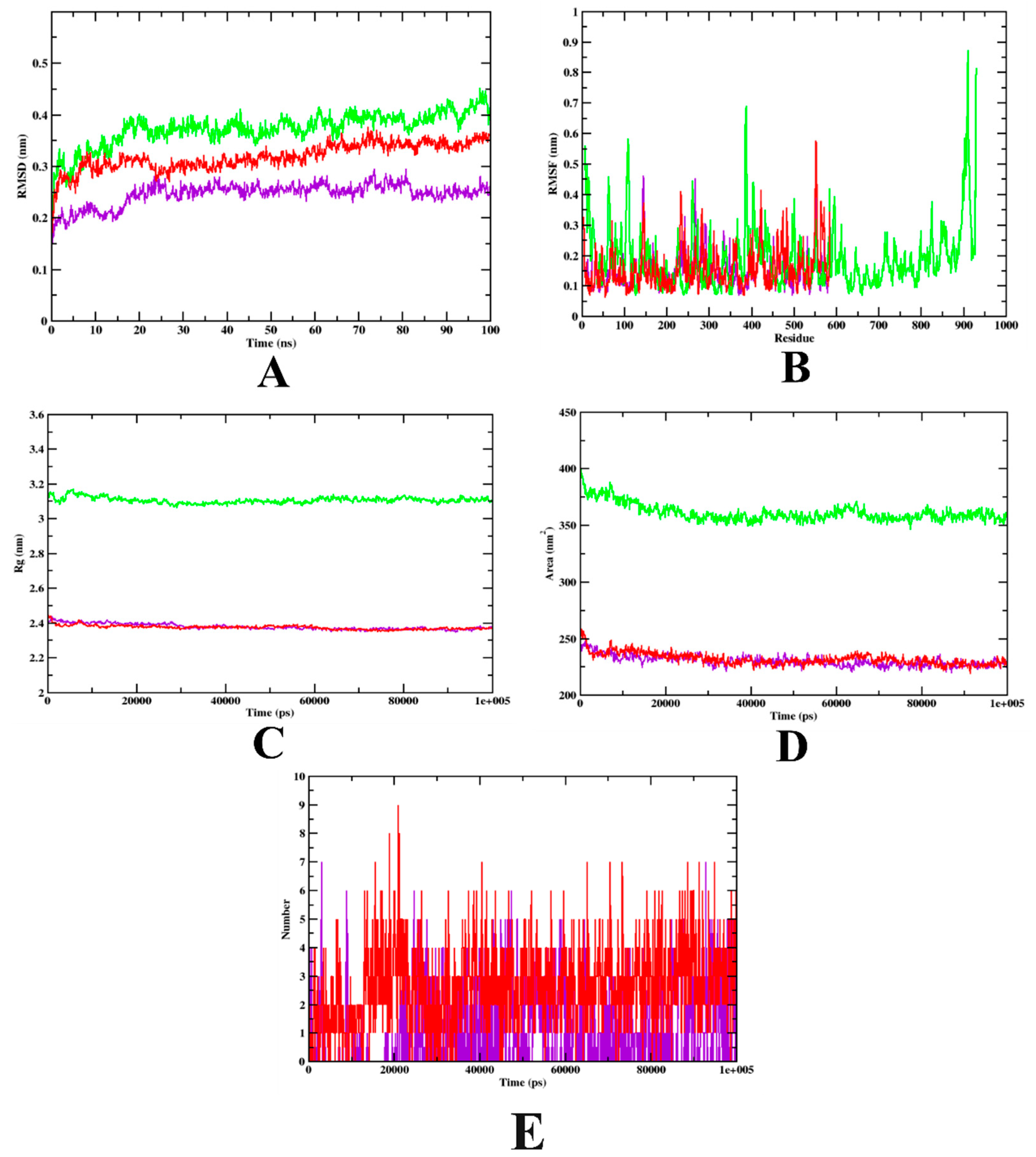
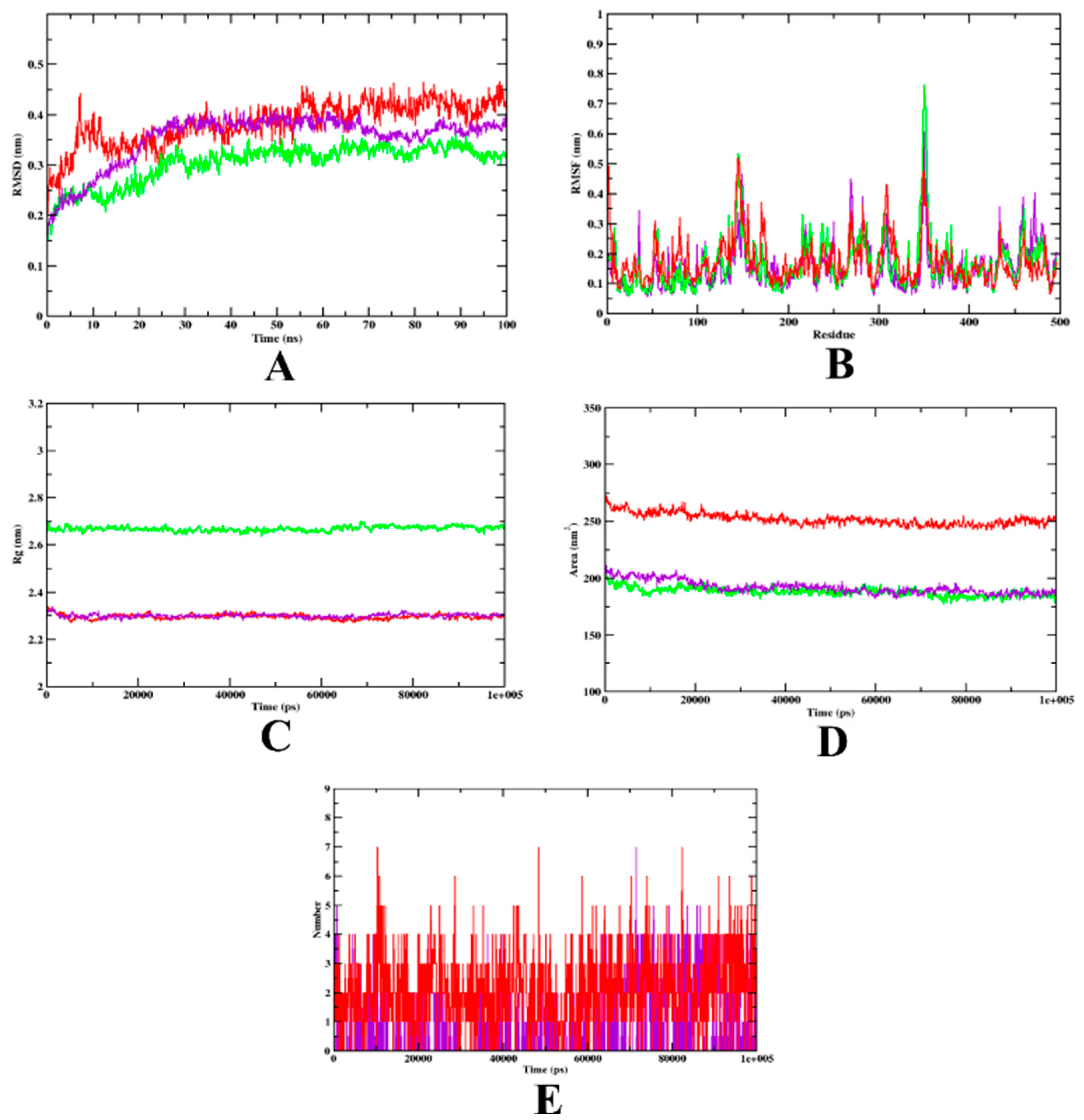
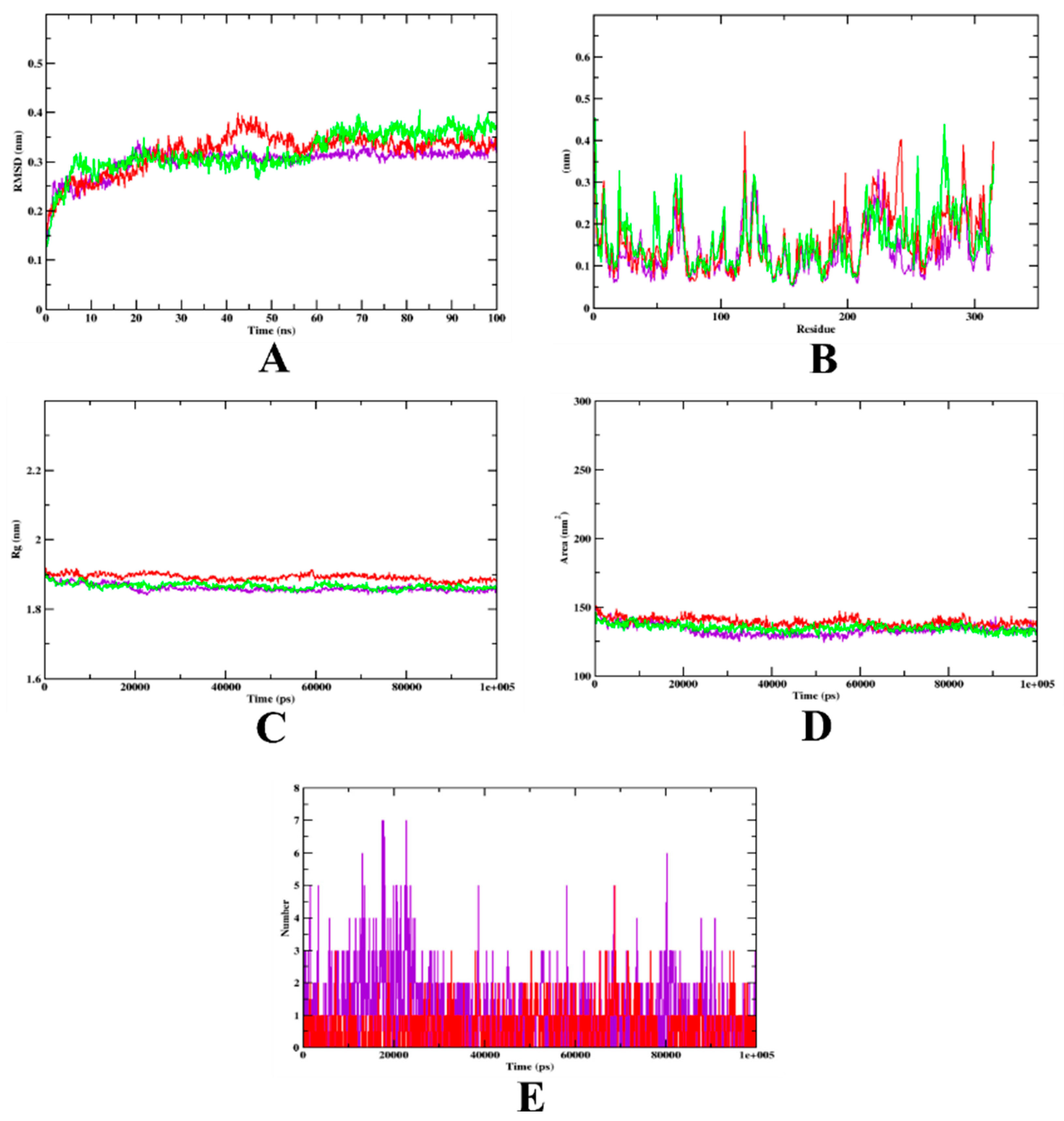
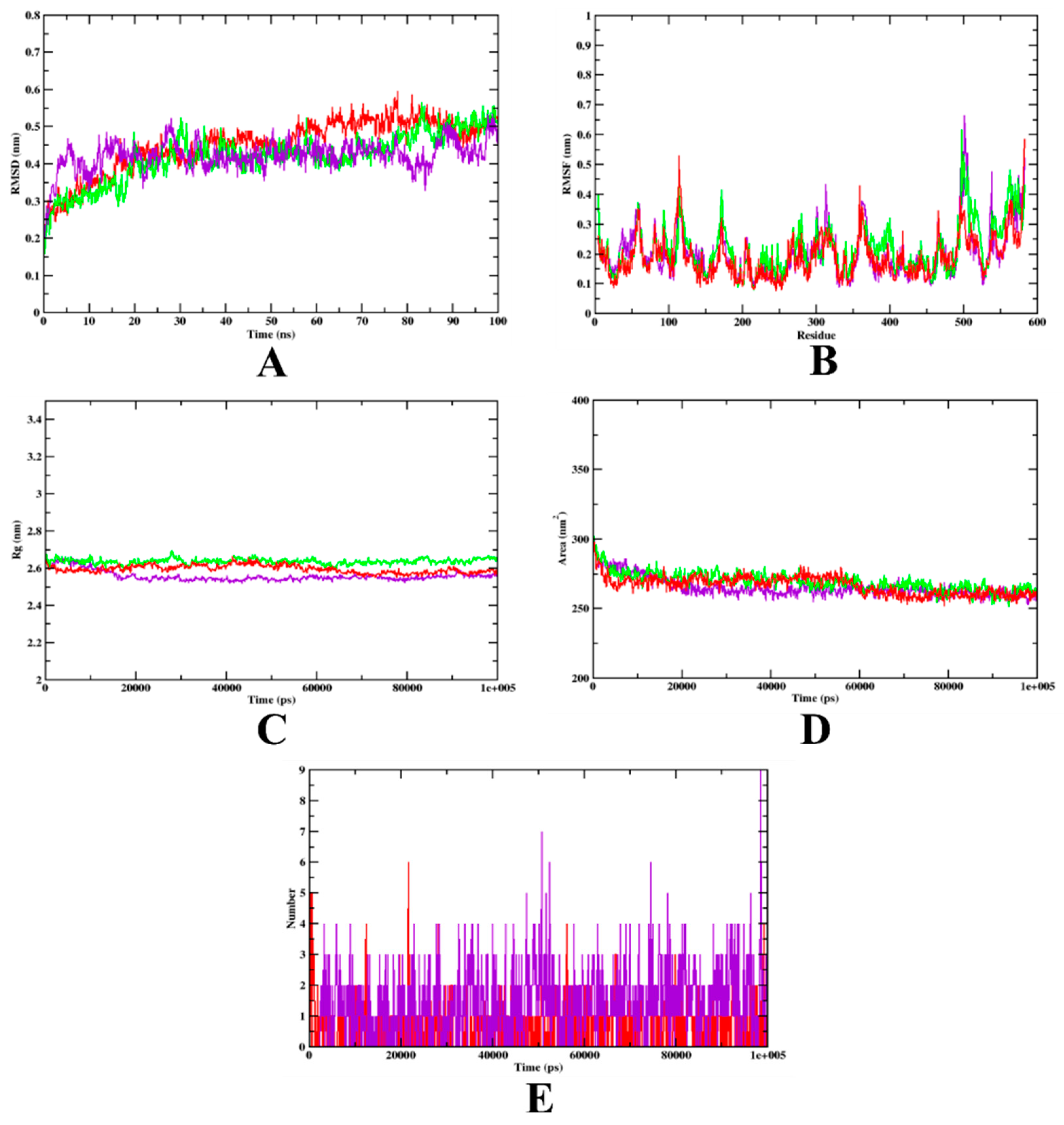
3.2. Molecular Dynamics Simulation
3.3. Binding Free Energy Calculations
3.4. Druglikeliness, Pharmacokinetic, and PASS Analysis of the Representative Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patil, S.M.; Shirahatti, P.S.; Ramu, R. Azadirachta indica A. Juss (neem) against diabetes mellitus: A critical review on its phytochemistry, pharmacology, and toxicology. J. Pharm. Pharmacol. 2022, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Ramu, R.; Shirahatti, P.S.; Zameer, F.; Ranganatha, L.V.; Prasad, M.N. Inhibitory effect of banana (Musa sp. var. Nanjangud rasa bale) flower extract and its constituents Umbelliferone and Lupeol on α-glucosidase, aldose reductase and glycation at multiple stages. S. Afr. J. Bot. 2014, 95, 54–63. [Google Scholar] [CrossRef]
- Ramu, R.; Shirahatti, P.S.; Zameer, F.; Lakkappa Dhananjaya, B.; Prasad, M.N. Assessment of in vivo antidiabetic properties of umbelliferone and lupeol constituents of banana (Musa sp. var. Nanjangud Rasa Bale) flower in hyperglycaemic rodent model. PLoS ONE 2016, 11, e0151135. [Google Scholar]
- Ramu, R.; Shirahatti, P.S.; Th, D.; Bajpe, S.N.; SreepathI, N.; Patil, S.M.; Mn, N.P. Investigating Musa paradisiaca (Var. Nanjangud rasa bale) pseudostem in preventing hyperglycemia along with improvement of diabetic complications. J. Appl. Biol. Biotechnol. 2022, 10, 56–65. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Satish, A.M.; Shbeer, A.M.; Ageel, M.; Al-Ghorbani, M.; Ranganatha, L.; Parameswaran, S.; Ramu, R. Discovery of Novel Coumarin Derivatives as Potential Dual Inhibitors against α-Glucosidase and α-Amylase for the Management of Post-Prandial Hyperglycemia via Molecular Modelling Approaches. Molecules 2022, 27, 3888. [Google Scholar] [CrossRef]
- Tang, W.; Martin, K.A.; Hwa, J. Aldose reductase, oxidative stress, and diabetic mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef]
- Maradesha, T.; Patil, S.M.; Al-Mutairi, K.A.; Ramu, R.; Madhunapantula, S.V.; Alqadi, T. Inhibitory Effect of Polyphenols from the Whole Green Jackfruit Flour against α-Glucosidase, α-Amylase, Aldose Reductase and Glycation at Multiple Stages and Their Interaction: Inhibition Kinetics and Molecular Simulations. Molecules 2022, 27, 1888. [Google Scholar] [CrossRef]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced Glycation End Products and Diabetic Complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.-J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- Hossain, U.; Das, A.K.; Ghosh, S.; Sil, P.C. An overview on the role of bioactive α-glucosidase inhibitors in ameliorating diabetic complications. Food Chem. Toxicol. 2020, 145, 111738. [Google Scholar] [CrossRef]
- Sharma, P.; Hajam, Y.A.; Kumar, R.; Rai, S. Complementary and alternative medicine for the treatment of diabetes and associated complications: A review on therapeutic role of polyphenols. Phytomed. Plus 2021, 2, 100188. [Google Scholar] [CrossRef]
- Mondal, S.; Mirdha, B.R.; Mahapatra, S.C. The science behind sacredness of Tulsi (Ocimum sanctum Linn.). Indian J. Physiol. Pharmacol. 2010, 53, 291–306. [Google Scholar]
- Mohan, L.; Amberkar, M.V.; Kumari, M. Ocimum sanctum linn.(TULSI)-an overview. Int. J. Pharm. Sci. Rev. Res. 2011, 7, 51–53. [Google Scholar]
- Jyothi, M.; Khamees, H.A.; Patil, S.M.; Ramu, R.; Khanum, S.A. Microwave-Assisted Synthesis, Characterization, Docking Studies and Molecular Dynamic of Some Novel Phenyl Thiazole Analogs as Xanthine Oxidase Inhibitor. J. Iran. Chem. Soc. 2022, 19, 3919–3933. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.P.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry And Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef]
- Patil, S.M.; Manu, G.; Shivachandra, J.C.; Kumar, K.A.; Vigneswaran, J.; Ramu, R.; Shirahatti, P.S.; Ranganatha, V.L. Computational screening of benzophenone integrated derivatives (BIDs) targeting the NACHT domain of the potential target NLRP3 inflammasome. Adv. Cancer Biol. Metastasis 2022, 5, 100056. [Google Scholar] [CrossRef]
- Sajal, H.; Patil, S.M.; Raj, R.; Shbeer, A.M.; Ageel, M.; Ramu, R. Computer-aided screening of phytoconstituents from Ocimum tenuiflorum against diabetes mellitus targeting DPP4 inhibition: A combination of molecular docking, molecular dynamics, and pharmacokinetics approaches. Molecules 2022, 27, 5133. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Kumar, B.R.P.; Kumar, N. Evaluation of flavonoids from banana pseudostem and flower (quercetin and catechin) as potent inhibitors of α-glucosidase: An in silico perspective. J. Biomol. Struct. Dyn. 2021, 7, 1–15. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Chandra, J.S.; Ranganatha, L.V. In silico identification of novel benzophenone-coumarin derivatives as SARS-CoV-2 RNAdependent RNA polymerase (RdRp) inhibitors. J. Biomol. Struct. Dyn. 2021, 11, 1–17. [Google Scholar]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. Des. Sel. 1999, 12, 439–446. [Google Scholar] [CrossRef]
- Kumar, V.; Shetty, P.; Chandra, K.S.; Ramu, R.; Patil, S.M.; Baliga, A.; Rai, V.M.; Udupi, V.; Poojary, V.; Poojary, B.; et al. Potential fluorinated anti-MRSA thiazolidinone derivatives with antibacterial, antitubercular activity and molecular docking studies. Chem. Biodivers. 2022, 19, e202100532. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Maruthi, K.R.; Bajpe, N.S.; Vyshali, V.M.; Sushmitha, S.; Chagalamari, A.; Ramith, R. Comparative molecular docking and simulation analysis of molnupiravir and remdesivir with SARS-CoV-2 RNA dependent RNA polymerase (RdRp). Bioinformation 2021, 7, 932–939. [Google Scholar]
- Kumar, V.; Ramu, R.; Shirahatti, P.S.; Kumari, V.C.; Sushma, P.; Mandal, S.P.; Patil, S.M. α-Glucosidase; α-Amylase inhibition; kinetics and docking studies of novel (2-chloro-6-(trifluoromethyl) benzyloxy) arylidene) based rhodanine and rhodanine acetic acid derivatives. Chem. Sel. 2021, 6, 9637–9644. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Patil, S.M.; Sujay, S.; Tejaswini, M.; Sushma, P.; Prithvi, S.; Ramu, R. Bioactive peptides: Its production and potential role on health. Innov. Food Sci. Emerg. Technol. 2020, 7, 167–182. [Google Scholar]
- Patil, S.M.; Al-Mutairi, K.A.; Firdose, N.; Ramu, R.; Martiz, R.M.; Ashwini, P. Pharmacoinformatics based screening discovers swertianolin from Lavandula angustifolia as a novel neuromodulator targeting epilepsy, depression, and anxiety. S. Afr. J. Bot. 2022, 149, 712–730. [Google Scholar] [CrossRef]
- Kumari, V.C.; Patil, S.M.; Shirahatti, P.S.; Sujay, S.; Tejaswini, M.; Ranganatha, L.V.; Jayanthi, M.K.; Ramu, R. The current status and perspectives for the emerging pandemic: COVID-19. Int. J. Pharm. Pharm. Sci. 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Gurupadaswamy, H.D.; Ranganatha, V.L.; Ramu, R.; Patil, S.M.; Khanum, S.A. Competent synthesis of biaryl analogs via asymmetric Suzuki–Miyaura cross-coupling for the development of anti-inflammatory and analgesic agents. J. Iran. Chem. Soc. 2022, 19, 2421–2436. [Google Scholar] [CrossRef]
- Ganavi, D.; Ramu, R.; Kumar, V.; Patil, S.M.; Martiz, R.M.; Shirahatti, P.S.; Sathyanarayana, R.; Poojary, B.; Holla, B.S.; Poojary, V.; et al. In vitro and in silico studies of fluorinated 2,3-disubstituted thiazolidinone-pyrazoles as potential α-amylase inhibitors and antioxidant agents. Arch. Pharm. 2021, 12, e2100342. [Google Scholar] [CrossRef]
- Martiz, R.M.; Patil, S.M.; Abdulaziz, M.; Babalghith, A.; Al-Areefi, M.; Al-Ghorbani, M.; Kumar, J.M.; Prasad, A.; Nagalingaswamy, N.P.M.; Ramu, R. Defining the Role of Isoeugenol from Ocimum tenuiflorum against Diabetes Mellitus-Linked Alzheimer’s Disease through Network Pharmacology and Computational Methods. Molecules 2022, 27, 2398. [Google Scholar] [CrossRef]
- Filimonov, D.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the Biological Activity Spectra of Organic Compounds Using the Pass Online Web Resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Martiz, R.M.; Patil, S.M.; Ramu, R.; Jayanthi, M.K.; Ashwini, P.; Ranganatha, L.V.; Khanum, S.A.; Silina, E.; Stupin, V.; Achar, R.R. Discovery of novel benzophenone integrated derivatives as anti-Alzheimer’s agents targeting presenilin-1 and prese-nilin-2 inhibition: A computational approach. PLoS ONE 2022, 17, e0265022. [Google Scholar] [CrossRef]
- Shivanna, C.; Patil, S.M.; Mallikarjunaswamy, C.; Ramu, R.; Akhileshwari, P.; Nagaraju, L.R.; Sridhar, M.A.; Khanum, S.A.; Ranganatha, V.L.; Silina, E.; et al. Synthesis, characterization, Hirshfeld surface analysis, crystal structure and molecular modeling studies of 1-(4-(Methoxy (phenyl) methyl)-2-methylphenoxy) butan-2-one derivative as a novel α-glucosidase inhibitor. Crystals 2022, 12, 960. [Google Scholar] [CrossRef]
- Lolok, N.; Sumiwi, S.A.; Muhtadi, A.; Susilawati, Y.; Hendriani, R.; Ramadhan, D.S.; Levita, J.; Sahidin, I. Molecular docking and molecular dynamics studies of bioactive compounds contained in noni fruit (Morinda citrifolia L.) against human pancreatic α-amylase. J. Biomol. Struct. Dyn. 2021, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Isogai, H.; Hirayama, N. In Silico Prediction of Interactions between Site II on Human Serum Albumin and Profen Drugs. ISRN Pharm. 2013, 2013, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural Basis of the Drug-binding Specificity of Human Serum Albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Pushpa, V.H.; Jayanthi, M.K.; Rashmi, H.R.; Shivamurthy, V.K.; Patil, S.M.; Shirahatti, P.S.; Ramu, R. New insights on the phytochemical intervention for the treatment of neuropsychiatric disorders using the leaves of Michelia champaca: An in vivo and in silico approach. Pharm Biol. 2022, 60, 1656–1668. [Google Scholar]
- Tanawattanasuntorn, T.; Thongpanchang, T.; Rungrotmongkol, T.; Hanpaibool, C.; Graidist, P.; Tipmanee, V. (−)-Kusunokinin as a Potential Aldose Reductase Inhibitor: Equivalency Observed via AKR1B1 Dynamics Simulation. ACS Omega 2020, 6, 606–614. [Google Scholar] [CrossRef]
- Soltanabadi, O.; Atri, M.S.; Bagheri, M. Spectroscopic analysis, docking and molecular dynamics simulation of the interaction of cinnamaldehyde with human serum albumin. J. Incl. Phenom. Macrocycl. Chem. 2018, 91, 189–197. [Google Scholar] [CrossRef]
- Radibratovic, M.; Minic, S.; Stanic-Vucinic, D.; Nikolic, M.; Milcic, M.; Velickovic, T.C. Stabilization of Human Serum Albumin by the Binding of Phycocyanobilin, a Bioactive Chromophore of Blue-Green Alga Spirulina: Molecular Dynamics and Experimental Study. PLoS ONE 2016, 11, e0167973. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, D.; Xu, F.; Pan, Q.; Chai, P.; Liu, B.; Chen, C. The binding affinity of human serum albumin and paclitaxel through MMPBSA based on docked complex. Mol. Simul. 2016, 42, 1460–1467. [Google Scholar] [CrossRef]
- Cyndya, Shibao, Acarbose. In Primer on the Autonomic Nervous System, 3rd ed.; Robertson, D.; Biaggioni, I.; Burnstock, G.; Phillip, A.L.; Julian, F.R.P. (Eds.) Academic Press: London, UK, 2012; pp. 639–641. [Google Scholar]
- Rosak, C.; Mertes, G. Critical evaluation of the role of acarbose in the treatment of diabetes: Patient considerations. Diabetes Metab. Syndr. Obes. Targets Ther. 2012, 5, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Dabeek, W.M.; Marra, M.V. Dietary Quercetin and Kaempferol: Bioavailability and Potential Cardiovascular-Related Bioactivity in Humans. Nutrients 2019, 11, 2288. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, G. Toxicology of acarbose, with special reference to long-term carcinogenicity studies. In Acarbose for the Treatment of Diabetes Mellitus, 1st ed.; Creutzfeldt, W., Ed.; Springer: Berlin, Germany, 1988; pp. 5–14. [Google Scholar]
- Chen, R.; Lin, J.; Hong, J.; Han, D.; Zhang, A.D.; Lan, R.; Fu, L.; Wu, Z.; Lin, J.; Zhang, W.; et al. Potential toxicity of quercetin: The repression of mitochondrial copy number via decreased POLG expression and excessive TFAM expression in irradiated murine bone marrow. Toxicol. Rep. 2014, 1, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Ramu, R.; Patil, S.M. A Perspective on the Effective Conduction of Functional-based Coaching Program on Diabetic Indonesian Communities. Oman Med. J. 2021, 36, e281. [Google Scholar] [CrossRef]
- Patil, S.M.; Shirahatti, P.S.; Ramu, R. Azadirachta indica A. Juss (neem) as a contraceptive: An evidence-based review on its pharmacological efficiency. Phytomedicine 2021, 88, 153596. [Google Scholar] [CrossRef]
- Patil, S.M.; Ramu, R.; Shirahatti, P.S.; Shivamallu, C.; Amachawadi, R.G. A systematic review on ethnopharmacology, phytochemistry and pharmacological aspects of Thymus vulgaris Linn. Heliyon 2021, 7, e07054. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
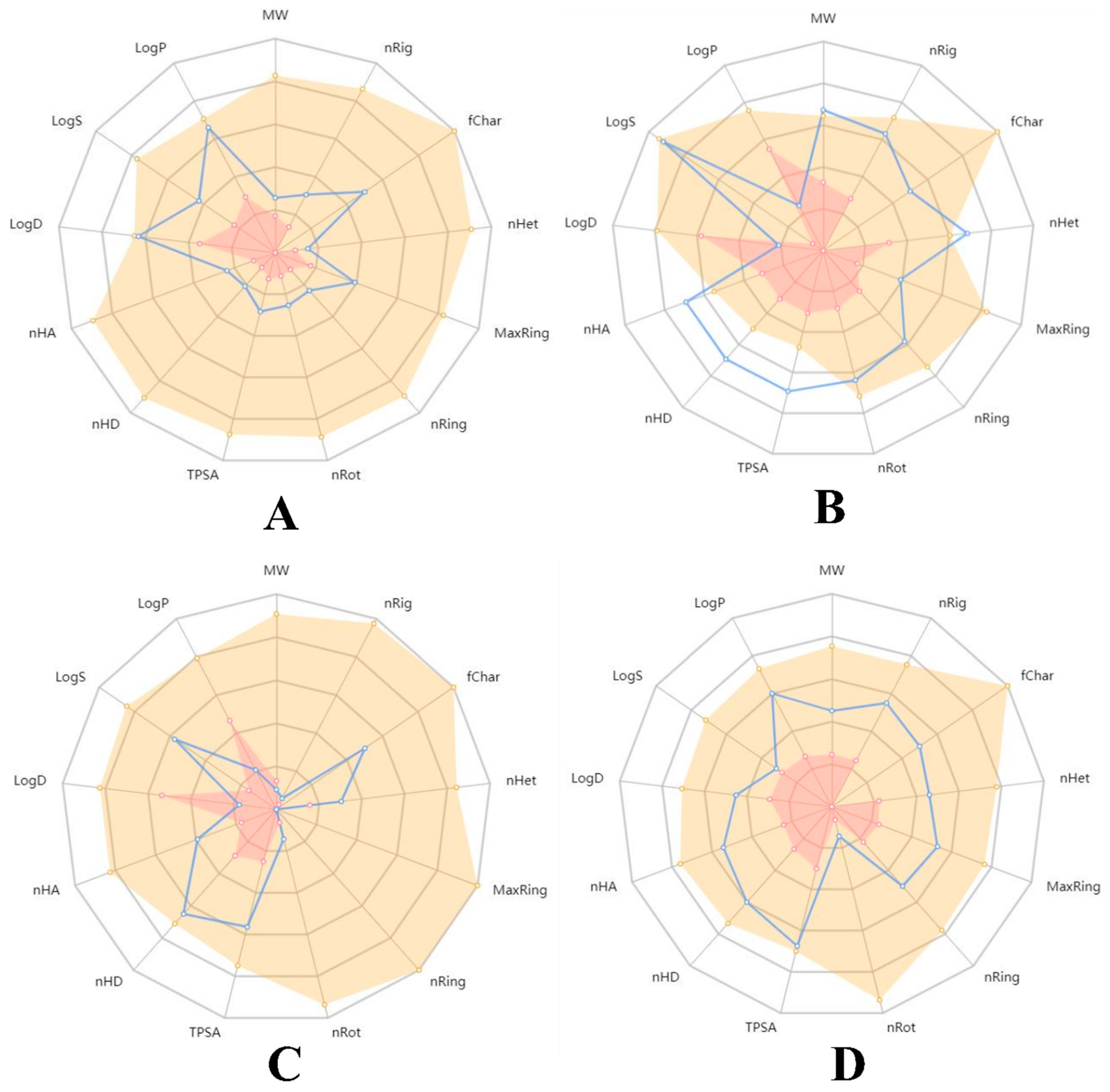{kind=link}
| Enzyme Targets | Coordinates of the Grid Box | Size of the Grid Box | ||
|---|---|---|---|---|
| x | y | z | ||
| α-glucosidase | −17.48 Å | −8.62 Å | −19.65 Å | 40 Å × 40 Å × 40 Å |
| α-amylase | 103.46 Å | 37.17 Å | 19.60 Å | 40 Å × 40 Å × 40 Å |
| HSA | 8.24 Å | 2.58 Å | −14.75 Å | 40 Å × 40 Å × 40 Å |
| HAR | −5.06 Å | 0.19 Å | 9.94 Å | 40 Å × 40 Å × 40 Å |
| Sl. No | Compound Names | OB (OB ≥ 30%) | BBB | DHL (HL < 3 h) | LR5 | IEP (Caco-2 Cells) | DILI | CL (CL > 15 mL/min/kg) | MW (100~600) | HBA (0~12) | HBD (0~7) | TPSA (0~140) | PAINS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | (−)-Alloaromadendrene | Pass | Pass | 0.671 | Accepted | −4.711 | Negative | 5.356 | 212.150 | 4 | 2 | 58.200 | 0 |
| 2 | (−)-Camphene | Pass | Pass | 0.013 | Accepted | −4.756 | Negative | 16.686 | 414.390 | 1 | 1 | 20.230 | 0 |
| 3 | (−)-Linalool | Pass | Pass | 0.040 | Accepted | −4.577 | Negative | 13.563 | 204.190 | 0 | 0 | 0.000 | 0 |
| 4 | (+)-α-Phellandrene | Pass | Pass | 0.077 | Accepted | −4.463 | Negative | 9.346 | 136.130 | 0 | 0 | 0.000 | 0 |
| 5 | (+)-Endo-β-bergamotene | Pass | Pass | 0.493 | Accepted | −4.375 | Negative | 9.738 | 154.140 | 1 | 1 | 20.230 | 0 |
| 6 | (1S)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-one | Pass | Pass | 0.617 | Accepted | −4.383 | Negative | 12.660 | 136.130 | 0 | 0 | 0.000 | 0 |
| 7 | (1S,2R,4S)-(−)-Bornyl acetate | Pass | Pass | 0.063 | Accepted | −4.466 | Negative | 16.946 | 204.190 | 0 | 0 | 0.000 | 0 |
| 8 | (E)-β-ocimene | Pass | Pass | 0.243 | Accepted | −4.552 | Moderate | 6.063 | 196.150 | 2 | 0 | 26.300 | 0 |
| 9 | 1S-α-Pinene | Pass | Pass | 0.701 | Accepted | −4.582 | Negative | 13.808 | 152.120 | 1 | 0 | 17.070 | 0 |
| 10 | 2,3-Dimethylaniline | Pass | Pass | 0.678 | Accepted | −4.434 | Negative | 14.171 | 136.130 | 0 | 0 | 0.000 | 0 |
| 11 | 3-Carene | Pass | Pass | 0.114 | Accepted | −4.303 | Negative | 15.022 | 136.130 | 0 | 0 | 0.000 | 0 |
| 12 | 4-Terpineol | Pass | Pass | 0.583 | Accepted | −4.255 | Negative | 10.496 | 121.090 | 1 | 2 | 26.020 | 0 |
| 13 | Acetyleugenol | Pass | Pass | 0.132 | Accepted | −4.307 | Negative | 16.061 | 136.130 | 0 | 0 | 0.000 | 0 |
| 14 | α-Fenchene | Pass | Pass | 0.447 | Accepted | −4.217 | Negative | 14.345 | 154.140 | 1 | 1 | 20.230 | 0 |
| 15 | α-Terpineol | Pass | Pass | 0.843 | Accepted | −4.453 | Moderate | 8.457 | 206.090 | 3 | 0 | 35.530 | 0 |
| 16 | β-caryophyllene | Pass | Pass | 0.099 | Accepted | −4.460 | Negative | 10.559 | 136.130 | 0 | 0 | 0.000 | 0 |
| 17 | β-Pinene | Pass | Pass | 0.527 | Accepted | −4.193 | Negative | 8.942 | 154.140 | 1 | 1 | 20.230 | 0 |
| 18 | Cis-Anethole | Pass | Pass | 0.048 | Accepted | −4.517 | Negative | 9.943 | 204.190 | 0 | 0 | 0.000 | 0 |
| 19 | Cyclo(L-Val-L-Leu) | Pass | Pass | 0.107 | Accepted | −4.460 | Negative | 10.097 | 136.130 | 0 | 0 | 0.000 | 0 |
| 20 | Dehydro-p-cymene | Pass | Moderate | 0.638 | Accepted | −4.440 | Negative | 11.146 | 148.090 | 1 | 0 | 9.230 | 0 |
| 21 | Eucalyptol | Pass | Pass | 0.568 | Accepted | −4.344 | Moderate | 10.755 | 132.090 | 0 | 0 | 0.000 | 0 |
| 22 | γ-Selinene | Pass | Pass | 0.352 | Accepted | −4.414 | Negative | 8.066 | 154.140 | 1 | 0 | 9.230 | 0 |
| 23 | Geranyl acetate | Pass | Pass | 0.088 | Accepted | −4.577 | Negative | 13.350 | 204.190 | 0 | 0 | 0.000 | 0 |
| 24 | Isoeugenol | Pass | Pass | 0.506 | Accepted | −4.420 | Moderate | 9.707 | 196.150 | 2 | 0 | 26.300 | 0 |
| 25 | Myrcene | Pass | Moderate | 0.880 | Accepted | −4.579 | Negative | 13.435 | 164.080 | 2 | 1 | 29.460 | 0 |
| 26 | Phytosterols | Pass | Pass | 0.453 | Accepted | −4.402 | Moderate | 13.108 | 136.130 | 0 | 0 | 0.000 | 0 |
| 27 | Acarbose | Fail | Fail | 0.546 | Rejected | −6.149 | Positive | 0.373 | 645.250 | 19 | 14 | 321.170 | 0 |
| 28 | Aminoguanidine | Pass | Moderate | 0.714 | Accepted | 5.448 | Negative | 5.857 | 74.060 | 4 | 6 | 87.920 | 0 |
| 29 | Quercetin | Fail | Pass | 0.929 | Accepted | −5.204 | Positive | 8.284 | 302.040 | 7 | 8 | 131.360 | 1 |
| Sl. No. | Compound Names | Binding Affinity (kcal/mol) | Total No. of Intermolecular Interactions | Total No. of Hydrogen Bonds | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AG | AM | HSA | HAR | AG | AM | HSA | HAR | AG | AM | HSA | HAR | ||
| 1 | (−)-Alloaromadendrene | −7.3 | −6.8 | −6.3 | −5.1 | 2 | 7 | 5 | 8 | - | - | - | - |
| 2 | (−)-Camphene | −5.3 | −5.4 | −5.7 | −6.2 | 3 | 3 | 3 | 7 | - | - | - | - |
| 3 | (−)-Linalool | −5.8 | −4.9 | −5.2 | −6.2 | 10 | 7 | 6 | 9 | 3 | - | - | - |
| 4 | (+)-α-Phellandrene | −5.8 | −5.6 | −5.9 | −6.5 | 5 | 4 | 6 | 7 | - | - | - | - |
| 5 | (+)-Endo-β-bergamotene | −7.3 | −6.1 | −6.2 | −7.3 | 6 | 5 | 6 | 7 | - | - | - | - |
| 6 | (1S)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-one | −5.9 | −5.5 | −5.7 | −6.3 | 2 | 4 | 3 | 3 | - | 2 | - | - |
| 7 | (1S,2R,4S)-(−)-Bornyl acetate | −6.9 | −5.7 | −6.4 | −6.4 | 2 | 3 | 3 | 3 | 1 | - | 1 | 2 |
| 8 | (E)-β-ocimene | −5.7 | −5.0 | −5.8 | −6.4 | 8 | 6 | 6 | 11 | - | - | - | - |
| 9 | 1S-α-Pinene | −5.5 | −5.4 | −6.2 | −6.1 | 4 | 9 | 6 | 9 | - | - | - | - |
| 10 | 2,3-Dimethylaniline | −5.1 | −5.3 | −5.6 | −5.9 | 3 | 5 | 7 | 3 | 3 | 3 | 1 | - |
| 11 | 3-Carene | −5.4 | −5.5 | −6.0 | −6.5 | 3 | 4 | 6 | 6 | - | - | - | - |
| 12 | 4-Terpineol | −5.8 | −5.7 | −6.2 | −6.2 | 6 | 3 | 5 | 7 | 1 | - | - | - |
| 13 | Acetyleugenol | −6.3 | −5.7 | −6.5 | −6.6 | 7 | 4 | 8 | 6 | 2 | 1 | 2 | 1 |
| 14 | α-Fenchene | −5.6 | −5.3 | −5.8 | −6.1 | 4 | 5 | 4 | 8 | - | - | - | - |
| 15 | α-Terpineol | −6.2 | −6.0 | −6.2 | −6.5 | 5 | 5 | 5 | 7 | 2 | - | - | - |
| 16 | β-caryophyllene | −7.3 | −6.0 | −6.1 | −7.0 | 1 | 1 | 2 | 3 | - | - | - | - |
| 17 | β-Pinene | −5.4 | −5.6 | −6.3 | −6.1 | 3 | 5 | 6 | 8 | - | - | - | - |
| 18 | Cis-Anethole | −5.7 | −5.4 | −6.3 | −5.9 | 7 | 4 | 6 | 6 | - | - | - | - |
| 19 | Cyclo(L-Val-L-Leu) | −6.6 | −5.6 | −6.4 | −6.3 | 4 | 3 | 1 | 1 | - | - | - | - |
| 20 | Dehydro-p-cymene | −6.0 | −5.8 | −6.1 | −6.8 | 5 | 6 | 7 | 10 | - | - | - | - |
| 21 | Eucalyptol | −5.5 | −5.3 | −6.2 | −6.2 | 1 | 5 | 4 | 7 | 1 | - | 2 | - |
| 22 | γ-Selinene | −7.3 | −6.7 | −6.8 | −7.4 | 1 | 4 | 5 | 8 | - | - | 1 | 1 |
| 23 | Geranyl acetate | −6.3 | −5.3 | −6.3 | −7.0 | 7 | 7 | 5 | 7 | 2 | 2 | 2 | - |
| 24 | Isoeugenol | −7.6 | −6.9 | −6.8 | −7.4 | 9 | 7 | 8 | 11 | 4 | 4 | 2 | 1 |
| 25 | Myrcene | −5.6 | −4.9 | −5.9 | −6.2 | 8 | 6 | 9 | 9 | - | - | - | - |
| 26 | Phytosterols | −7.2 | −6.6 | −6.8 | −4.0 | 6 | 7 | 3 | 2 | - | - | - | - |
| 27 | Acarbose | −8.2 | −7.4 | - | - | 7 | 4 | - | - | 6 | 4 | - | - |
| 28 | Aminoguanidine | - | - | −8.0 | - | - | - | 7 | - | - | - | 3 | - |
| 29 | Quercetin | - | - | - | −7.8 | - | - | - | 8 | - | - | - | 1 |
| MD Trajectory Values | Apo-Protein | Protein-Acarbose Complex | Protein-Isoeugenol Complex |
|---|---|---|---|
| RMSD | 0.30–0.40 nm | 0.25–0.32 nm | 0.20–0.25 nm |
| Rg | 3.10–3.14 nm | 2.39–2.45 nm | 2.39–2.45 nm |
| SASA | 350–370 nm2 | 240–250 nm2 | 240–250 nm2 |
| Ligand H-bonds | - | 9 | 7 |
| MD Trajectory Values | Apo-Protein | Protein-Acarbose Complex | Protein-Isoeugenol Complex |
|---|---|---|---|
| RMSD | 0.20–0.30 nm | 0.25–0.32 nm | 0.20–0.25 nm |
| Rg | 3.10–3.14 nm | 2.39–2.45 nm | 2.39–2.45 nm |
| SASA | 350–370 nm2 | 240–250 nm2 | 240–250 nm2 |
| Ligand H-bonds | - | 7 | 7 |
| MD Trajectory Values | Apo-Protein | Protein-Quercetin Complex | Protein-Isoeugenol Complex |
|---|---|---|---|
| RMSD | 0.20–0.36 nm | 0.20–0.35 nm | 0.20–0.30 nm |
| Rg | 1.70–1.90 nm | 1.90–1.92 nm | 1.70–1.90 nm |
| SASA | 140–150 nm2 | 140–150 nm2 | 140–150 nm2 |
| Ligand H-bonds | - | 5 | 7 |
| MD Trajectory Values | Apo-Protein | Protein-Aminoguanidine Complex | Protein-Isoeugenol Complex |
|---|---|---|---|
| RMSD | 0.25–0.45 nm | 0.25–0.45 nm | 0.25–0.45 nm |
| Rg | 2.60–2.61 nm | 2.60–2.61 nm | 2.58–2.60 nm |
| SASA | 270–280 nm2 | 270–280 nm2 | 270–280 nm2 |
| Ligand H-bonds | - | 6 | 7 |
| Protein-Ligand Complexes | Types of Binding Free Energies | ||||
|---|---|---|---|---|---|
| Van Der Waal’s Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | SASA Energy (kJ/mol) | Binding Energy (kJ/mol) | |
| α-Glucosidase-isoeugenol | −224.811 | −10.382 | 83.618 | −26.746 | −186.222 |
| α-Glucosidase-acarbose | −134.192 | −4.813 | 62.125 | −9.310 | −90.102 |
| α-Amylase-isoeugenol | −218.568 | −29.891 | 62.172 | −21.886 | −180.194 |
| α-Amylase-acarbose | −130.161 | −2.106 | 39.340 | −9.564 | −87.109 |
| HSA-isoeugenol | −189.601 | −15.288 | 56.9638 | −9.149 | −98.169 |
| HSA-aminoguanidine | −150.719 | −5.127 | 47.498 | −7.981 | −81.872 |
| HAR-isoeugenol | −184.951 | −10.105 | 87.107 | −25.191 | −99.171 |
| HAR-quercetin | −171.669 | −3.291 | 81.102 | −8.781 | −85.768 |
| Properties | Isoeugenol | Acarbose | Aminoguanidine | Quercetin |
|---|---|---|---|---|
| Oral bioavailability (OB ≥ 30%) | Pass | Fail | Pass | Fail |
| Blood–brain barrier (BBB) | Moderate | Fail | Moderate | Pass |
| Drug half-life (HL < 3 h) | 0.880 | 0.546 | 0.714 | 0.929 |
| Lipinski’s rule (LR) of five | Accepted | Rejected | Accepted | Accepted |
| Intestinal epithelial permeability (Caco-2 cells) | −4.579 | −6.149 | 5.448 | −5.204 |
| Drug-induced liver injury (DILI) | Negative | Positive | Negative | Positive |
| Clearness (CL > 15 mL/min/kg) | 13.435 | 0.373 | 5.857 | 8.284 |
| Molecular weight (MW 100~600) | 164.080 | 645.250 | 74.060 | 302.040 |
| Hydrogen bond acceptor (0~12) | 2 | 19 | 4 | 7 |
| Hydrogen bond donor (0~7) | 1 | 14 | 6 | 8 |
| TPSA (0~140) | 29.460 | 321.170 | 87.920 | 131.360 |
| PAINS | 0 | 0 | 0 | 1 |
| Compounds | α-Glucosidase | α-Amylase | AGE-Related Disorder | HAR | ||||
|---|---|---|---|---|---|---|---|---|
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | |
| Isoeugenol | 0.107 | 0.025 | 0.184 | 0.065 | 0.371 | 0.011 | 0.064 | 0.041 |
| Acarbose | 0.451 | 0.009 | 0.943 | 0.000 | - | - | - | - |
| Aminoguanidine | - | - | - | - | - | - | - | - |
| Quercetin | 0.139 | 0.058 | - | - | - | - | 0.466 | 0.003 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martiz, R.M.; Patil, S.M.; Thirumalapura Hombegowda, D.; Shbeer, A.M.; Alqadi, T.; Al-Ghorbani, M.; Ramu, R.; Prasad, A. Phyto-Computational Intervention of Diabetes Mellitus at Multiple Stages Using Isoeugenol from Ocimum tenuiflorum: A Combination of Pharmacokinetics and Molecular Modelling Approaches. Molecules 2022, 27, 6222. https://doi.org/10.3390/molecules27196222
Martiz RM, Patil SM, Thirumalapura Hombegowda D, Shbeer AM, Alqadi T, Al-Ghorbani M, Ramu R, Prasad A. Phyto-Computational Intervention of Diabetes Mellitus at Multiple Stages Using Isoeugenol from Ocimum tenuiflorum: A Combination of Pharmacokinetics and Molecular Modelling Approaches. Molecules. 2022; 27(19):6222. https://doi.org/10.3390/molecules27196222
Chicago/Turabian StyleMartiz, Reshma Mary, Shashank M. Patil, Deepika Thirumalapura Hombegowda, Abdullah M. Shbeer, Taha Alqadi, Mohammed Al-Ghorbani, Ramith Ramu, and Ashwini Prasad. 2022. "Phyto-Computational Intervention of Diabetes Mellitus at Multiple Stages Using Isoeugenol from Ocimum tenuiflorum: A Combination of Pharmacokinetics and Molecular Modelling Approaches" Molecules 27, no. 19: 6222. https://doi.org/10.3390/molecules27196222
APA StyleMartiz, R. M., Patil, S. M., Thirumalapura Hombegowda, D., Shbeer, A. M., Alqadi, T., Al-Ghorbani, M., Ramu, R., & Prasad, A. (2022). Phyto-Computational Intervention of Diabetes Mellitus at Multiple Stages Using Isoeugenol from Ocimum tenuiflorum: A Combination of Pharmacokinetics and Molecular Modelling Approaches. Molecules, 27(19), 6222. https://doi.org/10.3390/molecules27196222