

Recent Advances in Natural Product-Based Hybrids as Anti-Cancer Agents

Abstract
:1. Introduction
2. Hybrids of Natural Products with Small Molecules
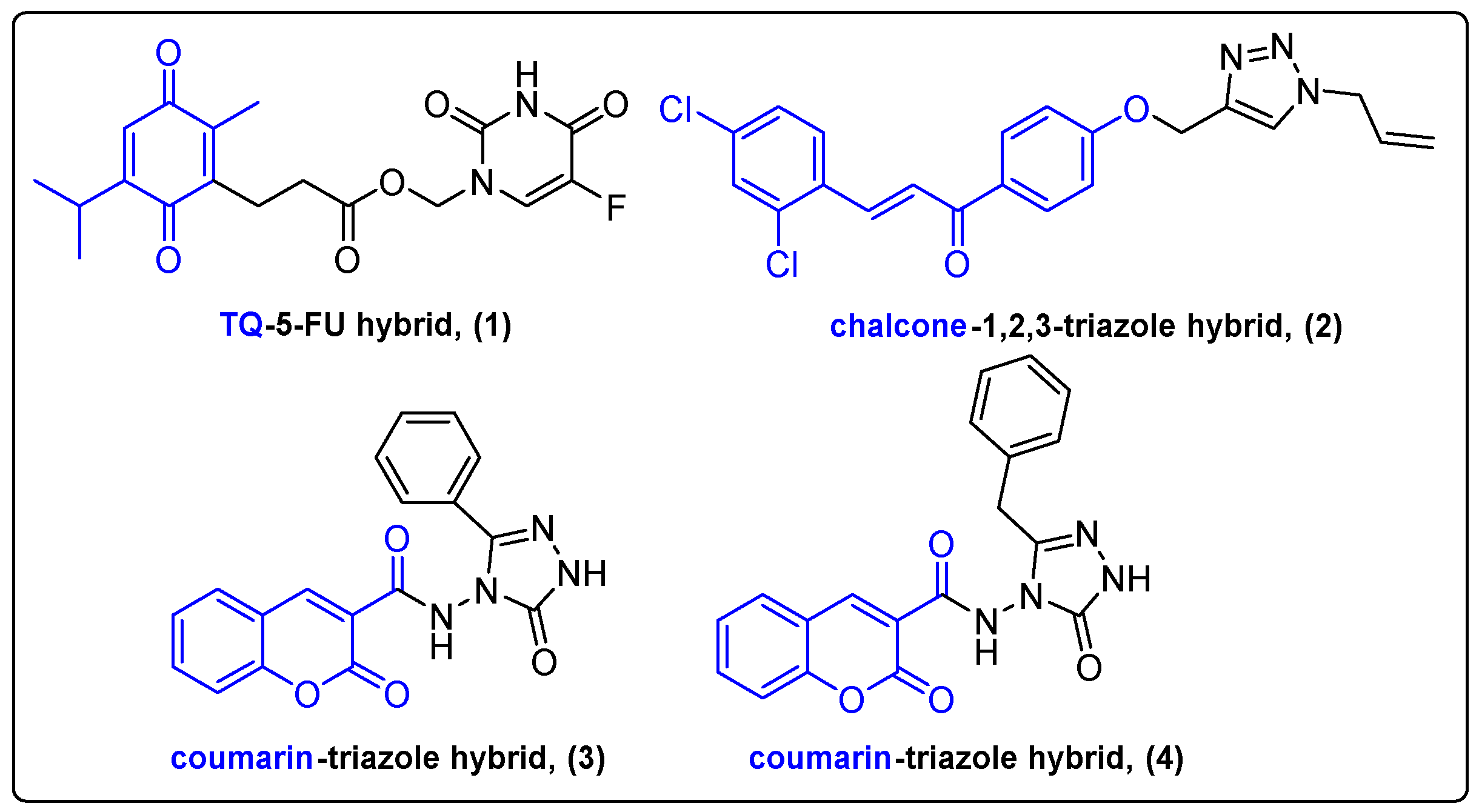
2.1. Hybrids of Natural Products with Heterocyclic Compounds
2.2. Hybrids of Natural Products with Phenyl Moieties
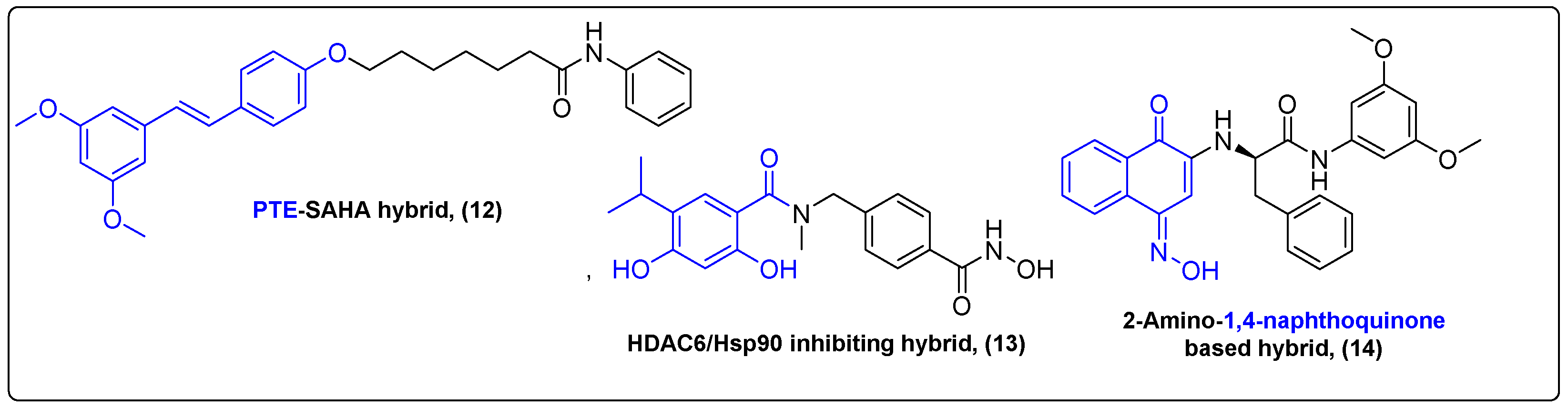
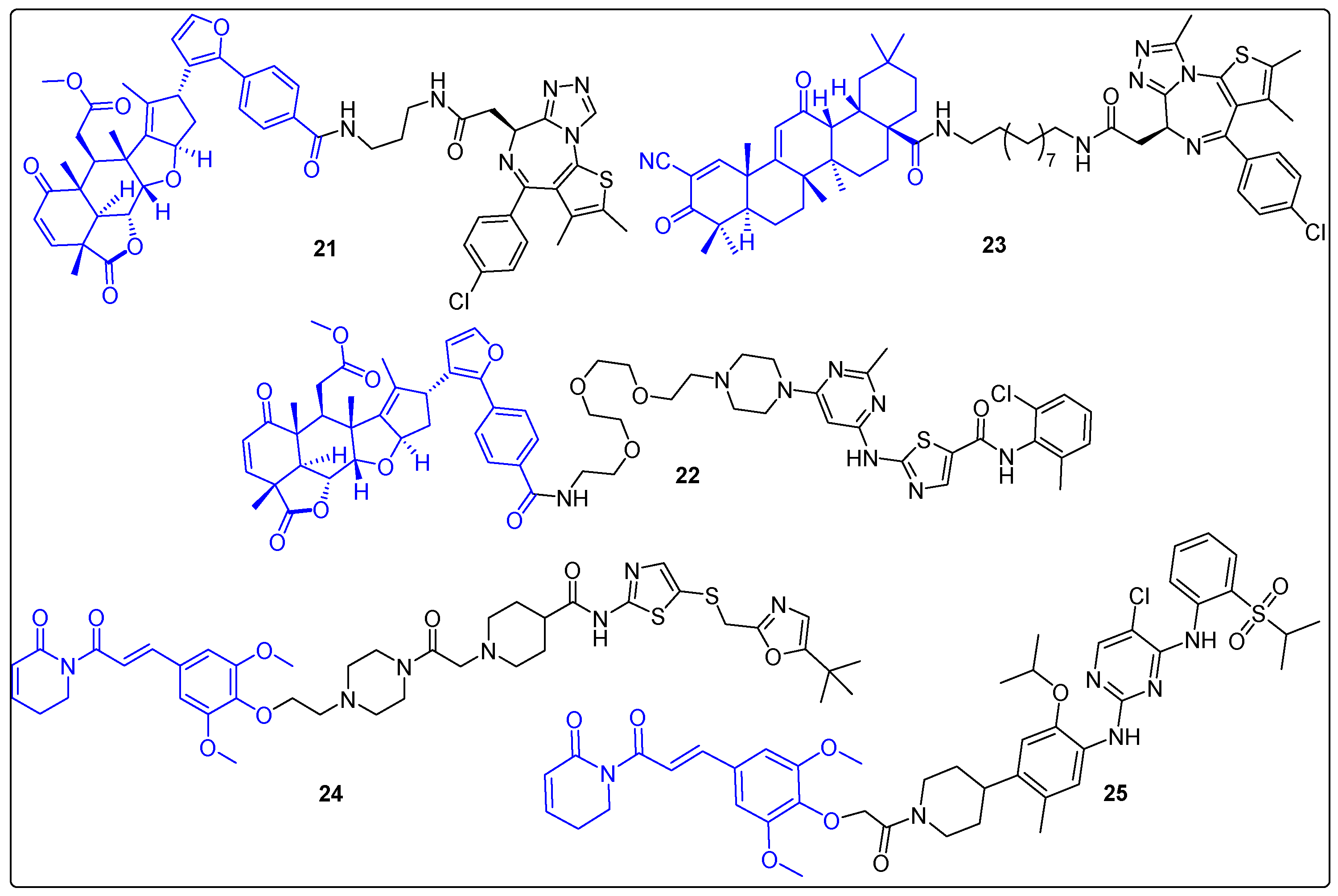
2.3. Hybrids of Natural Products with Small Molecule Proteolysis Inducers
2.4. Hybrids of Natural Products with Small Molecules Autophagy Inducers
2.5. Folate Hybrids of Natural Products
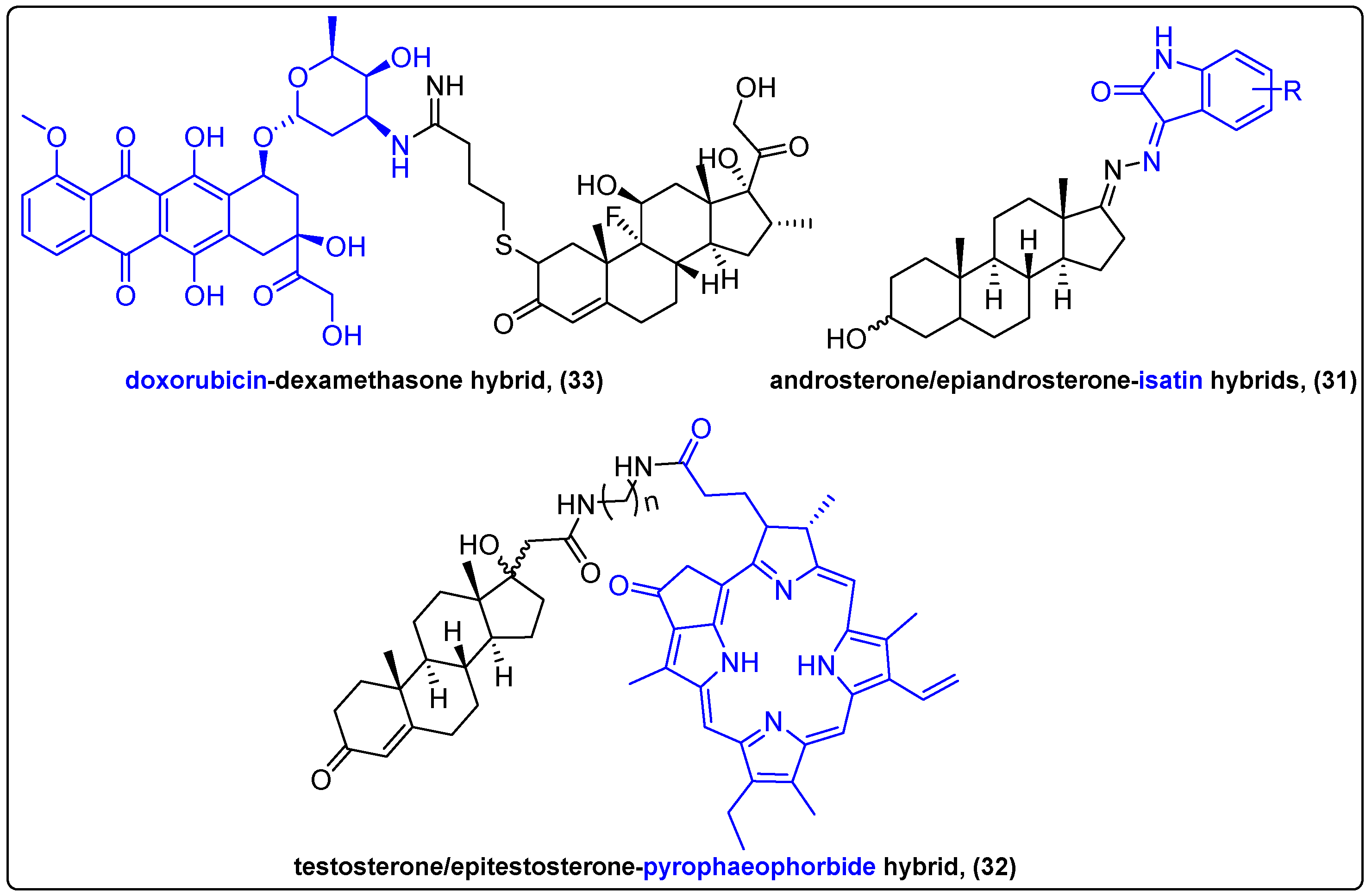
2.6. Steroidal Hybrids of Natural Products
2.7. Hybrids Incorporating Two Natural Products
3. Antibody–Drug Conjugates Based on Natural Products
4. Peptide Hybrids with Natural Products
5. Aptamer–Drug Conjugate Based on Natural Products
6. Hybrids of Natural Products with Metal Complexes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, S.; Zhu, W.; Tompson, P.; Hannun, Y.A. Evaluating intrinsic and non-intrinsic cancer risk factors. Nat. Commun. 2018, 9, 3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garodia, P.; Ichikawa, H.; Malani, N.; Sethi, G.; Aggarwal, B.B. From Ancient Medicine to Modern Medicine: Ayurvedic Concepts of Health and Their Role in Inflammation and Cancer. J. Soc. Integr. Oncol. 2007, 5, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaid, H.; Rayan, A.; Said, O.; Saad, B. Cancer Treatment by Greco-Arab and Islamic Herbal Medicine. Open Nutraceuticals J. 2010, 3, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Shultz, M.D. Two decades under the influence of the rule of five and the changing properties of approved oral drugs. J. Med. Chem. 2019, 62, 1701–1714. [Google Scholar] [CrossRef]
- Kharat, M.; Du, Z.; Zhang, G.; McClements, D.J. Physical and chemical stability of curcumin in aqueous solutions and emulsions: Impact of pH, temperature, and molecular environment. J. Agric. Food Chem. 2017, 65, 1525–1532. [Google Scholar] [CrossRef]
- Cragg, G.M.; Schepartz, S.A.; Suffness, M.; Grever, M.R. The taxol supply crisis. New NCI policies for handling the large-scale production of novel natural product anticancer and anti-HIV agents. J. Nat. Prod. 1993, 56, 1657–1668. [Google Scholar] [CrossRef]
- Colone, Μ.; Calcabrini, A.; Stringaro, A. Drug Delivery Systems of Natural Products in Oncology. Molecules 2020, 25, 4560. [Google Scholar] [CrossRef]
- Rani, K.; Paliwal, S. A review on targeted drug delivery: Its entire focus on advanced therapeutics and diagnostics. Sch. J. Appl. Med. Sci. 2014, 2, 328–331. [Google Scholar]
- Dubikovskaya, E.A.; Thorne, S.H.; Pillow, T.H.; Contag, C.H.; Wender, P.A. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc. Natl. Acad. Sci. USA 2008, 105, 12128–12133. [Google Scholar] [CrossRef] [Green Version]
- Csermely, P.; Agoston, V.; Pongor, S. The efficiency of multi-target drugs: The network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Lone, N.M.; Al-Othman, Z.; Al-Warthan, A.; Sanagi, M. Heterocyclic scaffolds: Centrality in the anticancer drug development. Curr. Drug Targets 2015, 16, 711–734. [Google Scholar] [CrossRef] [PubMed]
- Ndreshkjana, B.; Çapci, A.; Klein, V.; Chanvorachote, P.; Muenzner, J.K.; Huebner, K.; Steinmann, S.; Erlenbach-Wuensch, K.; Geppert, C.I.; Agaimy, A.; et al. Combination of 5-fluorouracil and thymoquinone targets stem cell gene signature in colorectal cancer cells. Cell Death Dis. 2019, 10, 379–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C.; Dick, J.E. Cancer stem cells: Lessons from leukemia. Trends Cell Biol. 2005, 15, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Tagle, R.; Escobar, C.; Romero, V.; Montorfano, I.; Armisén, R.; Borgna, V.; Jeldes, E.; Pizarro, L.; Simon, F.; Echeverria, C. Chalcone-induced apoptosis through caspase-dependent intrinsic pathways in human hepatocellular carcinoma cells. Int. J. Mol. Sci. 2016, 17, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chen, X.; Li, Y.; Wang, Y.; Xu, F. An orally antitumor chalcone hybrid inhibited HepG2 cells growth and migration as the tubulin binding agent. Investig. New Drugs 2019, 37, 784–790. [Google Scholar]
- Penthala, N.R.; Madhukuri, L.; Thakkar, S.; Madadi, N.R.; Lamture, G.; Eoff, R.L.; Crooks, P.A. Synthesis and anti-cancer screening of novel heterocyclic-(2H)-1,2,3-triazoles as potential anti-cancer agents. Med. Chem. Comm. 2015, 6, 1535–1543. [Google Scholar] [CrossRef] [Green Version]
- Kamal, A.; Prabhakar, S.; Ramaiah, M.J.; Reddy, P.V.; Reddy, C.R.; Mallareddy, A.; Shankaraiah, N.; Lakshmi Narayan Reddy, T.; Pushpavalli, S.N.; Pal-Bhadra, M. Synthesis and anticancer activity of chalcone-pyrrolobenzodiazepine conjugates linked via 1,2,3-triazole ring side-armed with alkane spacers. Eur. J. Med. Chem. 2011, 46, 3820–3831. [Google Scholar] [CrossRef]
- Güner, A.; Bektaş, H.; Menteşe, E. Novel coumarin compounds potentiate the effect of cisplatin on lung cancer cells by enhancing pro-apoptotic gene expressions, G2/M cell arrest, oxidative and antiangiogenic effects. Anticancer Agents Med. Chem. 2022, 22, 2429–2438. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Venigalla, M.; Gyengesi, E.; Münch, G. Curcumin and Apigenin—Novel and promising therapeutics against chronic neuroinflammation in Alzheimer’s disease. Neural Regen. Res. 2015, 10, 1181–1185. [Google Scholar] [PubMed]
- Long, H.; Hu, X.; Wang, B.; Wang, Q.; Wang, R.; Liu, S.; Xiong, F.; Jiang, Z.; Zhang, X.Q.; Ye, W.C.; et al. Discovery of Novel Apigenin−Piperazine Hybrids as Potent and Selective Poly (ADP-Ribose) Polymerase-1 (PARP-1) Inhibitors for the Treatment of Cancer. J. Med. Chem. 2021, 64, 12089–12108. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Chandra, V.; Jain, P.K.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive review on current developments of quinoline-based anticancer agents. Arab. J. Chem. 2019, 12, 4920–4946. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, X.; Li, B.; Gao, C.; Jiang, Y. Acridine and its derivatives: A patent review (2009–2013). Expert. Opin. Ther. Pat. 2014, 24, 647–664. [Google Scholar] [CrossRef]
- Lisboa, T.; Silva, D.; Duarte, S.; Ferreira, R.; Andrade, C.; Lopes, A.L.; Ribeiro, J.; Farias, D.; Moura, R.; Reis, M.; et al. Toxicity and Antitumor Activity of a Thiophene–Acridine Hybrid. Molecules 2020, 25, 64. [Google Scholar] [CrossRef] [Green Version]
- Strickland, S.A.; Podoltsev, N.A.; Mohan, S.R.; Zeidan, A.M.; Childress, M.A.; Ayers, G.D.; Byrne, M.T.; Gore, S.D.; Stuart, R.K.; Savona, M.R. The VITAL Trial: Phase II Trial of Vosaroxin and Infusional Cytarabine for Frontline Treatment of acute Myeloid Leukemia. Blood 2019, 134, 180. [Google Scholar] [CrossRef]
- Goodman, V.L.; Rock, E.P.; Dagher, R.; Ramchandani, R.P.; Abraham, S.; Gobburu, J.V.; Booth, B.P.; Verbois, S.L.; Morse, D.E.; Liang, C.Y.; et al. Approval summary: Sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin. Cancer Res. 2007, 13, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- National Institute for Health and Care Excellence. Nintedanib for Treating Idiopathic Pulmonary Fibrosis. Available online: https://www.nice.org.uk/guidance/ta379 (accessed on 26 August 2022).
- Panda, P.; Chakroborty, S. Navigating the Synthesis of Quinoline Hybrid Molecules as Promising Anticancer Agents. Chem. Sel. 2020, 5, 10187–10199. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Z. Coumarin-containing hybrids and their anticancer activities. Eur. J. Med. Chem. 2019, 181, 111587–111606. [Google Scholar] [CrossRef]
- Singh, H.; Singh, J.V.; Bhagat, K.; Gulati, H.K.; Sanduja, M.; Kumar, N.; Kinarivala, N.; Sharma, S. Rational approaches, design strategies, structure activity relationship and mechanistic insights for therapeutic coumarin hybrids. Bioorg. Med. Chem. 2019, 27, 3477–3510. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wen, X.; Gong, Y.; Wang, X. Current scenario of indole derivatives with potential anti-drugresistant cancer activity. Eur. J. Med. Chem. 2020, 200, 112359–112380. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Indole: A privileged scaffold for the design of anti-cancer agents. Eur. J. Med. Chem. 2019, 183, 9–29. [Google Scholar] [CrossRef]
- Chen, Z.; Clark, S.; Birkeland, M.; Sung, C.-M.; Lago, A.; Liu, R.; Kirkpatrick, R.; Johanson, K.; Winkler, J.D.; Hu, E. Induction and Superinduction of Growth Arrest and DNA Damage Gene 45 (Gadd45) Alpha and Beta Messenger Rnas by Histone Deacetylase Inhibitors Trichostatin a (Tsa) and Butyrate in Sw620 Human Colon Carcinoma Cells. Cancer Lett. 2002, 188, 127–140. [Google Scholar] [CrossRef]
- Sato, N.; Ohta, T.; Kitagawa, H.; Kayahara, M.; Ninomiya, I.; Fushida, S.; Fujimura, T.; Nishimura, G.; Shimizu, K.; Miwa, K. Fr901228, a Novel Histone Deacetylase Inhibitor, Induces Cell Cycle Arrest and Subsequent Apoptosis in Refractory Human Pancreatic Cancer Cells. Int. J. Oncol. 2004, 24, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, Y.-I.; Shimada, M.; Harimoto, N.; Rikimaru, T.; Shirabe, K.; Tanaka, S.; Sugimachi, K. Histone, Deacetylase Inhibitor Trichostatin a Induces Cell-Cycle Arrest/Apoptosis and Hepatocyte Differentiation in Human Hepatoma Cells. Int. J. Cancer 2003, 103, 572–576. [Google Scholar] [CrossRef]
- Zeng, H.; Qu, J.; Jin, N.; Xu, J.; Lin, C.; Chen, Y.; Yang, X.; He, X.; Tang, S.; Lan, X.; et al. Feedback Activation of Leukemia Inhibitory Factor Receptor Limits Response to Histone Deacetylase Inhibitors in Breast Cancer. Cancer Cell 2016, 30, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Ren, Υ.; Li, S.; Zhu, R.; Wan, C.; Song, D.; Zhu, J.; Cai, G.; Long, S.; Kong, L.; Yu, W. Discovery of STAT3 and Histone Deacetylase (HDAC) Dual-Pathway Inhibitors for the Treatment of Solid Cancer. J. Med. Chem. 2021, 64, 7468–7482. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Chiosis, G.; Neckers, L. Tumor selectivity of Hsp90 inhibitors: The explanation remains elusive. ACS Chem. Biol. 2006, 1, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Solit, D.B.; Chiosis, G. Development and application of Hsp90 inhibitors. Drug Discov. Today 2008, 13, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tu, G.; Hu, Y.; Jiang, Q.; Liu, J.; Lin, S.; Yu, Z.; Li, G.; Wu, X.; Tang, Y.; et al. Discovery of BP3 as an efficacious proteolysis targeting chimera (PROTAC) degrader of HSP90 for treating breast cancer. Eur. J. Med. Chem. 2002, 228, 114013–114029. [Google Scholar] [CrossRef]
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.Y.; Park, S.Y.; Jha, S.; Gupta, S.K.; Kim, M.; Ha, E.; Seo, Y.H. Design, synthesis, and biological evaluation of bifunctional inhibitors against Hsp90-HDAC6 interplay. Eur. J. Med. Chem. 2022, 240, 114582–114596. [Google Scholar] [CrossRef] [PubMed]
- Chai, R.C.; Vieusseux, J.L.; Lang, B.J.; Nguyen, C.H.; Kouspou, M.M.; Britt, K.L.; Price, J.T. Histone deacetylase activity mediates acquired resistance towards structurally diverse HSP90 inhibitors. Mol. Oncol. 2017, 11, 567–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Hughes, P.E.; Caenepeel, S.; Wu, L.C. Targeted therapy and checkpoint immunotherapy combinations for the treatment of cancer. Trends Immunol. 2016, 37, 462–476. [Google Scholar] [CrossRef]
- Wong, D.Y.Q.; Yeo, C.H.F.; Ang, W.H. Immunochemotherapeutic platinum (IV) prodrugs of cisplatin as multimodal anticancer agents. Angew. Chem. Int. Ed. 2014, 53, 6752–6756. [Google Scholar] [CrossRef]
- Huang, R.; Jing, X.; Huang, X.; Pan, Y.; Fang, Y.; Liang, G.; Liao, Z.; Wang, H.; Chen, Z.; Zhang, Y. Bifunctional Naphthoquinone Aromatic Amide-Oxime Derivatives Exert Combined Immunotherapeutic and Antitumor Effects through Simultaneous Targeting of Indoleamine-2,3-dioxygenase and Signal Transducer and Activator of Transcription 3. J. Med. Chem. 2020, 63, 1544–1563. [Google Scholar] [CrossRef]
- Shuai, W.; Wang, G.; Zhang, Y.; Bu, F.; Zhang, S.; Miller, D.D.; Li, W.; Ouyang, L.; Wang, Y. Recent Progress on Tubulin Inhibitors with Dual Targeting Capabilities for Cancer Therapy. J. Med. Chem. 2021, 64, 7963–7990. [Google Scholar] [CrossRef]
- Szumilak, M.; Wiktorowska-Owczarek, A.; Stanczak, A. Hybrid Drugs—A Strategy for Overcoming Anticancer Drug Resistance? Molecules 2021, 26, 2601. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhong, H.; Xia, X.; Qi, Z.; Wang, C.; Li, S. Potential Application of Proteolysis Targeting Chimera (PROTAC) Modification Technology in Natural Products for Their Targeted Protein Degradation. Food Sci. Hum. Wellness 2022, 11, 199–207. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, L.; Wu, J.; Liu, M.; Wang, G.; Liu, B.; Zhang, L. Transcriptional Cyclin-Dependent Kinases: Potential Drug Targets in Cancer Therapy. Eur. J. Med. Chem. 2022, 229, 114056–114079. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Ren, J.; Li, Y.; Wang, J.; Xu, X.; Feng, Y.; Tang, H.; Wang, Y.; Li, Z. Discovery of Wogonin-Based PROTACs against CDK9 and Capable of Achieving Antitumor Activity. Bioorg. Chem. 2018, 81, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Long, J.; Wang, Y.; Li, Y.; Zhang, X.; Tang, L.; Chang, Q.; Chen, Z.; Hu, G.; Hu, S.; et al. Targeted Degradation of CD147 Proteins in Melanoma. Bioorg. Chem. 2020, 105, 104453–104464. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Yang, G.; Deng, T.; Wang, J.; Zhou, H.; Popov, S.A.; Shults, E.E.; Wang, C. Design and Linkage Optimization of Ursane-Thalidomide-Based PROTACs and Identification of Their Targeted-Degradation Properties to MDM2 Protein. Bioorg. Chem. 2021, 111, 104901–104910. [Google Scholar] [CrossRef] [PubMed]
- Gerometta, E.; Grondin, I.; Smadja, J.; Frederich, M.; Gauvin-Bialecki, A. A Review of Traditional Uses, Phytochemistry and Pharmacology of the Genus Indigofera. J. Ethnopharmacol. 2020, 253, 112608–112685. [Google Scholar] [CrossRef]
- Cao, Z.; Gu, Z.; Lin, S.; Chen, D.; Wang, J.; Zhao, Y.; Li, Y.; Liu, T.; Li, Y.; Wang, Y.; et al. Attenuation of NLRP3 Inflammasome Activation by Indirubin-Derived PROTAC Targeting HDAC6. ACS Chem. Biol. 2021, 16, 2746–2751. [Google Scholar] [CrossRef]
- Fujioka, T.; Kashiwada, Y.; Kilkuskie, R.E.; Cosentino, L.M.; Ballas, L.M.; Jiang, J.B.; Janzen, W.P.; Chen, I.S.; Lee, K.H. Anti-AIDS Agents, 11. Betulinic Acid and Platanic Acid as Anti-HIV Principles from Syzigium Claviflorum, and the Anti-HIV Activity of Structurally Related Triterpenoids. J. Nat. Prod. 1997, 57, 243–247. [Google Scholar] [CrossRef]
- Nakano, N.; Fukuda, K.; Tashiro, E.; Ishikawa, H.; Nagano, W.; Kawamoto, R.; Mori, A.; Watanabe, M.; Yamazaki, R.; Nakane, T.; et al. Hybrid Molecule between Platanic Acid and LCL-161 as a Yes-Associated Protein Degrader. J. Biochem. 2022, 171, 631–640. [Google Scholar] [CrossRef]
- Spradlin, J.N.; Hu, X.; Ward, C.C.; Brittain, S.M.; Jones, M.D.; Ou, L.; To, M.; Proudfoot, A.; Ornelas, E.; Woldegiorgis, M.; et al. Harnessing the Anti-Cancer Natural Product Nimbolide for Targeted Protein Degradation. Nat. Chem. Biol. 2019, 15, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.; Spradlin, J.N.; Novaes, L.F.; Zhang, E.; Hu, X.; Moeller, M.; Brittain, S.M.; McGregor, L.M.; McKenna, J.M.; Tallarico, J.A.; et al. Nimbolide-Based Kinase Degrader Preferentially Degrades Oncogenic BCR-ABL. ACS Chem. Biol. 2020, 15, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Crews, C.M. Recent Developments in PROTAC-Mediated Protein Degradation: From Bench to Clinic. ChemBioChem 2022, 23, e202100270. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1–NRF2 System in Cancer. Front. Oncol. 2017, 7, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Tong, B.; Luo, M.; Xie, Y.; Spradlin, J.N.; Tallarico, J.A.; McKenna, J.M.; Schirle, M.; Maimone, T.J.; Nomura, D.K. Bardoxolone Conjugation Enables Targeted Protein Degradation of BRD4. Sci. Rep. 2020, 10, 15543. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Liu, X.; Wang, Y.; Chang, J.; Zhang, X.; Mackintosh, S.G.; Tackett, A.J.; He, Y.; Lv, D.; et al. Oxidation Resistance 1 Is a Novel Senolytic Target. Aging Cell 2018, 17, 12780–12794. [Google Scholar] [CrossRef]
- Ma, H.; Wu, Y.; Zhang, W.; Zhang, H.; Miao, Z.; Zhuang, C. Radiosensitization of Human Pancreatic Cancer by Piperlongumine Analogues. Chin. Chem. Lett. 2020, 32, 1197–1201. [Google Scholar] [CrossRef]
- Pei, J.; Xiao, Y.; Liu, X.; Hu, W.; Sobh, A.; Yuan, Y.; Zhou, S.; Hua, N.; Mackintosh, S.G.; Zhang, X.; et al. Identification of Piperlongumine (PL) as a New E3 Ligase Ligand to Induce Targeted Protein Degradation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning house: Selective autophagy of organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.H.; Kim, H.Y.; Lee, M.J.; Heo, A.J.; Park, D.Y.; Lim, S.; Shin, S.; Ganipisetti, S.; Yang, W.S.; Jung, C.A.; et al. The AUTOTAC Chemical Biology Platform for Targeted Protein Degradation via the Autophagy-Lysosome System. Nat. Commun. 2022, 13, 904. [Google Scholar] [CrossRef] [PubMed]
- Chatzisideri, T.; Leonidis, G.; Sarli, V. Cancer-targeted delivery systems based on peptides. Future Med. Chem. 2018, 10, 2201–2226. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.; Turk, M.J.; Westrick, E.; Lewis, J.D.; Low, P.S.; Leamon, C.P. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 2005, 338, 284–293. [Google Scholar] [CrossRef] [PubMed]
- O’Shannessy, D.J.; Yu, G.; Smale, R.; Fu, Y.S.; Singhal, S.; Thiel, R.P.; Somers, E.B.; Vachani, A. Folate receptor alpha expression in lung cancer: Diagnostic and prognostic significance. Oncotarget 2012, 3, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Scaranti, M.; Cojocaru, E.; Banerjee, S.; Banerji, U. Exploiting the folate receptor α in oncology. Nat. Rev. Clin. Oncol. 2020, 17, 349–359. [Google Scholar] [CrossRef]
- Reddy, J.A.; Dorton, R.; Bloomfield, A.; Nelson, M.; Dircksen, C.; Vetzel, M.; Kleindl, P.; Santhapuram, H.; Vlahov, I.R.; Leamon, C.P. Pre-clinical evaluation of EC1456, a folate-tubulysin anti-cancer therapeutic. Sci. Rep. 2018, 8, 8943. [Google Scholar] [CrossRef] [Green Version]
- Levine, P.M.; Michael, J.; Garabedian, M.J.; Kirshenbaum, K. Targeting the androgen receptor with steroid conjugates. J. Med. Chem. 2014, 57, 8224–8237. [Google Scholar] [CrossRef] [Green Version]
- Bérubé, G. An overview of molecular hybrids in drug discovery. Expert Opin. Drug Discov. 2016, 11, 281–305. [Google Scholar] [CrossRef]
- Ahmad, N.; Kumar, R. Steroid hormone receptors in cancer development: A target for cancer therapeutics. Cancer Lett. 2011, 300, 1–9. [Google Scholar] [CrossRef]
- Ke, S.; Shi, L.; Yang, Z. Discovery of novel isatin–dehydroepiandrosterone conjugates as potential anticancer agents. Bioorg. Med. Chem. Lett. 2015, 25, 4628–4631. [Google Scholar] [CrossRef]
- Ke, S.; Zhang, Z.; Liu, M.; Fang, W.; Huang, D.; Wan, Z.; Zhou, R.; Wang, K.; Shi, L. Synthesis and bioevaluation of novel steroidal isatin conjugates derived from epiandrosterone/androsterone. J. Enzyme. Inhib. Med. Chem. 2019, 34, 1607–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolottsev, V.A.; Ponomarev, G.V.; Taratynova, M.O.; Morozevich, G.E.; Novikov, R.A.; Timofeev, V.P.; Solyev, P.N.; Zavialova, M.G.; Zazulina, O.V.; Tkachev, Y.V.; et al. Conjugates of 17-substituted testosterone and epitestosterone with pyropheophorbide a differing in the length of linkers. Steroids 2018, 138, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Chaikomon, K.; Chattong, S.; Chaiya, T.; Tiwawech, D.; Sritana-Anant, Y.; Sereemaspun, A.; Manotham, K. Doxorubicin-conjugated dexamethasone induced MCF-7 apoptosis without entering the nucleus and able to overcome MDR-1-induced resistance. Drug Des. Devel. Ther. 2018, 12, 2361–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Liu, X.; Zhang, C.; Zeng, X. Food macromolecule based nanodelivery systems for enhancing the bioavailability of polyphenols. J. Food Drug Anal. 2017, 25, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; McClements, D.J. Formulation of More Efficacious Curcumin Delivery Systems Using Colloid Science: Enhanced Solubility, Stability, and Bioavailability. Molecules 2020, 25, 2791. [Google Scholar] [CrossRef]
- Micale, Ν.; Molonia, M.S.; Citarella, A.; Cimino, F.; Saija, A.; Cristani, M.; Speciale, A. Natural Product-Based Hybrids as Potential Candidates for the Treatment of Cancer: Focus on Curcumin and Resveratrol. Molecules 2021, 26, 4665. [Google Scholar] [CrossRef]
- Shen, J.L.; Man, K.M.; Huang, P.H.; Chen, W.C.; Chen, D.C.; Cheng, Y.W.; Liu, P.L.; Chou, M.C.; Chen, Y.H. Honokiol and magnolol as multifunctional antioxidative molecules for dermatologic disorders. Molecules 2010, 15, 6452–6465. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.F. Sulforaphane (SFN): An isothiocyanate in a cancer chemoprevention paradigm. Medicines 2015, 2, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Tao, C.; Chen, J.; Huang, X.; Chen, Z.; Li, X.; Li, Y.; Xu, Y.; Ma, M.; Wu, Z. CT1-3, a novel magnolol-sulforaphane hybrid suppresses tumorigenesis through inducing mitochondria-mediated apoptosis and inhibiting epithelial mesenchymal transition. Eur. J. Med. Chem. 2020, 199, 112441. [Google Scholar] [CrossRef]
- Crespo-Ortiz, M.P.; Wei, M.Q. Antitumor activity of artemisinin and its derivatives: From a well-known antimalarial agent to a potential anticancer drug. J. Biomed. Biotechnol. 2012, 2012, 247597–247615. [Google Scholar] [CrossRef] [Green Version]
- Das, A.K. Anticancer Effect of antimalarial artemisinin compounds. Ann. Med. Health Sci. Res. 2015, 5, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. Cancer combination therapies with artemisinin-type drugs. Biochem. Pharmacol. 2017, 139, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.E.; Peh, H.Y.; Chan, T.K.; Wong, W.S. Artemisinins: Pharmacological actions beyond anti-malarial. Pharmacol. Ther. 2014, 142, 126–139. [Google Scholar] [CrossRef]
- Ricci, J.; Kim, M.; Chung, W.Y.; Park, K.K.; Jung, M. Discovery of artemisinin-glycolipid hybrids as anti-oral cancer agents. Chem. Pharm. Bull. 2011, 59, 1471–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, G.T.; Lee, S.K.; Park, K.-K.; Park, J.; Son, S.H.; Jung, M.; Chung, W.-Y. Artemisinin-Daumone Hybrid Inhibits Cancer Cell-Mediated Osteolysis by Targeting Cancer Cells and Osteoclasts. Cell Physiol. Biochem. 2018, 49, 1460–1475. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, D.; Ge, M.; Li, Z.; Jiang, J.; Li, Y. Formononetin inhibits enterovirus 71 replication by regulating COX-2/PGE2 expression. Virol. J. 2015, 12, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Dang, Y.; Zhou, X.; Huang, B.; Huang, X.; Zhang, Z.; Kwan, Y.W.; Chan, S.W.; Leung, G.P.; Lee, S.M.; et al. Formononetin promotes angiogenesis through the estrogen receptor alpha enhanced ROCK pathway. Sci. Rep. 2015, 5, 16815. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Wu, K.; Zhu, Q.; Xiao, W.; Shan, Q.; Yan, Z.; Wu, J.; Deng, B.; Xue, Y.; Gong, W.; et al. Formononetin administration ameliorates dextran sulfate sodium-induced acute colitis by inhibiting NLRP3 Inflammasome signaling pathway. Mediat. Inflamm. 2018, 2018, 3048532–3048544. [Google Scholar]
- Yao, J.N.; Zhang, X.X.; Zhang, Y.Z.; Li, J.H.; Zhao, D.Y.; Gao, B.; Zhou, H.N.; Gao, S.L.; Zhang, L.F. Discovery and anticancer evaluation of a formononetin derivative against gastric cancer SGC7901 cells. Investig. New Drugs 2019, 37, 1300–1308. [Google Scholar] [CrossRef]
- Gonzalez, M.M.; Cabrerizo, F.M.; Baiker, A.; Erra-Balsells, R.; Osterman, A.; Nitschko, H.; Vizoso-Pinto, M.G. β-Carboline derivatives as novel antivirals for herpes simplex viruses. Int. J. Antimicrob. Agents 2018, 52, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Sathish, M.; Kavitha, B.; Nayak, V.L.; Tangella, Y.; Ajitha, A.; Nekkanti, S.; Alarifi, A.; Shankaraiah, N.; Nagesh, N.; Kamal, A. Synthesis of podophyllotoxin linked β-car- boline congeners as potential anticancer agents and DNA topoisomerase II inhibitors. Eur. J. Med. Chem. 2018, 144, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Tokala, R.; Thatikonda, S.; Vanteddu, U.S.; Sana, S.; Godugu, C.; Shankaraiah, N. Design and synthesis of DNA-interactive β-carboline-oxindole hybrids as cytotoxic and apoptosis-inducing agents. Chem. Med. Chem. 2018, 13, 1909–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Chen, X.; Chen, W.; Ma, Q.; Fan, W.; Zhang, J.; Dai, B. Molecular hybrid design, synthesis, in vitro and in vivo anticancer evaluation, and mechanism of action of N-acylhydrazone linked, heterobivalent β-carboline. Bioorg. Chem. 2020, 96, 103612–103624. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Singh, P.K.; Verma, H.; Singh, H.; Silakari, O. Success stories of natural product-based hybrid molecules for multi-factorial diseases. Eur. J. Med. Chem. 2018, 151, 62–97. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Oliveira, C.; Granja, P.L.; Sarmento, B. Antibodies and associates: Partners in targeted drug delivery. Pharmacol. Ther. 2017, 177, 129–145. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V. CPhI Annual Report 2018: ADCs Growth Driven by Lack of In-House Facilities, Oncology and Integrated CDMOs|Pharmaceutical Outsourcing—The Journal of Pharmaceutical & Biopharmaceutical Contract Services (pharmoutsourcing.com). Available online: https://www.pharmoutsourcing.com/Featured-Articles/354437-CPhI-Annual-Report-2018-ADCs-Growth-Driven-by-Lack-of-In-House-Facilities-Oncology-and-Integrated-CDMOs/ (accessed on 27 August 2022).
- Montemurro, F.; Delaloge, S.; Barrios, C.H.; Wuerstlein, R.; Anton, A.; Brain, E.; Hatschek, T.; Kelly, C.M.; Peña-Murillo, C.; Yilmaz, M.; et al. Trastuzumab emtansine (T-DM1) in patients with HER2-positive metastatic breast cancer and brain metastases: Exploratory final analysis of cohort 1 from KAMILLA, a single-arm phase IIIb clinical trial. Ann. Oncol. 2020, 31, 1350–1358. [Google Scholar] [CrossRef]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody–Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.H.; Sliwkowski, M.X.; et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef]
- Krop, I.E.; Kim, S.-B.; Martin, A.G.; Lorusso, P.M.; Ferrero, J.-M.; Badovinac-Crnjevic, T.; Hoersch, S.; Smitt, M.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. 2017, 18, 743–754. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Shustov ABrentuximab Vedotin (SGN-35) in Patients with Relapsed or Refractory Systemic Anaplastic Large-Cell Lymphoma: Results of a Phase II Study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; DeAngelo, D.J.; Advani, A.S.; Stelljes, M.; Kebriaei, P.; Cassaday, R.D.; Merchant, A.A.; Fujishima, N.; Uchida, T.; Calbacho, M.; et al. Hepatic adverse event profile of inotuzumab ozogamicin in adult patients with relapsed or refractory acute lymphoblastic leukemia: Results from the open-label, randomised, phase 3 INO-VATE study. Lancet Haematol. 2017, 4, e387–e398. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 1490. [Google Scholar]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef]
- Tilly, H.; Morschhauser, F.; Sehn, L.H.; Friedberg, J.W.; Trněný, M.; Sharman, J.P.; Herbaux, C.; Burke, J.M.; Matasar, M.; Rai, S.; et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 351–363. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 2592. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef]
- Sanchez, E.; Li, M.; Kitto, A.; Li, J.; Wang, C.S.; Kirk, D.T.; Yellin, O.; Nichols, C.M.; Dreyer, M.P.; Ahles, C.P.; et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br. J. Haematol. 2012, 158, 727–738. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Li, L.; Xu, M.Z.; Wang, L.; Jiang, J.; Dong, L.H.; Chen, F.; Dong, K.; Song, H.F. Conjugating MMAE to a novel antiHER2 antibody for selective targeted delivery. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 12929–12937. [Google Scholar] [PubMed]
- Sheng, X.; Yan, X.; Wang, L.; Shi, Y.; Yao, X.; Luo, H.; Shi, B.; Liu, J.; He, Z.; Yu, G.; et al. Open-label, multicenter, phase II study of RC48-ADC, a HER2-targeting antibody-drug conjugate, in patients with locally advanced or metastatic urothelial carcinoma. Clin. Cancer Res. 2021, 27, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhu, X.; Wei, X.; Tang, C.; Zhang, W. HER2-targeted therapies in gastric cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188549–188563. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, Y.; Gong, J.; Zhang, X.; Peng, Z.; Sheng, X.; Mao, C.; Fan, Q.; Bai, Y.; Ba, Y.; et al. Phase I study of the recombinant humanized anti-HER2 monoclonal antibody-MMAE conjugate RC48-ADC in patients with HER2-positive advanced solid tumors. Gastric Cancer 2021, 24, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Liu, Y.; Zhou, X.; Shen, P.; Xue, R.; Zhang, M. Disitamab vedotin: A novel antibody-drug conjugates for cancer therapy. Drug Deliv. 2022, 29, 1335–1344. [Google Scholar] [CrossRef]
- Poudel, Y.B.; Rao, C.; Kotapati, S.; Deshpande, M.; Thevanayagam, L.; Pan, C.; Cardarelli, J.; Chowdari, N.; Kaspady, M.; Samikannu, R.; et al. Design, synthesis and biological evaluation of phenol-linked uncialamycin antibody-drug conjugates. Bioorg. Med. Chem. Lett. 2020, 30, 126782–126789. [Google Scholar] [CrossRef]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Peptide–drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliv. Rev. 2017, 110, 112–126. [Google Scholar] [CrossRef] [Green Version]
- Israeli, R.S.; Powell, C.T.; Corr, J.G.; Fair, W.R.; Heston, W.D. Expression of the prostate-specific membrane antigen. Cancer Res. 1994, 54, 1807–1811. [Google Scholar]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulos, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumors. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef]
- Mahalingam, D.; Peguero, J.; Cen, P.; Arora, S.; Sarantopoulos, J.; Rowe, J.; Allgood, V.; Tubb, B.; Campos, L. A Phase II, Multicenter, Single-Arm Study of Mipsagargin (G-202) as a Second-Line Therapy Following Sorafenib for Adult Patients with Progressive Advanced Hepatocellular Carcinoma. Cancers 2019, 11, 833. [Google Scholar] [CrossRef] [Green Version]
- Whalen, K.A.; White, B.H.; Quinn, J.M.; Kriksciukaite, K.; Alargova, R.; Au Yeung, T.P. Targeting the Somatostatin Receptor 2 with the Miniaturized Drug Conjugate, PEN-221: A Potent and Novel Therapeutic for the Treatment of Small Cell Lung Cancer. Mol. Cancer Ther. 2019, 18, 1926–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, G.; Brown, A.; Mudd, G.; Huxley, P.; Van Rietschoten, K.; Pavan, S.; Chen, L.; Watcham, S.; Lahdenranta, J.; Keen, N. MMAE Delivery Using the Bicycle Toxin Conjugate BT5528. Mol. Cancer Ther. 2020, 19, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.; Knapp, D.W.; Cheng, L.; Snyder, P.W.; Mittal, S.K.; Bangari, D.S.; Kinch, M.; Wu, L.; Dhariwal, J.; Mohammed, S.I. Expression of EphA2 and Ephrin A-1 in carcinoma of the urinary bladder. Clin. Cancer Res. 2006, 12, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowland, C.; Berry, P.; Errington, J.; Jeffrey, P.; Bennett, G.; Godfrey, L. Development of a LC–MS/MS method for the quantification of toxic payload DM1 cleaved from BT1718 in a Phase I study. Bioanalysis 2021, 13, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Zoptarelin Doxorubicin (AEZS 108) as Second Line Therapy for Endometrial Cancer (ZoptEC): Identifier: NCT01767155. Available online: https://clinicaltrials.gov (accessed on 29 August 2022).
- ANGLeD. ANG1005 in Leptomeningeal Disease from Breast Cancer (ANGLeD): Identifier: NCT03613181. 2018. Available online: https://clinicaltrials.gov (accessed on 29 August 2022).
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.P.; Kelly, L.; Ahrens, D.P.; Barry, A.P.; Kratschmer, C.; Levy, M.; Sullenger, B.A. Tunable cytotoxic aptamer–drug conjugates for the treatment of prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 4761–4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.Y.; Kim, H.; Lee, M.; Hong, J.; Lee, J.H.; Kim, J. Development of HER2-Specific Aptamer-Drug Conjugate for Breast Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 9764. [Google Scholar] [CrossRef]
- He, J.; Peng, T.; Peng, Y.; Ai, L.; Deng, Z.; Wang, X.-Q.; Tan, W. Molecularly Engineer Triptolide with Aptamers for High Specifici-ty and Cytotoxicity for Triple-Negative Breast Cancer. J. Am. Chem. Soc. 2020, 142, 2699–2703. [Google Scholar] [CrossRef]
- Kim, D.-H.; Seo, J.-M.; Shin, K.-J.; Yang, S.-G. Design and clinical developments of aptamer-drug conjugates for targeted cancer therapy. Biomater. Res. 2021, 25, 42–54. [Google Scholar] [CrossRef]
- Shipra, Y. Potential of Metal Complexes for the Treatment of Cancer: Current Update and Future Prospective. In Chemistry of Biologically Potent Natural Products and Synthetic Compounds; Scrivener Publishing LLC: Beverly, MA, USA, 2021; pp. 183–203. [Google Scholar]
- Oliveira, K.M.; Corrêa, R.S.; Barbosa, M.I.; Ellena, J.; Cominetti, M.R.; Batista, A.A. Ruthenium(II)/Triphenylphosphine Complexes: An Effective Way to Improve the Cytotoxicity of Lapachol. Polyhedron 2017, 130, 108–114. [Google Scholar] [CrossRef]
- Beauperin, M.; Polat, D.; Roudesly, F.; Top, S.; Vessières, A.; Oble, J.; Jaouen, G.; Poli, G. Approach to Ferrocenyl-Podophyllotoxin Analogs and Their Evaluation as Anti-Tumor Agents. J. Organomet. Chem. 2017, 839, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Product | Bioactive Moiety | Conjugation | Preclinical/ Clinical Studies | Ref |
|---|---|---|---|---|
| Thymoquinone | 5-FU |  | Colorectal cancer | [13] |
| Chalcone | 1,2,3-triazole | Ether bond | Hepatocellular carcinoma | [17] |
| Coumarin | dihydrotriazole | Amide bond | Lung cancer | [20] |
| Apigenin | Piperazine | Methylene group | Lung and ovarian cancer | [23] |
| Acridine | Thiophene | Direct conjugation | Colorectal cancer | [26] |
| Quinolone | Thiazole | Direct conjugation | Phase I and II AML, myelodysplastic syndrome | [27] |
| Isatin | Piperazine |  | Phase II pancreatic cancer | [29] |
| Quinazoline | Furan | Direct | >10 active clinical trials | [30] |
| Pterostilbene | Vorinostat | Ether bond | Breast cancer | [39] |
| Isatin | Pyrrole | Direct conjugation | Phase II thymoma, glioblastoma osteosarcoma | [28] |
| Resorcinol | Phenyl | Amide bond | Lung cancer | [45] |
| 1,4-Napthoquinone | Phenyl | Direct conjugation | Melanoma | [50] |
| JH-VIII-49 | Pomalidomide |  | Leukemia | [54] |
| Wogonin | Pomalidomide |  | Breast cancer | [55] |
| Pseudolaric acid | Thalidomide | Ethylene diamine | Melanoma | [56] |
| Ursolic acid | Thalidomide | POE-4 linker | Lung, hepatoma | [57] |
| Indirubin | Pomalidomide | Diethylene glycol | Leukemia | [59] |
| Platanic acid | LCL-161 | Diethylene glycol | Lung cancer | [61] |
| Nimbolide | JQ1 | 1,3-diaminopropane | Breast cancer | [63] |
| Bardoxolone | JQ1 |  | Breast cancer | [66] |
| Piperlongumine | SNS-032 | Piperazine secondary amide | Leukemia | [69] |
| Fumagillin | YTK-105 | PEG linker | Renal cancer | [72] |
| DM1 | Folic acid | Disulfide bond | Lung cancer | [76] |
| Desacetylvinblastine monohydrazide | Folic acid | Peptide spacer | Phase I, II and III Ovarian, small cell lung cancer | [76] |
| Epothilone A | Folic acid | Saccharo-peptidic spacer | Phase I/IIa solid tumors | [76] |
| Tubulysin | Folic acid | Saccharo-peptidic spacer | Phase I Cervical cancer | [77] |
| Isatin | Epiandrosterone, androsterone |  | Gastric, melanoma Hepatocellular carcinoma | [81,82] |
| Pyropheophorbide | 17α-Testosterone, 17β-epitestosterone |  | Prostate cancer | [83] |
| Doxorubicin | Dexamethasone |  | Breast cancer | [84] |
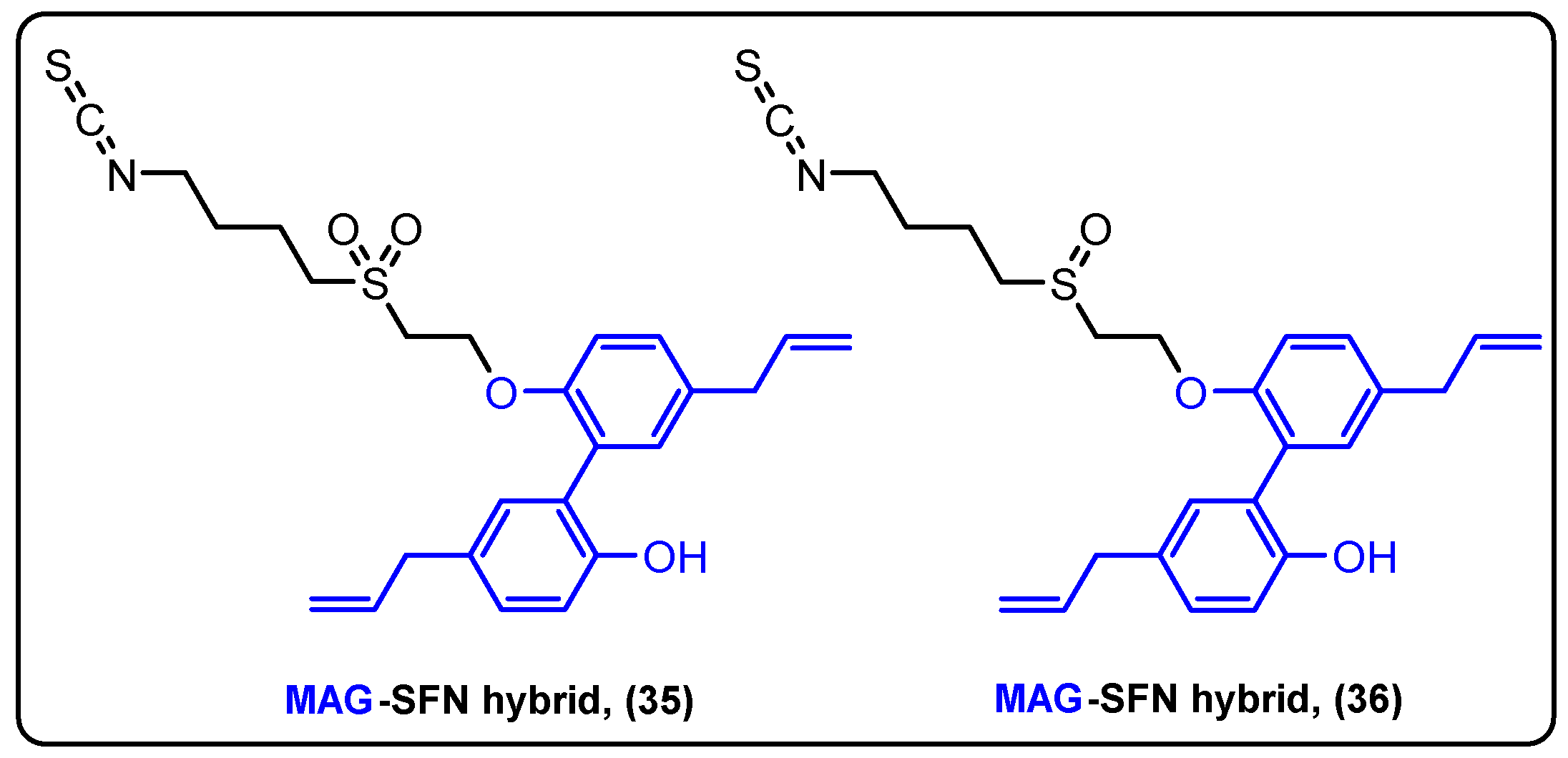
| Magnolol | Sulforaphane | Ether | Breast cancer | [90] |
| Artemisinin | Daumone | Carbamate | Breast cancer | [95] |
| Formonentin | Umbelliferone | 1,2,3-triazole | Gastric cancer | [100] |
| β-Carboline | β-carboline | N-acyl hydrazone | Sarcoma | [104] |
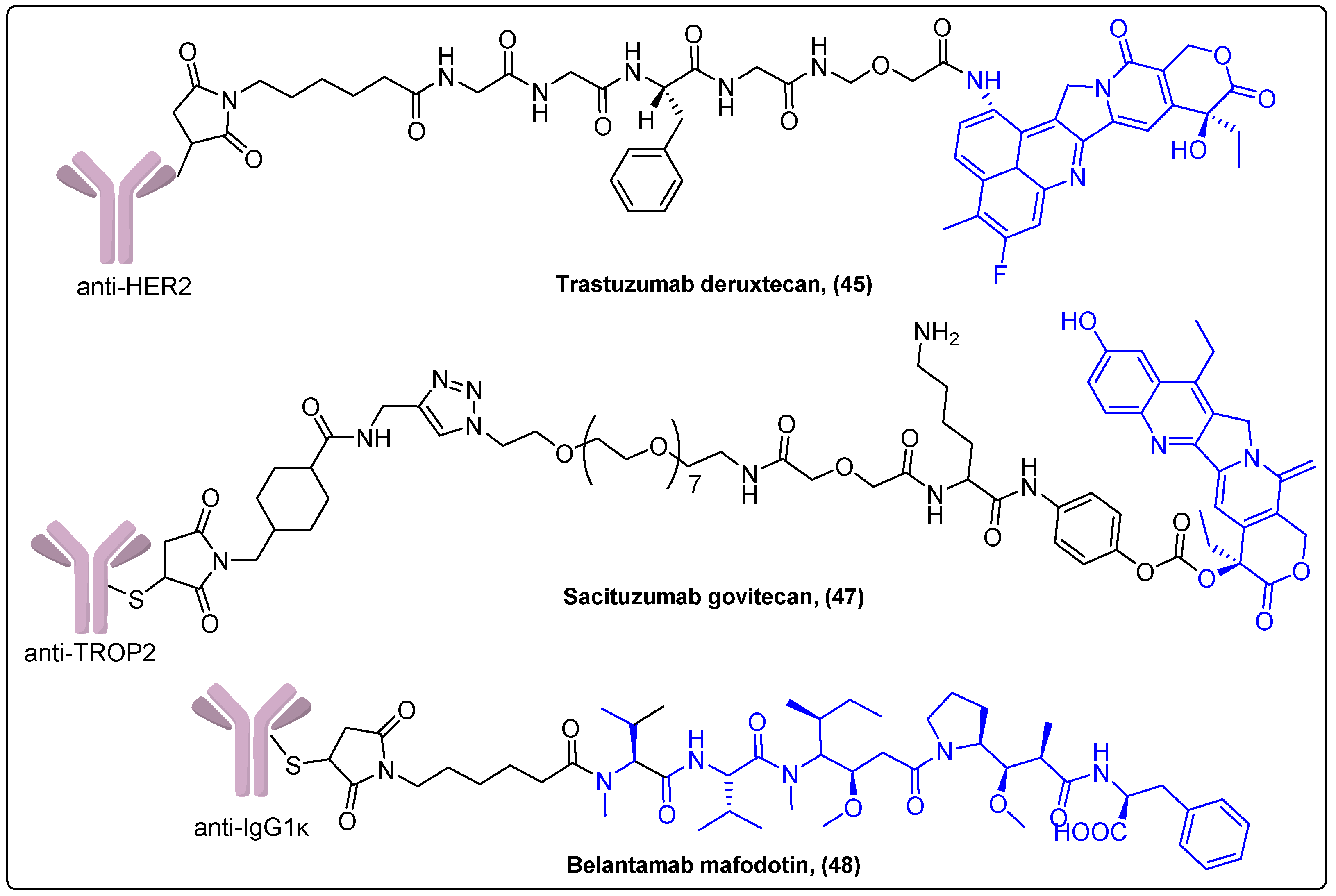
| Mertansine | Anti-HER2 | Maleimide-thiol | Approved for ErbB2-positive advanced breast cancer | [112] |
| MMAE | Anti-CD30 | Maleimide-thiol | Approved for relapsed or refractory systemic anaplastic large-cell lymphoma | [113] |
| N-acetyl gamma-calicheamicin-dimethyl hydrazide | Anti-CD22 | Hydrazone | Approved for relapsed or refractory CD22+ acute lymphoblastic leukemia | [114] |
| N-acetyl gamma-calicheamicin-dimethyl hydrazide | Anti-CD33 | Hydrazone | Approved for CD 33+ acute myeloid leukemia | [115] |
| MMAE | Anti-CD79b | Maleimide-thiol | Approved for relapsed or refractory diffuse large B cell lymphoma | [116] |
| Exatecan | Anti-HER2 | Maleimide-thiol | Approved for ErbB2-positive metastatic breast cancer | [118] |
| MMAE | Nectin-4 | Maleimide-thiol | Approved for advanced or metastatic urothelial cancer | [119] |
| SN-38 | TROP2 | Maleimide-thiol | Approved for metastatic triple-negative breast cancer | [120] |
| MMAF | IgG1κ | Maleimide-thiol | Approved for multiple myeloma | [122] |
| MMAE | Anti-HER2 | Valine-citruline | Phase I and II breast, urothelial, gastric cancer | [123] |
| Uncialamycin | Anti-mesothelin | Peptide | Lung cancer | [128] |
| Thapsigargin | Asp-γ-Glu-γ-Glu-γ-Glu-Glu | Amide bond | Phase II advanced refractory hepatocellular carcinoma | [131] |
| Mertansine | [Tyr3, Cys8] octreotate | Disulfide bond | Small-cell lung cancer | [133] |
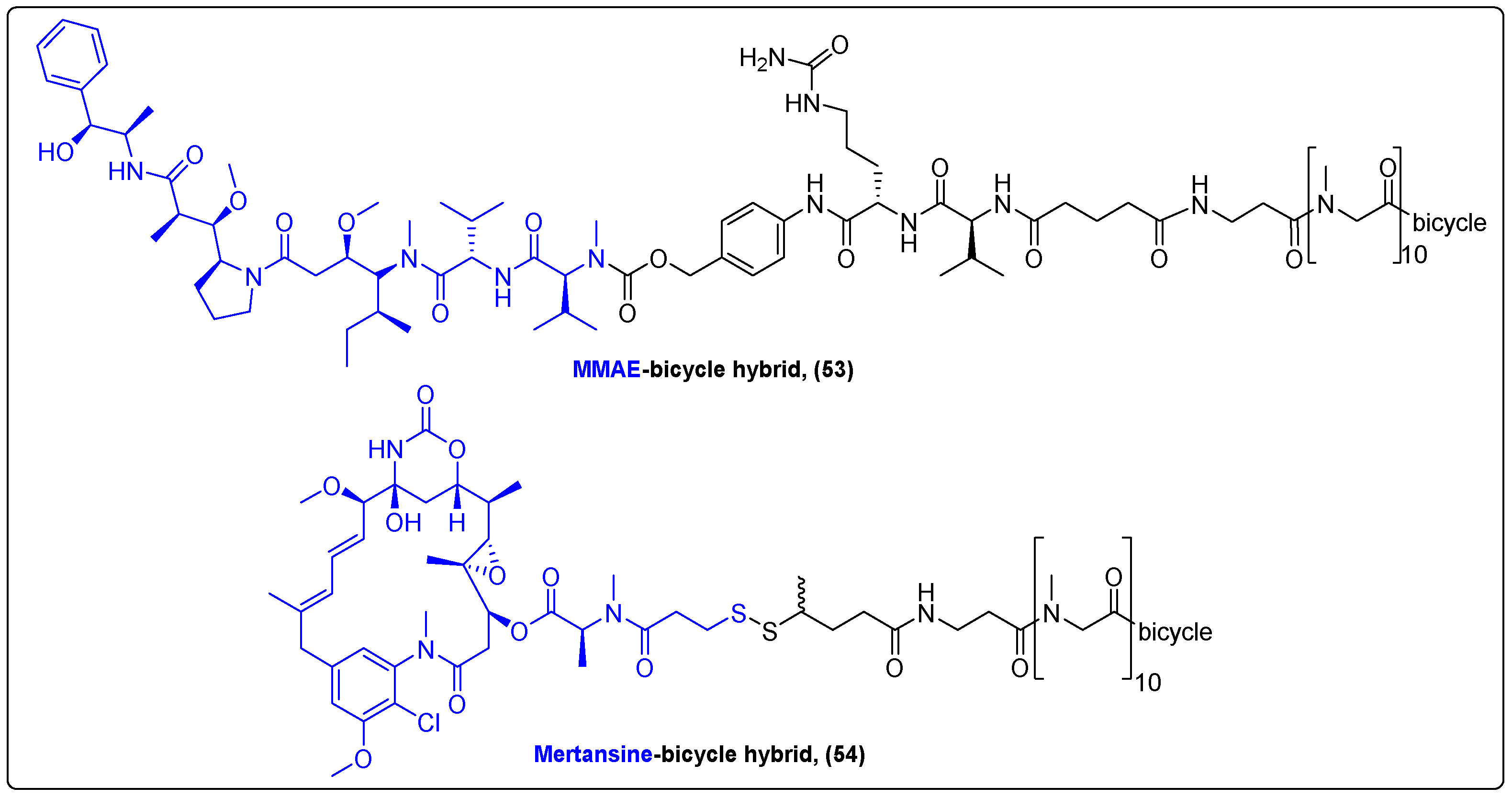
| MMAE | Bicyclic peptide | Glutaric acid/peptide | Prostate cancer | [134] |
| Mertansine | Bicyclic peptide | Disulfide bond | Phase I/IIa advanced solid tumors | [136] |
| Doxorubicin | GnRH analogue | Ester and glutaric acid | Phase IIIendometrial | [137] |
| Paclitaxel | Angiopep-2 | Ester bond | Phase I | [138] |
| MMAE, MMAF | E3 aptamer | Peptide spacer/aliphatic | Prostate cancer | [140] |
| Mertansine | HER2 RNA aptamer | Disulfide bond | Breast cancer | [141] |
| Triptolide | AS1411 | Amide bond | Triple-negative breast cancer | [142] |
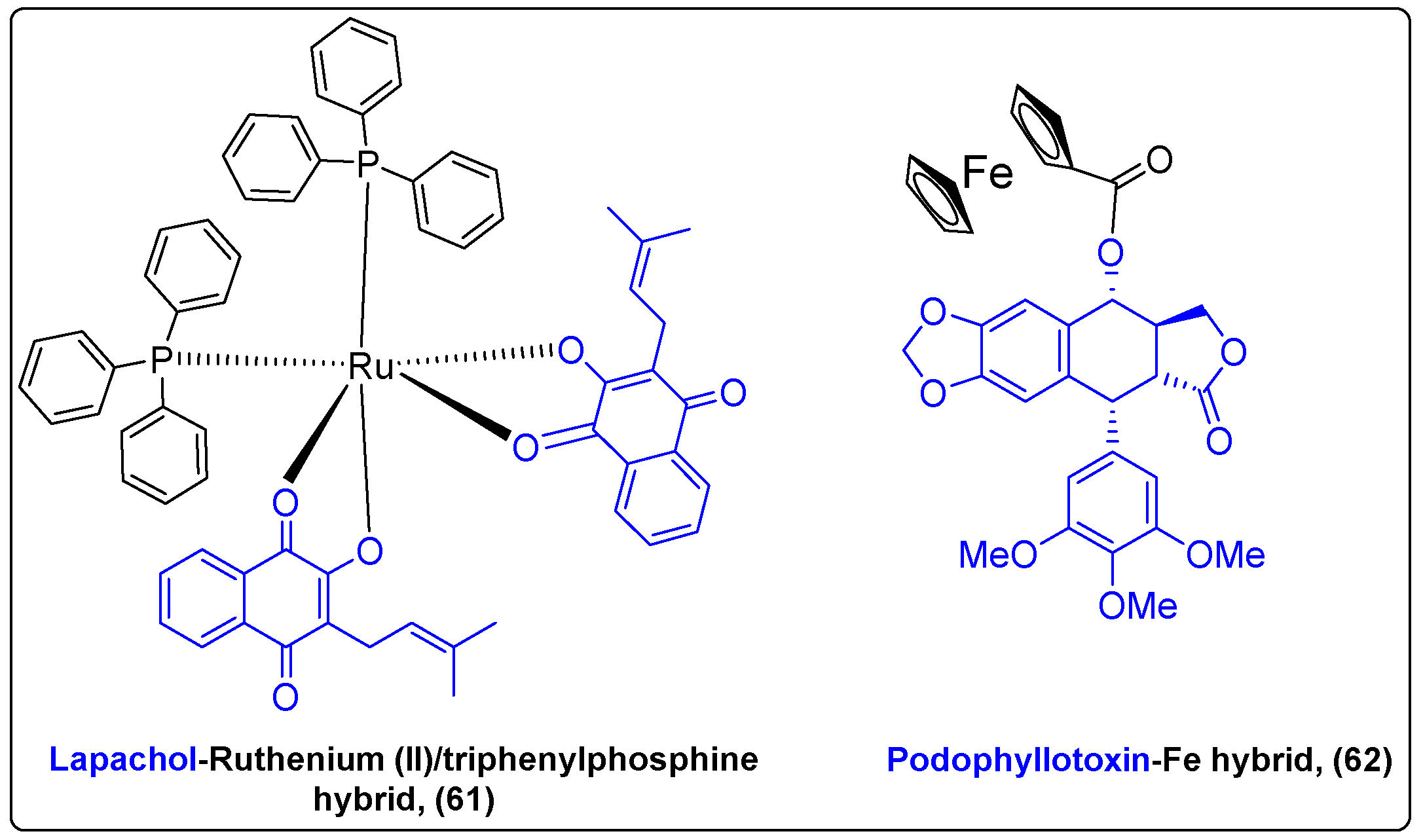
| Lapachol | Ruthenium (II)/PPh3 | Direct | Lung, breast cancer | [145] |
| Podophyllotoxin | Ferrocenyl moiety | Ester | Breast cancer | [146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sflakidou, E.; Leonidis, G.; Foroglou, E.; Siokatas, C.; Sarli, V. Recent Advances in Natural Product-Based Hybrids as Anti-Cancer Agents. Molecules 2022, 27, 6632. https://doi.org/10.3390/molecules27196632
Sflakidou E, Leonidis G, Foroglou E, Siokatas C, Sarli V. Recent Advances in Natural Product-Based Hybrids as Anti-Cancer Agents. Molecules. 2022; 27(19):6632. https://doi.org/10.3390/molecules27196632
Chicago/Turabian StyleSflakidou, Eleni, George Leonidis, Eirini Foroglou, Christos Siokatas, and Vasiliki Sarli. 2022. "Recent Advances in Natural Product-Based Hybrids as Anti-Cancer Agents" Molecules 27, no. 19: 6632. https://doi.org/10.3390/molecules27196632
APA StyleSflakidou, E., Leonidis, G., Foroglou, E., Siokatas, C., & Sarli, V. (2022). Recent Advances in Natural Product-Based Hybrids as Anti-Cancer Agents. Molecules, 27(19), 6632. https://doi.org/10.3390/molecules27196632