
Theoretical Study of the Structural Stability, Chemical Reactivity, and Protein Interaction for NMP Compounds as Modulators of the Endocannabinoid System

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
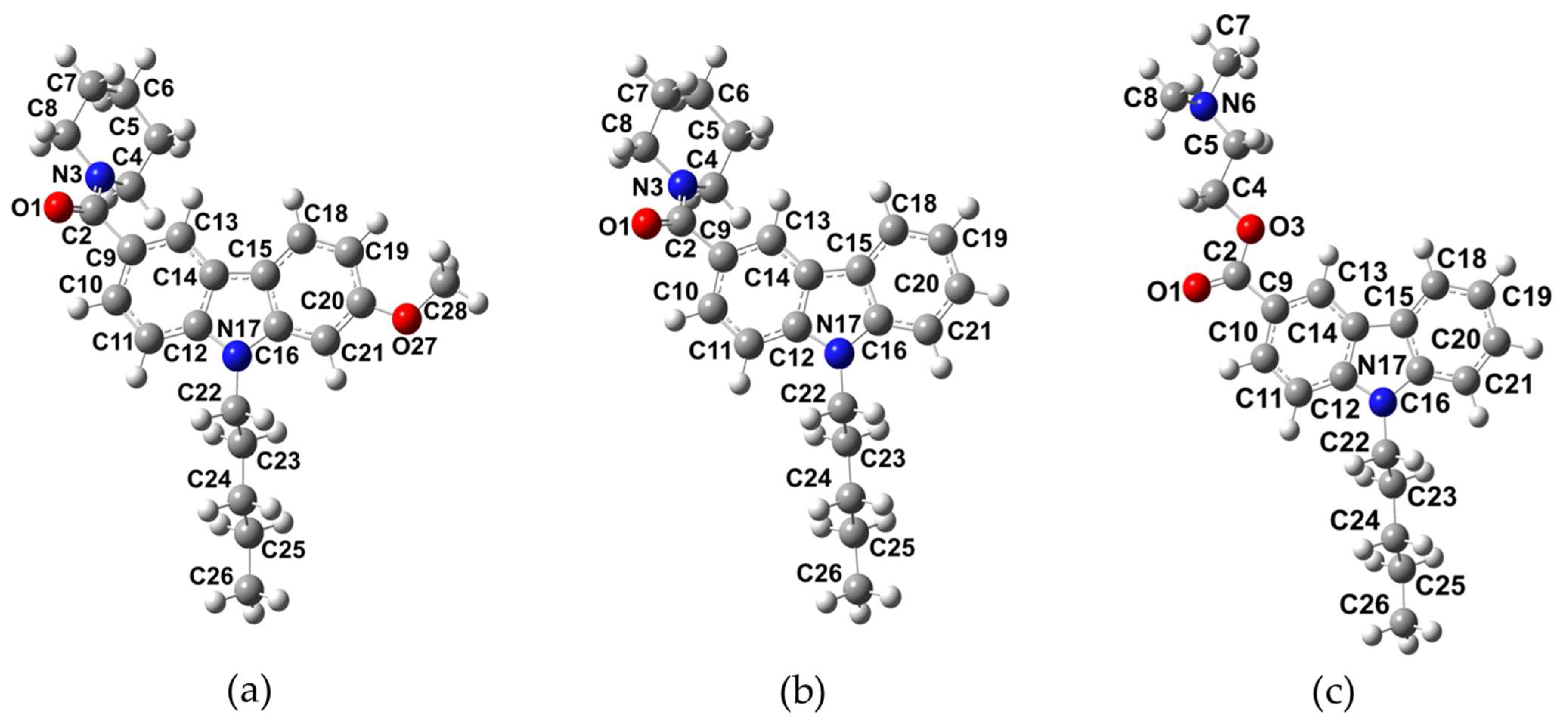
2.1. Molecular Structure of NMP Compounds
2.2. NMR and IR Calculations
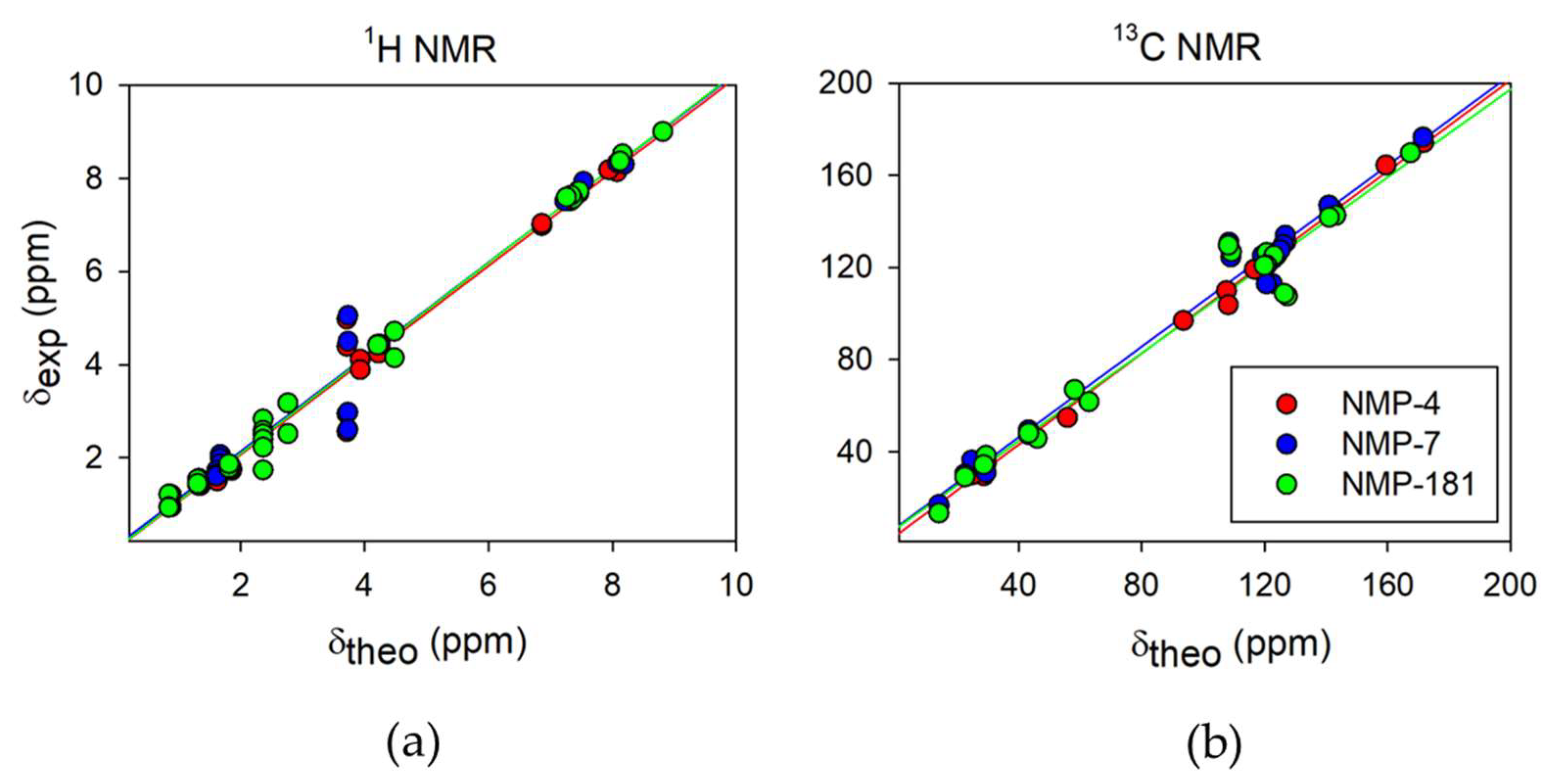
2.2.1. 1H and 13C NMR
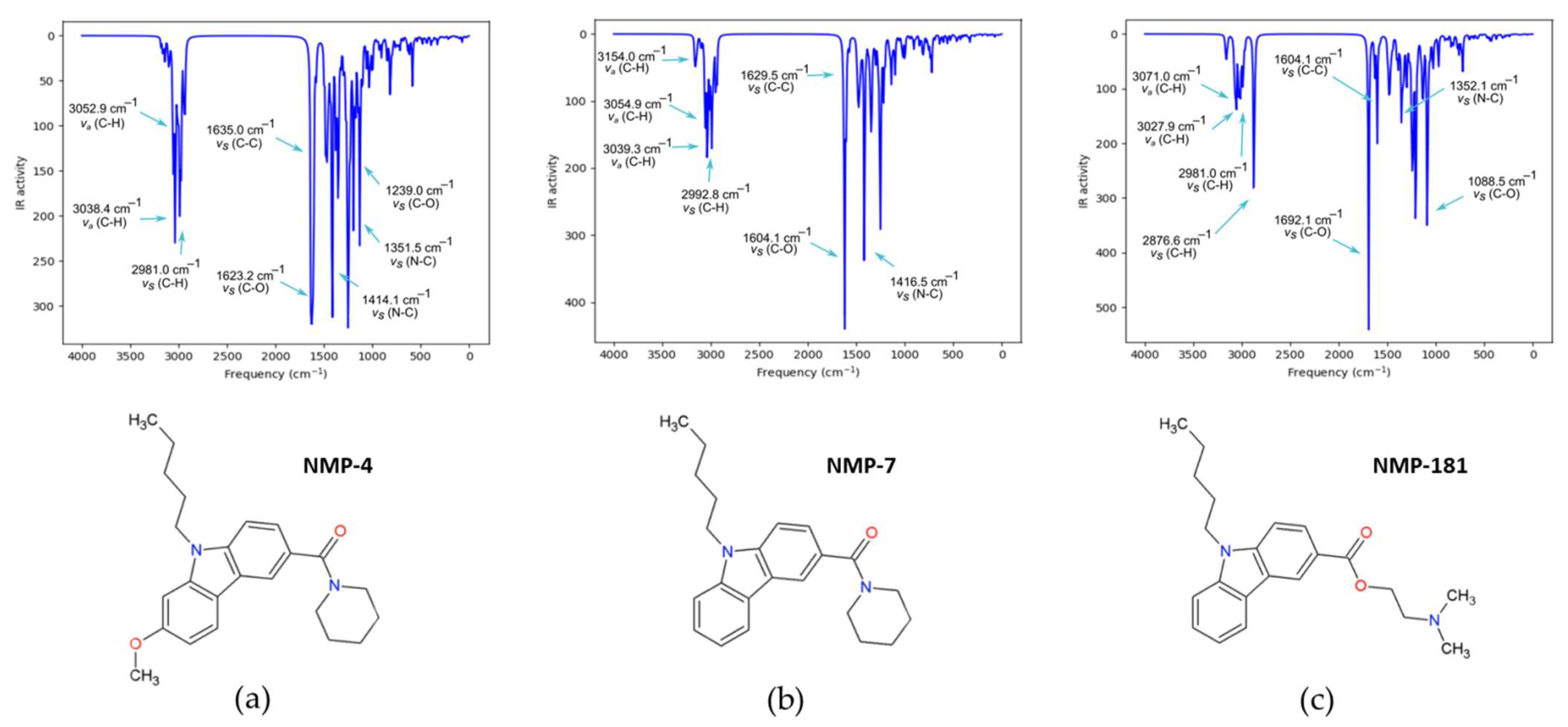
2.2.2. IR Characterization
2.3. Electronic Properties
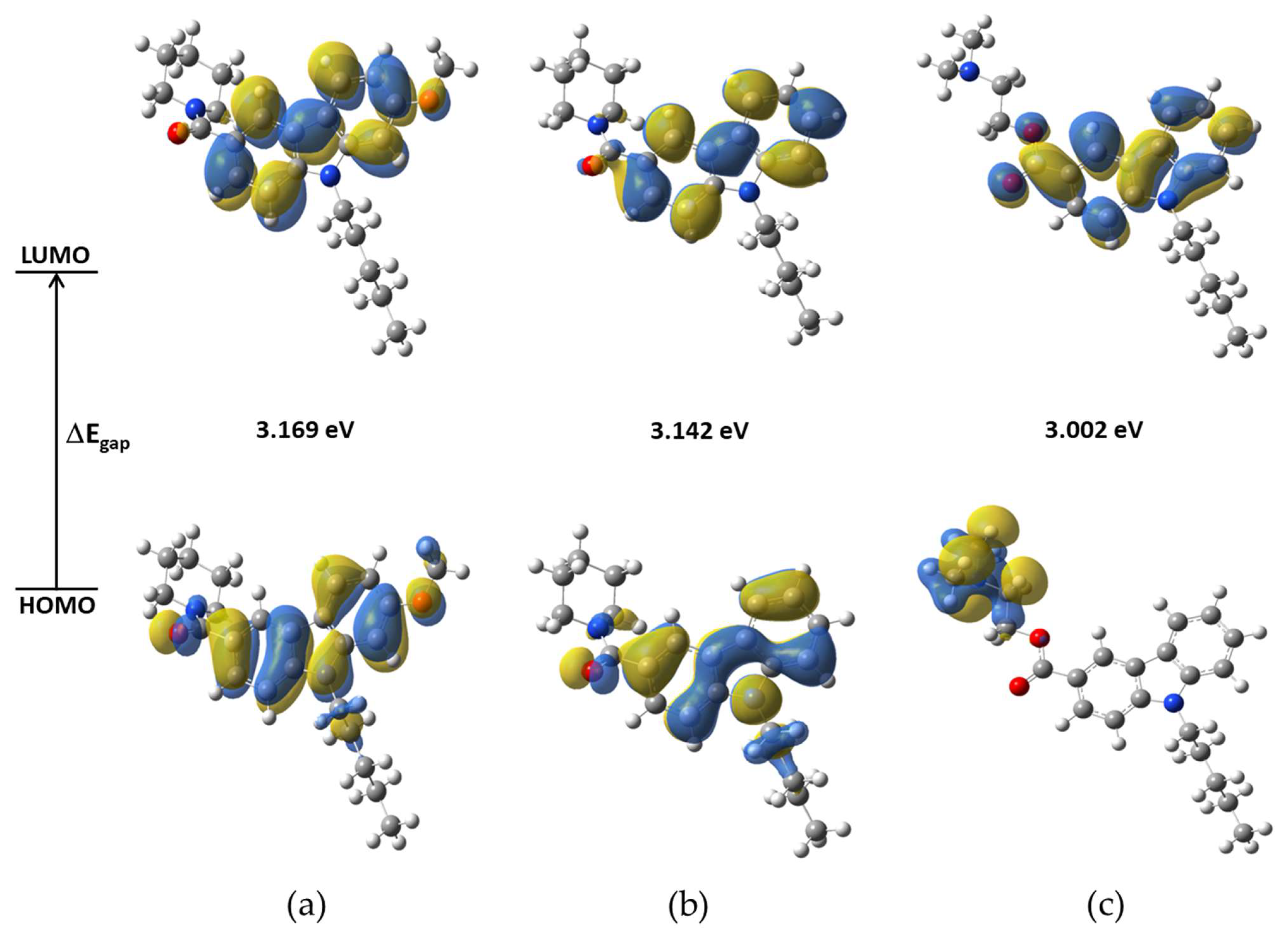
2.3.1. Frontier Molecular Orbitals (FMO)
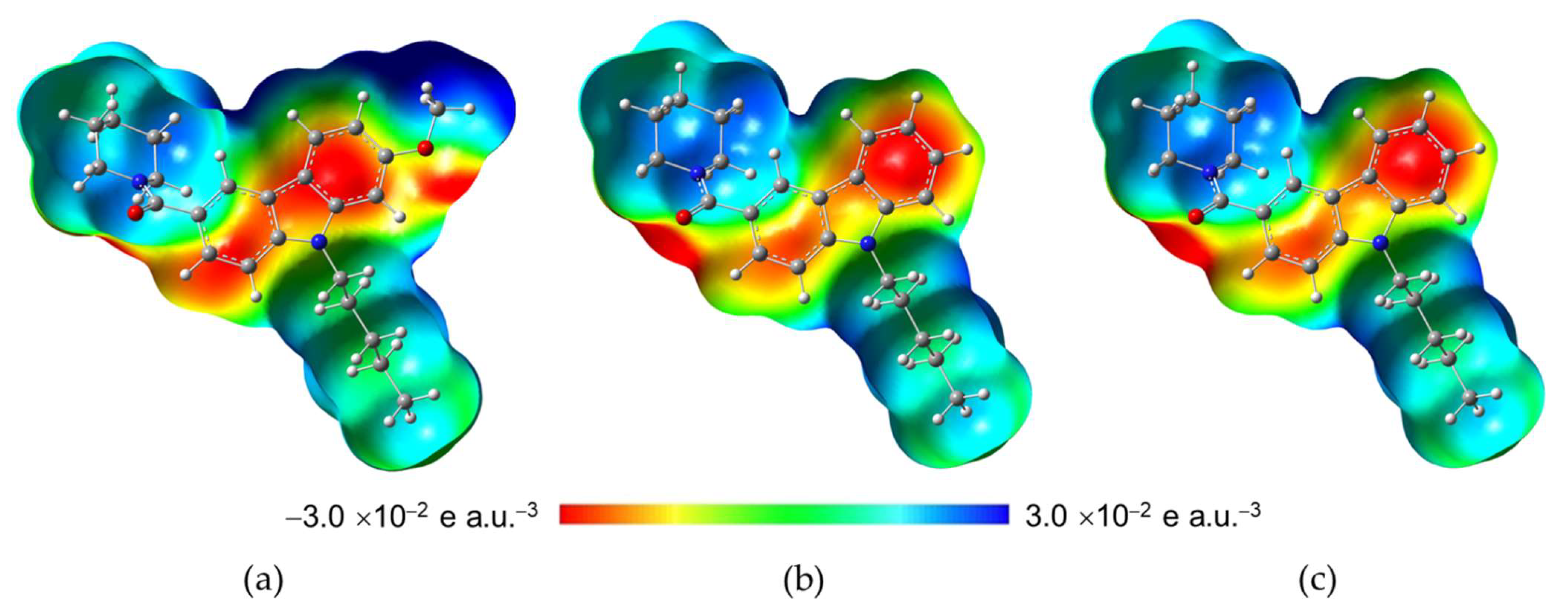
2.3.2. Molecular Electrostatic Potential (MEP)
2.3.3. Natural Bond Orbitals (NBO)
2.4. Reactivity Analysis
2.4.1. Global Reactivity Descriptors
2.4.2. Local Reactivity Descriptors
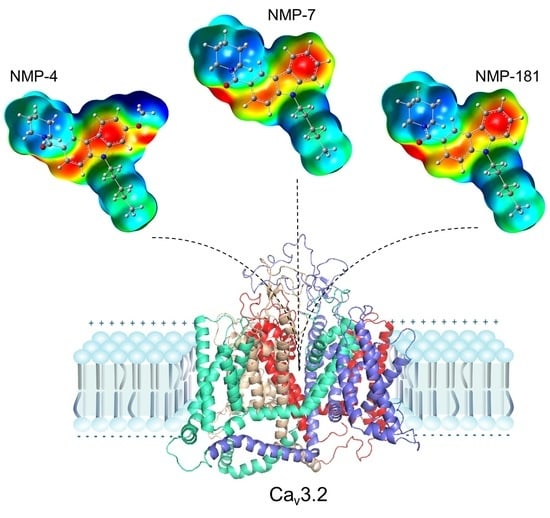
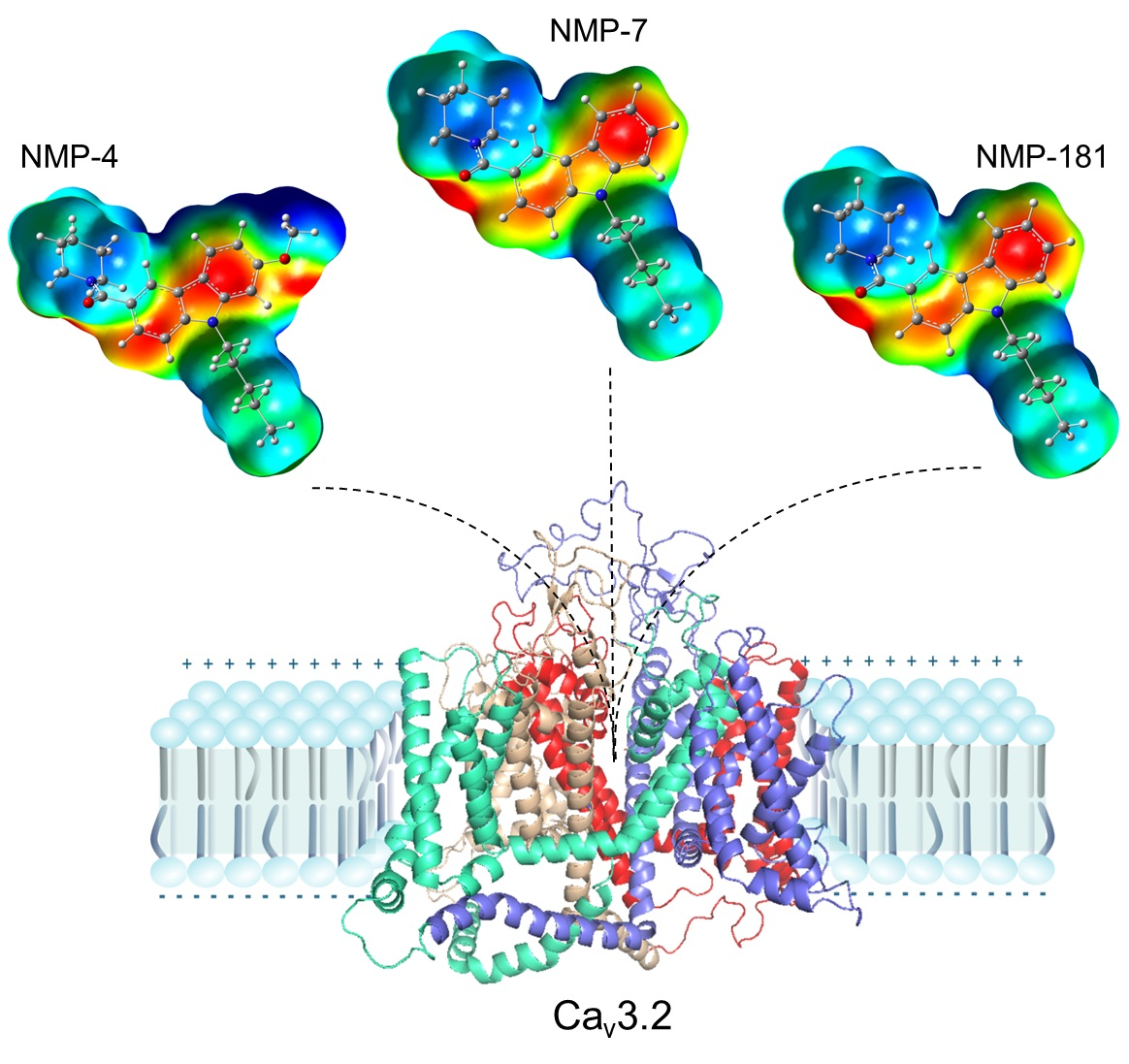
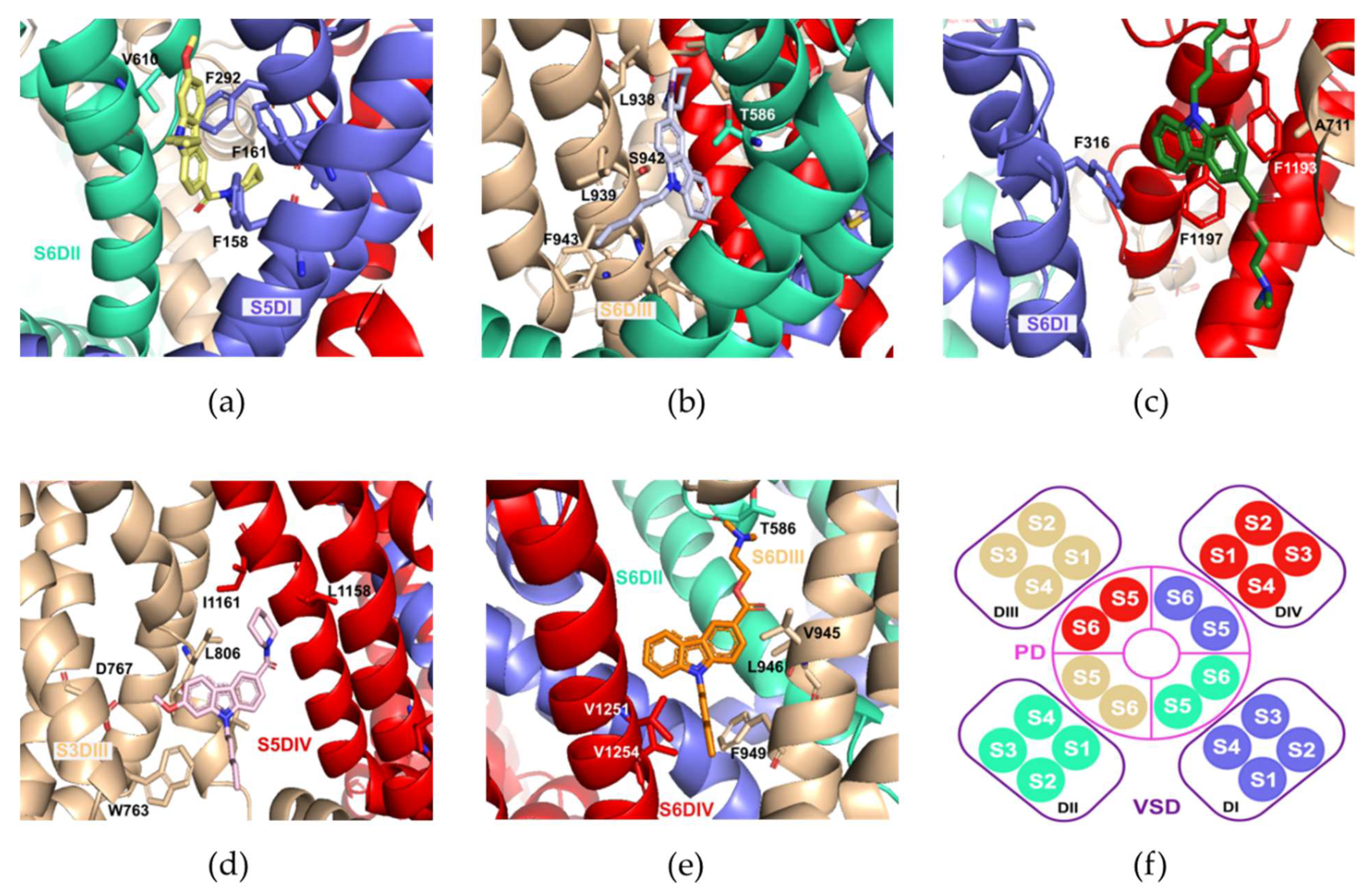
2.5. Molecular Docking Calculations
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, J. Cannabis, cannabinoid receptors, and endocannabinoid system: Yesterday, today, and tomorrow. Acta Pharmacol. Sin. 2019, 40, 297–299. [Google Scholar] [CrossRef]
- Pacher, P.; Bátkai, S.; Kunos, G. The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [Green Version]
- Colino, L.; Herranz-Herrer, J.; Gil-Benito, E.; Ponte-Lopez, T.; del Sol-Calderon, P.; Rodrigo-Yanguas, M.; Gil-Ligero, M.; Sánchez-López, A.J.; de Leon, J.; Blasco-Fontecilla, H. Cannabinoid Receptors, Mental Pain and Suicidal Behavior: A Systematic Review. Curr. Psychiatry Rep. 2018, 20, 19. [Google Scholar] [CrossRef]
- Todorovic, S.M.; Jevtovic-Todorovic, V. Neuropathic pain: Role for presynaptic T-type channels in nociceptive signaling. Pflugers Arch. Eur. J. Physiol. 2013, 465, 921–927. [Google Scholar] [CrossRef]
- Tibbs, G.R.; Posson, D.J.; Goldstein, P.A. Voltage-Gated Ion Channels in the PNS: Novel Therapies for Neuropathic Pain? Trends Pharmacol. Sci. 2016, 37, 522–542. [Google Scholar] [CrossRef]
- Snutch, T.P.; Zamponi, G.W. Recent advances in the development of T-type calcium channel blockers for pain intervention. Br. J. Pharmacol. 2018, 175, 2375–2383. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Cui, Y.; Sang, K.; Dong, Y.; Ni, Z.; Ma, S.; Hu, H. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 2018, 554, 317–322. [Google Scholar] [CrossRef]
- Knölker, H.J.; Reddy, K.R. Isolation and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2002, 102, 4303–4427. [Google Scholar] [CrossRef]
- Bashir, M.; Bano, A.; Ijaz, A.S.; Chaudhary, B.A. Recent developments and biological activities of n-substituted carbazole derivatives: A review. Molecules 2015, 20, 13496–13517. [Google Scholar] [CrossRef] [Green Version]
- Gadotti, V.M.; You, H.; Petrov, R.R.; Berger, N.D.; Diaz, P.; Zamponi, G.W. Analgesic Effect of a Mixed T-Type Channel Inhibitor/CB2 Receptor Agonist. Mol. Pain 2013, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Gadotti, V.M.; Petrov, R.R.; Zamponi, G.W.; Diaz, P. Functional characterization and analgesic effects of mixed cannabinoid receptor/T-type channel ligands. Mol. Pain 2011, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Berger, N.D.; Gadotti, V.M.; Petrov, R.R.; Chapman, K.; Diaz, P.; Zamponi, G.W. NMP-7 inhibits chronic inflammatory and neuropathic pain via block of Cav3.2 T-type calcium channels and activation of CB2 receptors. Mol. Pain 2014, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, R.; Webster, F. Spectrometric Identification of Organic Compounds, 7th ed.; John Wiley and Sons Inc.: New York, NY, USA, 2005. [Google Scholar]
- Pavia, D.; Lampman, G.; Kriz, G. Introduction to Spectroscopy: A Guide for Students of Organic Chemistry, 3rd ed.; Thomson Learning Inc.: Washington, DA, USA, 2001. [Google Scholar]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; John Wiley & Sons: Middlesex, UK, 2004. [Google Scholar]
- Abu-Dief, A.M.; El-khatib, R.M.; Aljohani, F.S.; Alzahrani, S.O.; Mahran, A.; Khalifa, M.E.; El-Metwaly, N.M. Synthesis and intensive characterization for novel Zn(II), Pd(II), Cr(III) and VO(II)-Schiff base complexes; DNA-interaction, DFT, drug-likeness and molecular docking studies. J. Mol. Struct. 2021, 1242, 130693. [Google Scholar] [CrossRef]
- Abdel-Rahman, L.H.; Adam, M.S.; Abu-Dief, A.M.; Ahmed, H.E.S.; Nafady, A. Non-Linear Optical Property and Biological Assays of Therapeutic Potentials Under In Vitro Conditions of Pd(II), Ag(I) and Cu(II) Complexes of 5-Diethyl amino-2-({2-[(2-hydroxy-Benzylidene)-amino]-phenylimino}-methyl)-phenol. Molecules 2020, 25, 5089. [Google Scholar] [CrossRef]
- Abdel-Rahman, L.H.; Abu-Dief, A.M.; Shehata, M.R.; Atlam, F.M.; Abdel-Mawgoud, A.A.H. Some new Ag(I), VO(II) and Pd(II) chelates incorporating tridentate imine ligand: Design, synthesis, structure elucidation, density functional theory calculations for DNA interaction, antimicrobial and anticancer activities and molecular docking studies. Appl. Organomet. Chem. 2019, 33, 1–24. [Google Scholar] [CrossRef]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons, Ltd.: Odense, Denmark, 2007; ISBN 978-0-470-01186-7. [Google Scholar]
- Serdaroğlu, G.; Ortiz, J.V. Ab Initio Calculations on some Antiepileptic Drugs such as Phenytoin, Phenbarbital, Ethosuximide and Carbamazepine. Struct. Chem. 2017, 28, 957–964. [Google Scholar] [CrossRef]
- Bladen, C.; McDaniel, S.W.; Gadotti, V.M.; Petrov, R.R.; Berger, N.D.; Diaz, P.; Zamponi, G.W. Characterization of novel cannabinoid based T-type calcium channel blockers with analgesic effects. ACS Chem. Neurosci. 2015, 6, 277–287. [Google Scholar] [CrossRef]
- Gómez-Jeria, J.S. A quantum-chemical analysis of the relationships between hCB2 cannabinoid receptor binding affinity and electronic structure in a family of 4-oxo-1,4-dihydroquinoline-3-carboxamide derivatives. Pharm. Lett. 2014, 6, 95–104. [Google Scholar]
- Gangadharana, R.P.; Krishnanb, S.S. Natural Bond Orbital (NBO) population analysis of 1-azanapthalene-8-ol. Acta Phys. Pol. A 2014, 125, 18–22. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [Green Version]
- Parthasarathi, R.; Subramanian, V.; Roy, D.R.; Chattaraj, P.K. Electrophilicity index as a possible descriptor of biological activity. Bioorg. Med. Chem. 2004, 12, 5533–5543. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, G.; Wu, Q.; Wu, K.; Li, R.; Lei, J.; Pan, X.; Yan, N. Cryo-EM structures of apo and antagonist-bound human Cav3.1. Nature 2019, 576, 492–497. [Google Scholar] [CrossRef]
- Mitterdorfer, J.; Grabner, M.; Kraus, R.L.; Hering, S.; Prinz, H.; Glossmann, H.; Striessnig, J. Molecular basis of drug interaction with L-type Ca2+ channels. J. Bioenerg. Biomembr. 1998, 30, 319–334. [Google Scholar] [CrossRef]
- Hughes, T.E.T.; Pumroy, R.A.; Yazici, A.T.; Kasimova, M.A.; Fluck, E.C.; Huynh, K.W.; Samanta, A.; Molugu, S.K.; Zhou, Z.H.; Carnevale, V.; et al. Structural insights on TRPV5 gating by endogenous modulators. Nat. Commun. 2018, 9, 4198. [Google Scholar] [CrossRef] [Green Version]
- Marksteiner, R.; Schurr, P.; Berjukow, S.; Margreiter, E.; Perez-Reyes, E.; Hering, S. Inactivation determinants in segment IIIS6 of Cav3.1. J. Physiol. 2001, 537, 27–34. [Google Scholar] [CrossRef]
- Rangel-Galván, M.; Rangel, A.; Romero-Méndez, C.; Dávila, E.M.; Castro, M.E.; Caballero, N.A.; Meléndez Bustamante, F.J.; Sanchez-Gaytan, B.L.; Meza, U.; Perez-Aguilar, J.M. Inhibitory Mechanism of the Isoflavone Derivative Genistein in the Human Cav3.3 Channel. ACS Chem. Neurosci. 2021, 12, 651–659. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Jamróz, M.H. Vibrational energy distribution analysis (VEDA): Scopes and limitations. Spectrochim. Acta—Part A Mol. Biomol. Spectrosc. 2013, 114, 220–230. [Google Scholar] [CrossRef]
- Alecu, I.M.; Zheng, J.; Zhao, Y.; Truhlar, D.G. Computational thermochemistry: Scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J. Chem. Theory Comput. 2010, 6, 2872–2887. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.O1; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Tood, A.K.; Jhon, M.M. GaussView; Version 6.1; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Khosravani, H.; Bladen, C.; Parker, D.B.; Snutch, T.P.; McRory, J.E.; Zamponi, G.W. Effects of Cav3.2 channel mutations linked to idiopathic generalized epilepsy. Ann. Neurol. 2005, 57, 745–749. [Google Scholar] [CrossRef]
- Peloquin, J.B.; Khosravani, H.; Barr, W.; Bladen, C.; Evans, R.; Mezeyova, J.; Parker, D.; Snutch, T.P.; McRory, J.E.; Zamponi, G.W. Functional analysis of Cav3.2 T-type calcium channel mutations linked to childhood absence epilepsy. Epilepsia 2006, 47, 655–658. [Google Scholar] [CrossRef]
- Arias-Olguín, I.I.; Vitko, I.; Fortuna, M.; Baumgart, J.P.; Sokolova, S.; Shumilin, I.A.; Van Deusen, A.; Soriano-García, M.; Gomora, J.C.; Perez-Reyes, E. Characterization of the gating brake in the I-II loop of Cav3.2 T-type Ca2+ channels. J. Biol. Chem. 2008, 283, 8136–8144. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L. The PyMOL Molecular Graphics System; Version 2.0; DeLano Scientific LCC: Palo Alto, CA, USA, 2017. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | NMP-4 | NMP-7 | NMP-181 |
|---|---|---|---|
| O1-C2 | 1.24 | 1.24 | 1.23 |
| N3-C2 | 1.38 | 1.38 | - |
| O3-C2 | - | - | 1.36 |
| N3-C4 | 1.47 | 1.47 | - |
| O3-C4 | - | - | 1.45 |
| N17-C22 | 1.46 | 1.46 | 1.46 |
| O1-C2-N3 | 122 | 122 | - |
| O1-C2-O3 | - | - | 123 |
| C2-N3-C4 | 119 | 119 | - |
| C2-O3-C4 | - | - | 115 |
| O1-C2-C9 | 120 | 119 | 125 |
| C2-C9-C10 | 118 | 117 | 118 |
| C22-C23-C24 | 112 | 112 | 112 |
| C28-O27-C20 | 118 | - | - |
| C6-C7-C8 | 111 | 111 | - |
| O3-C4-C5 | - | - | 107 |
| O1-C2-N3-C4 | −160 | −161 | - |
| O1-C2-O3-C4 | - | - | 0 |
| C5-C4-N3-C2 | −138 | −136 | - |
| C5-C4-O3-C2 | - | - | 174 |
| C9-C2-N3-C4 | 22 | 21 | - |
| C9-C2-O3-C4 | - | - | −180 |
| C23-C24-C25-C26 | 180 | 180 | −179 |
| C19-C20-O27-C28 | 0 | - | - |
| C6-C7-C8-N3 | 54 | 55 | - |
| O3-C4-C5-N6 | - | - | 179 |
| NMP-4 | NMP-7 | NMP-181 | |
|---|---|---|---|
| µ | −3.44 | −3.57 | −3.62 |
| χ | 3.44 | 3.57 | 3.62 |
| η | 3.17 | 3.14 | 3.00 |
| s | 0.32 | 0.32 | 0.33 |
| ω | 1.87 | 2.02 | 2.19 |
| Sites | NMP-4 | NMP-7 | NMP-181 |
|---|---|---|---|
| S6DI | −9.1 | −10.0 | −9.6 |
| S6DII | −9.5 | −10.0 | −9.1 |
| S6DIII | −9.5 | −10.1 | −9.2 |
| S5DIV | −7.9 | −8.1 | −8.2 |
| Pore-blocking | −9.4 | −9.1 | −8.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangel-Galván, M.; Castro, M.E.; Perez-Aguilar, J.M.; Caballero, N.A.; Rangel-Huerta, A.; Melendez, F.J. Theoretical Study of the Structural Stability, Chemical Reactivity, and Protein Interaction for NMP Compounds as Modulators of the Endocannabinoid System. Molecules 2022, 27, 414. https://doi.org/10.3390/molecules27020414
Rangel-Galván M, Castro ME, Perez-Aguilar JM, Caballero NA, Rangel-Huerta A, Melendez FJ. Theoretical Study of the Structural Stability, Chemical Reactivity, and Protein Interaction for NMP Compounds as Modulators of the Endocannabinoid System. Molecules. 2022; 27(2):414. https://doi.org/10.3390/molecules27020414
Chicago/Turabian StyleRangel-Galván, Maricruz, María Eugenia Castro, Jose Manuel Perez-Aguilar, Norma A. Caballero, Alejandro Rangel-Huerta, and Francisco J. Melendez. 2022. "Theoretical Study of the Structural Stability, Chemical Reactivity, and Protein Interaction for NMP Compounds as Modulators of the Endocannabinoid System" Molecules 27, no. 2: 414. https://doi.org/10.3390/molecules27020414
APA StyleRangel-Galván, M., Castro, M. E., Perez-Aguilar, J. M., Caballero, N. A., Rangel-Huerta, A., & Melendez, F. J. (2022). Theoretical Study of the Structural Stability, Chemical Reactivity, and Protein Interaction for NMP Compounds as Modulators of the Endocannabinoid System. Molecules, 27(2), 414. https://doi.org/10.3390/molecules27020414