
Replicates Number for Drug Stability Testing during Bioanalytical Method Validation—An Experimental and Retrospective Approach


Abstract
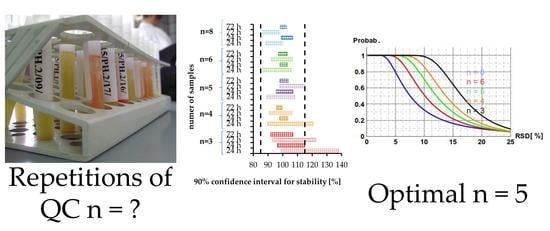
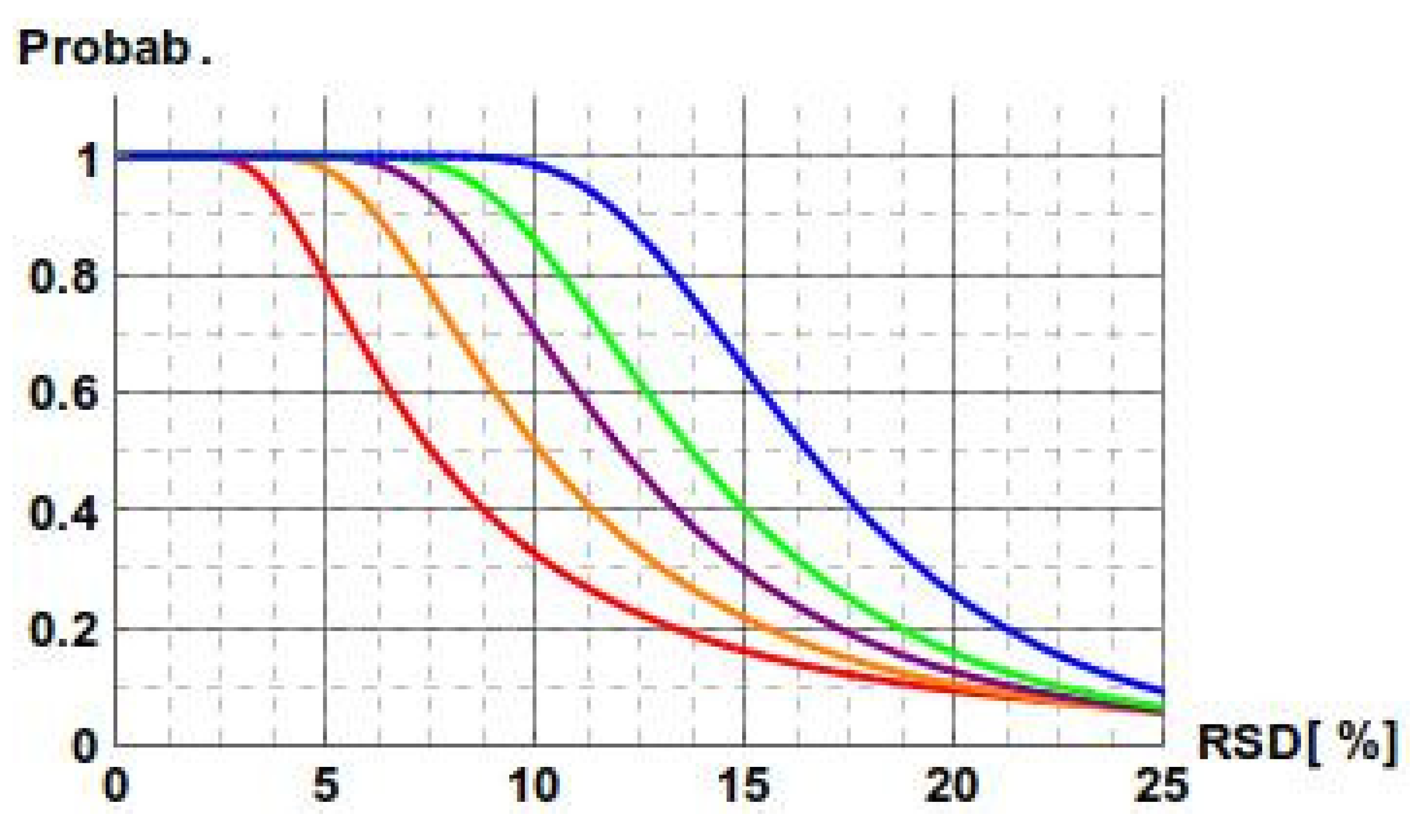
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Mass Spectrometric and Chromatographic Conditions
2.3. Stock Solution, Calibration Standards, and Quality Control Samples
2.4. Sample Preparation
2.5. Stability Evaluation and Statistical Methods
- χn−1—cumulative distribution function of the chi-square distribution for degrees of freedom (df) = n − 1;
- n—number of repetitions;
- k—the value of the Student t-distribution quantile at a 0.1 significance level for n − 1 degrees of freedom (df);
- —standard deviation in stability.
2.6. Retrospective Analysis
3. Results
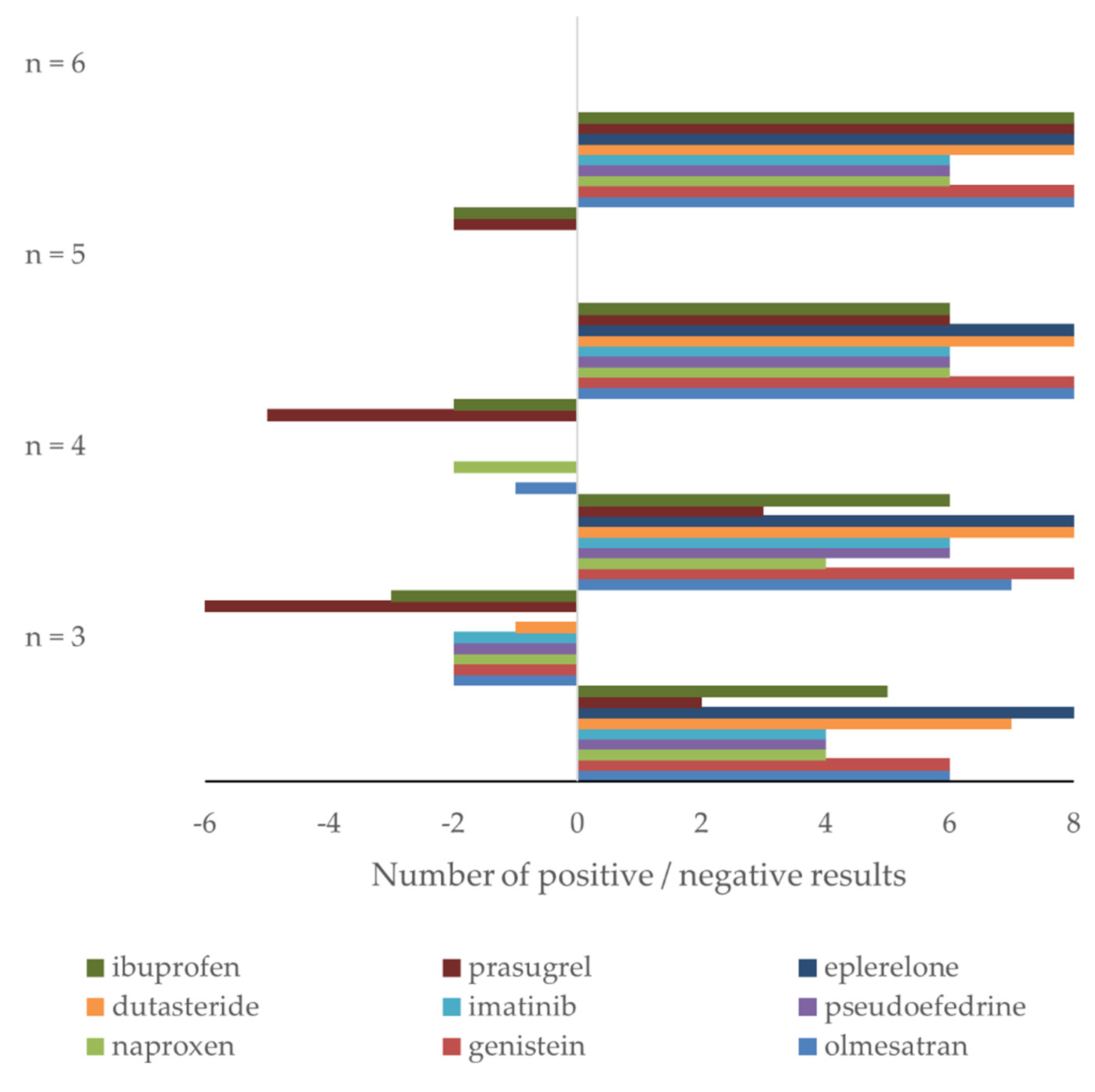
3.1. Experimental and Mathematical Studies
3.2. Retrospective Study
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Samples (n) | Low QC (ng/mL) | High QC (ng/mL) | ||||||
|---|---|---|---|---|---|---|---|---|
| Reference for 24 h | Tested 24 h | Reference for 72 h | Tested 72 h | Reference for 24 h | Tested 24 h | Reference for 72 h | Tested 72 h | |
| 8 | 14.9 | 14.5 | 14.2 | 14.6 | 573 | 567 | 590 | 640 |
| 15.2 | 14.7 | 14.6 | 14.6 | 582 | 569 | 594 | 627 | |
| 15.4 | 14.8 | 14.7 | 14.6 | 584 | 574 | 572 | 611 | |
| 15.5 | 15.4 | 14.9 | 14.9 | 586 | 575 | 589 | 578 | |
| 15.6 | 15.4 | 15.1 | 14.9 | 588 | 579 | 580 | 617 | |
| 15.8 | 15.5 | 15.3 | 14.9 | 590 | 587 | 609 | 609 | |
| 15.9 | 15.6 | 15.5 | 15.4 | 594 | 591 | 586 | 584 | |
| 16.0 | 16.3 | 15.7 | 16.2 | 597 | 615 | 582 | 589 | |
| 6 | 14.7 | 14.0 | 13.9 | 14.4 | 568 | 559 | 574 | 582 |
| 14.8 | 14.5 | 13.9 | 15.1 | 573 | 564 | 578 | 585 | |
| 15.1 | 14.9 | 14.4 | 15.3 | 577 | 567 | 583 | 587 | |
| 15.2 | 15.1 | 14.5 | 15.6 | 587 | 583 | 596 | 591 | |
| 15.7 | 15.3 | 14.5 | 15.7 | 598 | 591 | 603 | 594 | |
| 16.3 | 16.0 | 14.7 | 15.7 | 600 | 630 | 606 | 602 | |
| 5 | 14.7 | 14.8 | 14.9 | 14.5 | 555 | 572 | 553 | 569 |
| 14.9 | 14.9 | 15.0 | 14.8 | 559 | 579 | 585 | 581 | |
| 14.9 | 15.2 | 15.1 | 15.2 | 566 | 584 | 589 | 588 | |
| 15.0 | 15.4 | 15.2 | 15.8 | 578 | 601 | 590 | 596 | |
| 15.4 | 15.6 | 15.2 | 16.1 | 591 | 601 | 594 | 599 | |
| 4 | 14.3 | 14.3 | 13.8 | 14.3 | 594 | 571 | 579 | 559 |
| 14.9 | 14.3 | 14.9 | 14.7 | 594 | 582 | 579 | 567 | |
| 14.9 | 15.1 | 15.5 | 15.5 | 595 | 585 | 582 | 592 | |
| 15.0 | 15.7 | 15.7 | 15.8 | 606 | 588 | 597 | 597 | |
| 3 | 15.0 | 14.9 | 14.6 | 14.7 | 570 | 565 | 575 | 584 |
| 15.6 | 15.2 | 15.4 | 15.4 | 574 | 578 | 589 | 598 | |
| 16.4 | 15.5 | 16.2 | 15.6 | 580 | 642 | 592 | 603 | |
| Number of Samples (n) | Low QC (ng/mL) | High QC (ng/mL) | ||||||
|---|---|---|---|---|---|---|---|---|
| Reference for 24 h | Tested 24 h | Reference for 72 h | Tested 72 h | Reference for 24 h | Tested 24 h | Reference for 72 h | Tested 72 h | |
| 8 | 7.59 | 6.87 | 6.74 | 6.40 | 112 | 132 | 117 | 118 |
| 7.21 | 6.88 | 7.07 | 6.47 | 111 | 115 | 118 | 119 | |
| 7.65 | 6.90 | 7.55 | 6.66 | 112 | 117 | 118 | 121 | |
| 7.89 | 7.22 | 7.64 | 6.70 | 114 | 117 | 122 | 122 | |
| 7.93 | 7.41 | 7.79 | 6.95 | 117 | 117 | 122 | 122 | |
| 8.16 | 7.67 | 7.92 | 7.34 | 117 | 118 | 123 | 123 | |
| 8.36 | 8.02 | 7.94 | 7.68 | 119 | 119 | 123 | 124 | |
| 8.37 | 8.52 | 8.28 | 7.69 | 126 | 119 | 127 | 124 | |
| 6 | 7.89 | 6.37 | 7.16 | 6.61 | 114 | 117 | 117 | 125 |
| 7.35 | 6.44 | 7.21 | 6.81 | 116 | 118 | 118 | 116 | |
| 7.77 | 8.08 | 7.23 | 7.11 | 116 | 118 | 119 | 119 | |
| 8.11 | 8.19 | 7.46 | 7.45 | 120 | 120 | 124 | 122 | |
| 8.22 | 8.31 | 7.73 | 8.04 | 120 | 120 | 124 | 122 | |
| 8.25 | 8.36 | 7.82 | 8.32 | 124 | 121 | 125 | 123 | |
| 5 | 7.02 | 7.85 | 6.64 | 6.87 | 119 | 124 | 117 | 119 |
| 7.48 | 6.24 | 6.71 | 7.16 | 113 | 111 | 120 | 120 | |
| 7.52 | 7.70 | 6.72 | 7.33 | 113 | 114 | 121 | 121 | |
| 7.87 | 7.84 | 7.37 | 7.69 | 117 | 115 | 122 | 122 | |
| 8.20 | 7.84 | 7.68 | 8.22 | 122 | 127 | 124 | 127 | |
| 4 | 6.98 | 7.66 | 6.94 | 6.40 | 117 | 116 | 122 | 118 |
| 6.42 | 7.52 | 7.02 | 6.83 | 117 | 118 | 122 | 119 | |
| 8.39 | 7.55 | 7.09 | 6.97 | 119 | 119 | 124 | 120 | |
| 8.46 | 8.61 | 7.33 | 7.74 | 120 | 123 | 124 | 123 | |
| 3 | 7.05 | 7.64 | 6.70 | 6.41 | 123 | 117 | 115 | 120 |
| 6.39 | 8.54 | 6.84 | 7.54 | 116 | 125 | 127 | 122 | |
| 8.01 | 8.80 | 6.88 | 7.93 | 117 | 133 | 128 | 124 | |
| Drug | Method | Internal Standard | Low/High QC (ng/mL) | Type of Extraction | Source |
|---|---|---|---|---|---|
| Dutasteride | HPLC, ESI + | [13C6]-dutasteride | 0.3/2.8 | LLE | [24] |
| Eplerenon | HPLC-MS, ESI + | [2H3]-eplerenone | 50/1500 | LLE | [23] |
| Genistein | HPLC-MS, ESI − | [2H4]-genistein | 50/2000 | LLE | N/A |
| Ibuprofen | HPLC-UV, λ = 220 nm | naproxen | 900/24,000 | LLE | N/A |
| Imatinib | HPLC-UV, λ = 265 nm | propranolol hydrochloride | 120/3200 | LLE | [22] |
| Naproxen | HPLC-UV, λ = 265 nm | ibuprofen | 1500/60,000 | LLE | [20] |
| Olmesartan | HPLC-MS, ESI + | [2H6]-olmesarta | 15/2000 | LLE | [21] |
| Prasugrel | HPLC-MS/MS, ESI + | [13C6] R-138727 | 1.5/200 | LLE | N/A |
| Pseudoephedrine | HPLC-MS/MS, ESI + | [2H3][13C6]-pseudoephedrine | 4.5/240 | LLE | N/A |





References
- Guideline on Bioanalytical Method Validation; EMEA/CHMP/EWP/192217/2009; Committee for Medicinal Products for Human Use (CHMP), European Medicines Agency: London, UK, 2011.
- Guidance for Industry: Bioanalytical Method Validation; Food and Drug Administration; Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM): Rockville, MD, USA, 2018.
- Draft ICH Guideline M10 on Bioanalytical Method Validation; EMA/CHMP/ICH/172948/2019; Committee for Human Medicinal Products, European Medicines Agency: London, UK, 2019.
- Kaza, M.; Karaźniewicz-Łada, M.; Kosicka, K.; Siemiątkowska, A.; Rudzki, P.J. Bioanalytical method validation: New FDA guidance vs. EMA guideline. Better or worse? J. Pharm. Biomed. Anal. 2019, 165, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Health Canada’s: Notice Clarification of Bioanalytical Method Validation Procedures. 2015. Available online: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/announcements/notice-clarification-bioanalytical-method-validation-procedures.html (accessed on 6 December 2021).
- Timm, U.; Wall, M.; Dell, D. A New Approach for Dealing with the Stability of Drugs in Biological Fluids. J. Pharm. Sci. 1985, 74, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Rudzki, P.J.; Leś, A. Application of confidence intervals to bioanalytical method validation-drug stability in biological matrix testing. Acta Pol. Pharm. 2008, 65, 743–747. [Google Scholar] [PubMed]
- Guideline on the Investigation of Bioequivalence; CPMP/EWP/QWP/1401/98/Rev. 1/Corr**; Committee for Human Medicinal Products, European Medicines Agency: London, UK, 2010.
- Neyman, J. Outline of a theory of statistical estimation based on the classical theory of probability. Philos. Trans. R. Soc. Lond. Ser. A Math. Phys. Sci. 1937, 236, 333–380. [Google Scholar]
- Rudzki, P.J.; Jarus-Dziedzic, K.; Filist, M.; Gilant, E.; Buś-Kwaśnik, K.; Leś, A.; Sasinowska-Motyl, M.; Nagraba, Ł.; Bujalska-Zadrożny, M. Evaluation of tramadol human pharmacokinetics and safety after co-administration of magnesium ions in randomized, single- and multiple-dose studies. Pharmacol. Rep. 2021, 73, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Filist, M.; Szlaska, I.; Kaza, M.; Pawiński, T. Validated HPLC-UV method for determination of naproxen in human plasma with proven selectivity against ibuprofen and paracetamol. Biomed. Chromatogr. 2016, 30, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Piórkowska, E.; Musijowski, J.; Buś-Kwaśnik, K.; Rudzki, P.J. Is a deuterated internal standard appropriate for the reliable determination of olmesartan in human plasma? J. Chrom. B 2017, 1040, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Kaza, M.; Piorkowska, E.; Filist, M.; Rudzki, P.J. HPLC-UV assay of imatinib in human plasma optimized for bioequivalence studies. Acta Pol. Pharm. 2016, 73, 1495–1503. [Google Scholar] [PubMed]
- Buś-Kwaśnik, K.; Filist, M.; Rudzki, P.J. Environmentally friendly LC/MS determination of eplerenone in human plasma. Acta Pol. Pharm. 2016, 73, 1487–1493. [Google Scholar] [PubMed]
- Gniazdowska, E.; Kaza, M.; Buś-Kwaśnik, K.; Giebułtowicz, J. LC-MS/MS determination of dutasteride and its major metabolites in human plasma. J. Pharm. Biomed. Anal. 2021, 206, 114362. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, L.; Micheletti, E.; Carassa, R.; Bruttini, C.; Fausto, R.; Katsanos, A.; Riva, I. Efficacy and Safety of PreserFlo® MicroShunt After a Failed Trabeculectomy in Eyes with Primary Open-Angle Glaucoma: A Retrospective Study. Adv. Ther. 2021, 38, 4403–4412. [Google Scholar] [CrossRef] [PubMed]
- Wilde, H.; Dennis, J.M.; McGovern, A.P.; Vollmer, S.J.; Mateen, B.A. A national retrospective study of the association between serious operational problems and COVID-19 specific intensive care mortality risk. PLoS ONE 2021, 16, e0255377. [Google Scholar] [CrossRef] [PubMed]
- Monakhova, Y.B.; Diehl, B.W.K. Retrospective multivariate analysis of pharmaceutical preparations using (1)H nuclear magnetic resonance (NMR) spectroscopy: Example of 990 heparin samples. J. Pharm. Biomed. Anal. 2019, 173, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.; Jeon, W.; Choi, Y.J.; Yang, H.; Lee, J. Impact of drug formulation on outcomes of pharmaceutical poisoning in children aged 7 years or younger: A retrospective observational study in South Korea. Medicine 2021, 100, e27485. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, T.; Kudo, T.; Jinno, F.; Schmidt, E.R.; Kondo, T. Retrospective Data Analysis and Proposal of a Practical Acceptance Criterion for Inter-laboratory Cross-validation of Bioanalytical Methods Using Liquid Chromatography/Tandem Mass Spectrometry. AAPS J. 2014, 16, 1226–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, P.M.; Wietecha-Posłuszny, R.; Pawliszyn, J. White Analytical Chemistry: An approach to reconcile the principles of Green Analytical Chemistry and functionality. TrAC Trends Anal. Chem. 2021, 138, 116223. [Google Scholar] [CrossRef]
- Watanabe, K.; Varesio, E.; Hopfgartner, G. Parallel ultra high pressure liquid chromatography–mass spectrometry for the quantification of HIV protease inhibitors using dried spot sample collection format. J. Chrom. B 2014, 965, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Pihl, S.; Huusom, A.K.T.; Rohde, M.; Poulsen, M.N.; Jørgensen, M.; Kall, M.A. Evaluation of an isochronic study design for long-term frozen stability investigation of drugs in biological matrices. Bioanalysis 2010, 2, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Bourgogne, E.; Mathy, F.X.; Boucaut, D.; Boekens, H.; Laprevote, O. Simultaneous quantitation of histamine and its major metabolite 1-methylhistamine in brain dialysates by using precolumn derivatization prior to HILIC-MS/MS analysis. Anal. Bioanal. Chem. 2012, 402, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Barker, S.; Freisleben, A.; Laakso, S.; Staelens, L.; White, S.; Timmerman, P. European Bioanalysis Forum recommendation on the best practices to demonstrate processed sample stability. Bioanalysis 2019, 11, 7–11. [Google Scholar] [CrossRef] [PubMed]







| Retention Time (min) | MRM [m/z] | DP [V] | CE [V] | CXP [V] | |
|---|---|---|---|---|---|
| tramadol | 3.4 | 264.2 > 42.3 | 51 | 125 | 10 |
| tramadol-d6 | 3.4 | 270.3 > 252.2 | 66 | 17 | 16 |
| O-desmethyl-tramadol | 2.6 | 250.2 > 232.2 | 71 | 17 | 18 |
| O-desmethyl-tramadol-d6 | 2.6 | 256.0 > 238.3 | 61 | 17 | 14 |
| Low QC | High QC | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of Pairs | 3 | 4 | 5 | 6 | 8 | 3 | 4 | 5 | 6 | 8 |
| Experimental Data for Tramadol and O-desmethyl-tramadol (n = 4 of results at each column) | ||||||||||
| Mean | 23.8 | 17.7 | 12.4 | 10.2 | 8.5 | 14.2 | 4.8 | 7.2 | 5.5 | 5.1 |
| Geometric mean | 21.1 | 16.2 | 10.7 | 9.7 | 8.1 | 12.9 | 4.6 | 6.9 | 5.4 | 4.9 |
| Median | 22.1 | 15.1 | 12.8 | 9.9 | 8.6 | 16.3 | 4.7 | 6.0 | 5.5 | 4.6 |
| Min | 11.7 | 9.9 | 4.7 | 6.4 | 5.5 | 5.7 | 3.1 | 5.5 | 3.9 | 3.6 |
| Max | 39.1 | 30.8 | 19.5 | 14.5 | 11.4 | 18.5 | 6.8 | 11.4 | 7.3 | 7.7 |
| SD | 12.9 | 9.1 | 6.9 | 3.5 | 3.1 | 5.8 | 1.7 | 2.8 | 1.4 | 1.8 |
| RSD [%] | 54 | 51 | 56 | 34 | 37 | 41 | 35 | 39 | 26 | 36 |
| Retrospective Analysis (n = 33 of results at each column) | ||||||||||
| Mean | 21.5 | 14.9 | 11.4 | 9.1 | - | 11.9 | 8.4 | 6.4 | 5.1 | - |
| Geometric mean | 18.0 | 12.9 | 9.9 | 8.1 | - | 10.8 | 7.9 | 6.0 | 4.8 | - |
| Median | 18.5 | 12.8 | 10.1 | 7.7 | - | 10.9 | 7.6 | 5.8 | 4.6 | - |
| Min | 2.7 | 3.9 | 3.1 | 2.9 | - | 3.3 | 3.0 | 2.3 | 1.8 | - |
| Max | 54.3 | 37.6 | 28.2 | 23.2 | - | 28.8 | 19.5 | 14.6 | 12.9 | - |
| SD | 13.0 | 8.4 | 6.3 | 5.0 | - | 5.2 | 3.2 | 2.5 | 2.1 | - |
| RSD [%] | 57 | 54 | 52 | 53 | - | 43 | 38 | 38 | 40 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gniazdowska, E.; Goch, W.; Giebułtowicz, J.; Rudzki, P.J. Replicates Number for Drug Stability Testing during Bioanalytical Method Validation—An Experimental and Retrospective Approach. Molecules 2022, 27, 457. https://doi.org/10.3390/molecules27020457
Gniazdowska E, Goch W, Giebułtowicz J, Rudzki PJ. Replicates Number for Drug Stability Testing during Bioanalytical Method Validation—An Experimental and Retrospective Approach. Molecules. 2022; 27(2):457. https://doi.org/10.3390/molecules27020457
Chicago/Turabian StyleGniazdowska, Elżbieta, Wojciech Goch, Joanna Giebułtowicz, and Piotr J. Rudzki. 2022. "Replicates Number for Drug Stability Testing during Bioanalytical Method Validation—An Experimental and Retrospective Approach" Molecules 27, no. 2: 457. https://doi.org/10.3390/molecules27020457
APA StyleGniazdowska, E., Goch, W., Giebułtowicz, J., & Rudzki, P. J. (2022). Replicates Number for Drug Stability Testing during Bioanalytical Method Validation—An Experimental and Retrospective Approach. Molecules, 27(2), 457. https://doi.org/10.3390/molecules27020457