3.2. Experimental Procedures
General Procedure (A) for the Sonogashira coupling-intracellular cyclization to benzofuran intermediates. To a mixture of the appropriate o-halophenol (1 eq., 1 mmol), Pd(PPh3)4 (0.05 eq., 0.05 mmol), PPh3 (0.055 eq., 0.055 mmol), and CuI (0.05 eq., 0.05 mmol) under an N2 atmosphere, a degassed solution of the appropriate terminal alkyne (3 eq., 3 mmol) in anhydrous Et3N (6 mL) was added dropwise. The mixture was heated to 90 °C and stirred for 12 h. The resulting dark suspension was acidified with HCl (1 M) and extracted with EtOAc (3 × 5 mL). The combined organic phases were washed with NaHCO3 (s.s., 1 × 5 mL) and brine (1 × 5 mL), dried over Na2SO4, and evaporated under reduced pressure. The residue was purified by silica gel column chromatography.
General procedure (B) for the aldol condensation between substituted acetophenones and substituted aromatic aldehydes. To a solution of the appropriate acetophenone (1 eq., 1 mmol) in absolute EtOH (3 mL), cooled to 0 °C, NaOHaq. (5 M, 5 mmol) was added dropwise. The mixture was stirred at 0 °C for 30 min, and then a solution of the appropriate aromatic aldehyde (1.2 eq., 1.2 mmol) in absolute EtOH (1.5 mL) was added dropwise. The ice bath was removed, and the mixture was stirred at 25 °C for 48 h. The reaction was acidified with HCl (1 M) and extracted with EtOAc (3 × 5 mL). The combined organic phases were washed with NaHCO3 (s.s., 1 × 10 mL) and brine (1 × 5 mL), dried over Na2SO4, and evaporated under reduced pressure. The residue was purified by silica gel column chromatography.
General Procedure (C) for the methoxymethyl ether cleavage. To a 0 °C cooled solution of the appropriate chalcone (1 eq., 1 mmol) in MeOH/THF (1:1, 20 mL in total), HCl (12 M, 20 eq., 20 mmol) was added dropwise. The ice bath was removed, and the reaction mixture was heated to 40 °C for 3 h. After cooling, NaHCO3 (s.s.) was carefully added until pH = 7, and the aqueous phase was extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with brine (1 × 15 mL), dried over Na2SO4, and evaporated under reduced pressure. The residue was purified by silica gel column chromatography.
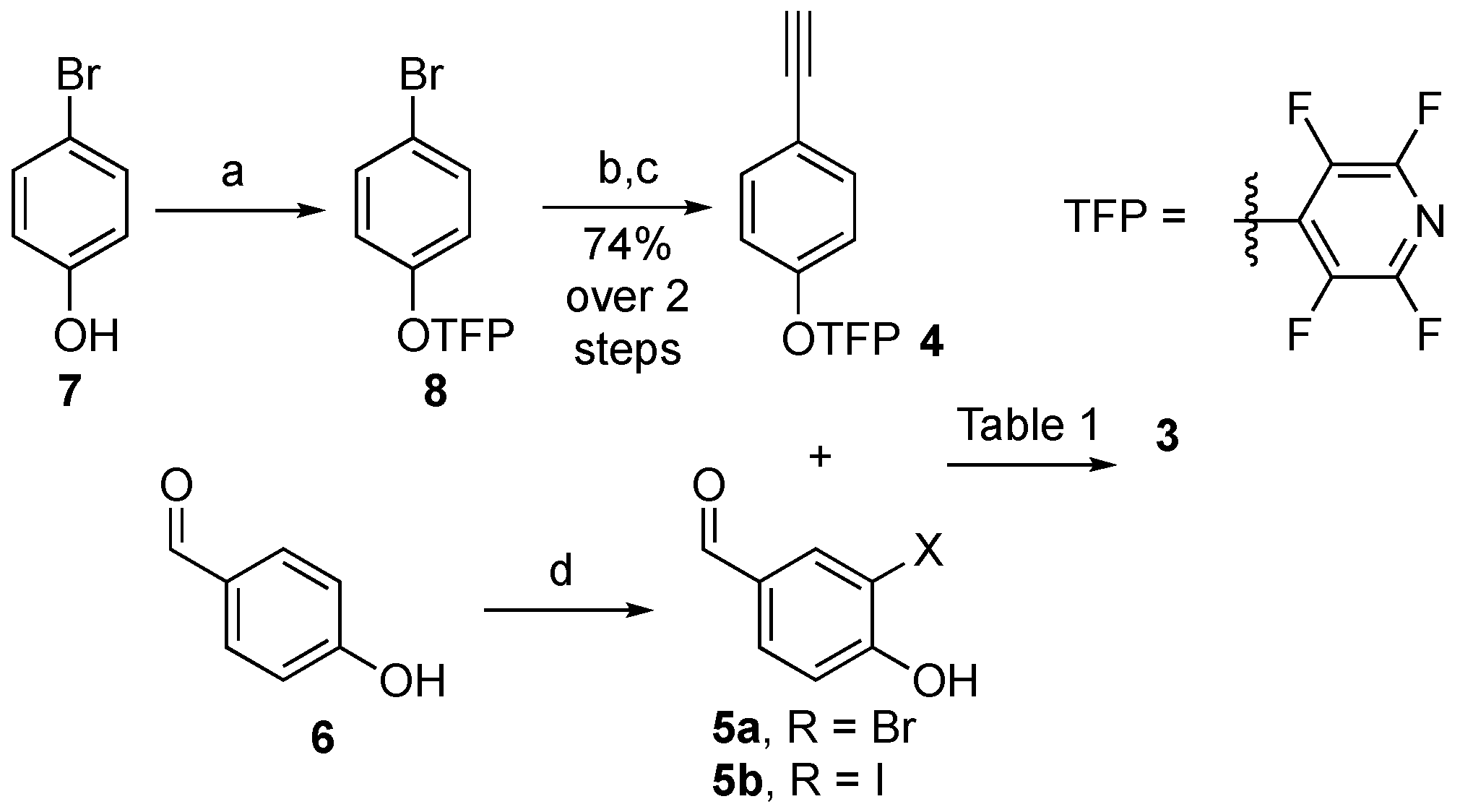
3-Bromo-4-hydroxybenzaldehyde (5a). To a stirred solution 4-hydroxybenzaldehyde (6, 1 eq., 1 mmol) in ACN (5 mL), a solution of N-bromosuccinimide (1.1 eq., 1.1 mmol) and thioacetamide (0.05 eq., 0.05 mmol) in ACN (5 mL) was added dropwise over 2 h using a dropping funnel. The mixture was stirred for 2 h, then quenched with Na2S2O3 (s.s., 10 mL) and extracted with EtOAc. (3 × 5 mL). The combined organic phases were washed with brine (5 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography (DCM/Hex 9:1) to afford the title compound (37% yield) as a white solid. 1H NMR (300 MHz, CDCl3) δ 9.83 (s, 1H, CHO), 8.04 (d, J = 1.9 Hz, 1H, C6-H), 7.77 (dd, J = 8.4, 1.9 Hz, 1H, C5-H), 7.15 (d, J = 8.4 Hz, 1H, C2-H), 6.30 (s, 1H, OH). ESI-MS, m/z [M-H]− 200 and 202 (1:1 ratio).
3-Iodo-4-hydroxybenzaldehyde (5b). To a stirred solution of 4-hydroxybenzaldehyde (6, 1 eq., 1 mmol) and AgNO3 (1 eq., 1 mmol) in MeOH (6 mL) at 25 °C, I2 (1.1 eq., 1.1 mmol) was added. The mixture was stirred for 24 h, quenched with Na2S2O3 (s.s., 10 mL), and extracted with EtOAc. (3 × 5 mL). The combined organic phases were washed with brine (5 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography (DCM/Hex 15:1) to afford the title compound (44% yield) as an off-white solid. 1H NMR (300 MHz, CDCl3) δ 9.83 (s, 1H CHO), 8.04 (d, J = 1.9 Hz, 1H. C6-H), 7.77 (dd, J = 8.4, 1.9 Hz, 1H, C5-H), 7.15 (d, J = 8.4 Hz, 1H, C2-H), 6.30 (s, 1H, OH). ESI-MS m/z 247 [M-H]−.
4-(4-Bromophenoxy)-2,3,5,6-tetrafluoropyridine (8). To a stirred suspension of 4-bromophenol (7, 1 eq., 1 mmol) and anhydrous K2CO3 (1.05 eq., 1.05 mmol) in can (5 mL) at 25 °C, pentafluoropyridine (1.05 eq., 1.05 mmol) was added. The reaction mixture was stirred for 12 h, quenched with HCl (1 M, 10 mL), and extracted with EtOAc (3 × 5 mL). The combined organic phases were washed with brine (5 mL), dried over Na2SO4, and evaporated under reduced pressure to afford the title compound (quantitative yield) as a colourless oil without further purification. 1H NMR (300 MHz, CDCl3) δ 7.50 (d, J = 9.1 Hz, 2H, C2-H and C6-H), 6.96 (d, J = 9.1 Hz, 2H, C3-H and C5-H). 19F NMR (282 MHz, CDCl3) δ −88.21 (m), −151.66–−157.75 (m). ESI-MS, m/z [M+H]+ 322 and 324 (1:1 ratio).
4-(4-Ethynylphenoxy)-2,3,5,6-tetrafluoropyridine (4). To a mixture of 4-(4-bromophenoxy)-2,3,5,6-tetrafluoropyridine (8, 1eq., 1 mmol), Pd(PPh3)4 (0.05 eq., 0.05 mmol), PPh3 (0.055 eq., 0.05 mmol), and CuI (0.05 eq., 0.05 mmol) under N2 atmosphere, a degassed solution of trimethylsilylacetylene (1.5 eq., 1.5 mmol) in anhydrous Et3N (6 mL) was added dropwise at 25 °C. The mixture was heated to 90 °C and stirred for 4 h. The dark suspension was acidified with HCl (1 M) and extracted with EtOAc (3 × 5 mL). The combined organic phases were washed with NaHCO3 (s.s., 5 mL) and brine (5 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography (Pet. Et./EtOAc 10:1) to afford 2,3,5,6-tetrafluoro-4-(4-((trimethylsilyl)ethynyl)phenoxy)pyridine (93% yield) as a transparent oil. 1H NMR (300 MHz, CDCl3) δ 7.48 (d, J = 8.9 Hz, 2H, C2-H and C6-H), 6.98 (d, J = 8.7 Hz, 2H, C3-H and C5-H), 0.25 (s, 9H, SiMe3). 19F NMR (282 MHz, CDCl3) δ −88.18–−88.41 (m), −153.93–−154.16 (m). ESI-MS, m/z [M+H]+ 340. To solution of the above-mentioned compound (1 eq., 1 mmol) in anhydrous THF (5 mL), cooled to −78 °C, TBAF (1.0 M solution in THF, 1 eq., 1 mmol) was added dropwise. After 5 min, the reaction was quenched with NaHCO3 (s.s., 10 mL) and extracted with EtOAc (3 × 5 mL). The combined organic phases were washed with brine (5 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography (Pet. Et./EtOAc 100:1) to afford the title compound (80% yield) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.50 (d, J = 8.9 Hz, 2H, C2-H and C6-H), 7.01 (d, J = 8.9 Hz, 2H, C3-H and C5-H), 3.08 (s, 1H). 19F NMR (282 MHz, CDCl3) δ −88.24 (m), −149.41–−162.97 (m). ESI-MS, m/z [M+H]+ 268.
2-(4-((Perfluoropyridin-4-yl)oxy)phenyl)benzofuran-5-carbaldehyde (3). Following the General Procedure A, starting from 3-iodo-4-hydroxybenzaldehyde (5b) and 4-(4-ethynylphenoxy)-2,3,5,6-tetrafluoropyridine (4), the title compound was obtained as a yellow solid (83% yield) after purification by silica gel column chromatography (Hex/Acetone 6:1). 1H NMR (300 MHz, CDCl3) δ 10.07 (s, 1H, CHO), 8.14 (d, J = 1.6 Hz, 1H, C4-H), 7.88 (m, 3H, C6-H and C2′-H, C6′-H), 7.64 (d, J = 8.5 Hz, 1H, C7-H), 7.17 (d, J = 9.0 Hz, 2H, C3′-H, C5′-H), 7.11 (s, 1H, C3-H). 19F NMR (282 MHz, CDCl3) δ −88.04 (m), −148.90–−162.97 (m). ESI-MS, m/z [M+H]+ 388.
1-(2,4-bis((Perfluoropyridin-4-yl)oxy)phenyl)ethanone (2). To a stirred suspension of 1-(2,4-dihydroxyphenyl)ethanone (10, 1 eq., 1 mmol) and anhydrous K2CO3 (2.5 eq., 2.5 mmol) in ACN (5 mL), pentafluoropyridine (2.5 eq., 2.5 mmol) was added. The reaction mixture was stirred at 25 °C for 12 h, quenched with HCl (1 M, 10 mL), and the aqueous phase was extracted with EtOAc (3 × 5 mL). The combined organic phases were washed with brine (5 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography (Pet.Et./DCM 2:1) to afford the title compound (74% yield) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.95 (d, J = 8.7 Hz, 1H, C6-H), 6.89 (dd, J = 8.7, 2.3 Hz, 1H, C5-H), 6.77 (d, J = 2.3 Hz, 1H, C3-H), 2.64 (s, 3H, CH3). 19F NMR (282 MHz, CDCl3) δ −85.04–−90.34 (m), −150.28–−158.26 (m). ESI-MS, m/z [M+H]+ 451.
(E)-1-(2,4-bis((Perfluoropyridin-4-yl)oxy)phenyl)-3-(2-(4-((perfluoropyridin-4-yl)oxy)phenyl)benzofuran-5-yl)prop-2-en-1-one (11). To a solution of 1-(2,4-bis((perfluoropyridin-4-yl)oxy)phenyl)ethanone (2, 1 eq., 1 mmol), 2-(4-((perfluoropyridin-4-yl)oxy)phenyl)benzofuran-5-carbaldehyde (3, 1.2 eq., 1.2 mmol), and anhydrous Et3N (1 eq., 1 mmol) in anhydrous DCM (5 mL), cooled to −20 °C, TMSOTf (1 eq., 1 mmol) was added dropwise. The cooling bath was removed, and the reaction was stirred at 25 °C for 2 h. The addition of TMSOTf (1 eq., 1 mmol) was repeated two more times at −20 °C every 2 h. The reaction was quenched with NaHCO3 (s.s., 10 mL) and extracted with DCM (3 × 5 mL). The combined organic phases were washed with brine (5 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography (Hex/Acetone 6:1) to afford the title compound (68% yield) as a yellow oil. 1H NMR (300 MHz, Acetone-d6) δ 8.07–8.00 (m, 3H, Ar-H), 7.98 (d, J = 8.6 Hz, 1H, Ar-H), 7.84–7.73 (m, 2H, Ar-H and (E)-CαH=CβH), 7.64 (d, J = 8.6 Hz, 1H, Ar-H), 7.49 (d, J = 16.0 Hz, 1H, (E)-CαH=CβH), 7.44 (m, 3H, Ar-H), 7.40–7.33 (m, 2H, Ar-H). 19F NMR (282 MHz, Acetone-d6) δ −88.09–−96.64 (m), −152.75–−165.21 (m). ESI-MS, m/z [M+H]+ 820.
(E)-1-(2,4-Dihydroxyphenyl)-3-(2-(4-hydroxyphenyl)benzofuran-5-yl)prop-2-en-1-one (1). To a suspension of (E)-1-(2,4-bis((perfluoropyridin-4-yl)oxy)phenyl)-3-(2-(4-((perfluoropyridin-4-yl)oxy)phenyl)benzofuran-5-yl)prop-2-en-1-one (11, 1 eq., 1 mmol), KF (3 eq., 3 mmol), 18-C-6 (3 eq., 3 mmol) in ACN/H2O (1 mL/20 μL), cooled to 0 °C, a solution of methyl thioglycolate (2 eq., 2 mmol) in ACN/H2O (1 mL/20 μL) was added dropwise. The cooling bath was removed, and the mixture was stirred for 1 h. The addition of methyl thioglycolate (2 eq., 2 mmol) in ACN/H2O (1 mL/20 μL) was repeated two more times at 1 h intervals. The solvent was removed under reduced pressure, the crude residue was suspended in H2O (2 mL), and the aqueous phase was extracted with EtOAc (3 × 2 mL). The combined organic phases were washed with brine (2 × 2 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by semi-preparative HPLC (Purosphere® STAR RP-18 endcapped 5 μm, 250-10), eluting with ACN/H2O (0.1% TFA v/v) (1:10 → 8:10 over 30 min, 8:10 for 5 min, 8:10 → 9.5:10 over 5 min) to afford the title compound (45% yield) as an orange solid. Spectroscopic data are identical to those reported for the natural compound. 1H NMR (300 MHz, Acetone-d6) δ 13.59 (s, 1H, C2′OH), 8.19 (d, J = 8.9 Hz, 1H, C6′-H), 8.10 (d, J = 1.6 Hz, 1H, C4-H), 8.01 (d, J = 15.5 Hz, 1H, (E)-CαH=CβH), 7.96 (d, J = 15.5 Hz, 1H, (E)-CαH=CβH), 7.89–7.78 (m, 3H, C6-H, C2″-H and C6″-H), 7.62 (d, J = 8.5 Hz, 1H, C7-H), 7.16 (d, J = 0.8 Hz, 1H, C3-H), 7.00 (d, J = 8.8 Hz, 2H, C3″-H and C5″-H), 6.53 (dd, J = 8.9, 2.4 Hz, 1H, C5′-H), 6.41 (d, J = 2.4 Hz, 1H, C3′-H). 13C NMR (75 MHz, Acetone-d6) δ 191.89, 166.78, 164.86, 158.54, 157.70, 155.92, 144.64, 132.62, 130.43, 130.35, 126.66 (2C), 124.82, 121.70, 121.52, 119.44, 115.83 (2C), 113.62, 111.29, 107.91, 102.85, 99.24. ESI-MS, m/z [M-H]− 371.
2-(4-(Methoxymethoxy)phenyl)benzofuran-5-carbaldehyde (13). Following the General Procedure A, starting from 3-iodo-4-hydroxybenzaldehyde (5b) and 1-ethynyl-4-(methoxymethoxy)benzene (12), the title compound was obtained as a yellow oil (89% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 8:1). 1H NMR (300 MHz, CDCl3) δ 10.05 (s, 1H, CHO), 8.08 (s, 1H, C4-H), 7.86–7.68 (m, 3H, C3′-H, C5′-H and C6-H), 7.60 (d, J = 8.5 Hz, 1H, C7-H), 7.13 (d, J = 8.7 Hz, 2H, C2′-H, C6′-H), 6.98 (s, 1H, C3-H), 5.24 (s, 2H, OCH2OCH3), 3.51 (s, 3H, OCH2OCH3). ESI-MS, m/z [M+H]+ 283.
1-(2-Hydroxy-4-(methoxymethoxy)phenyl)ethan-1-one (14a). To a solution of 1-(2,4-dihydroxyphenyl)ethan-1-one (10, 1 eq., 1 mmol) in anhydrous DCM (10 mL), DIPEA (1.5 eq., 1.5 mmol) was added, followed by chloromethyl methyl ether (1.5 eq., 1.5 mmol). The mixture was stirred at 25 °C for 2 h. The reaction mixture was washed with HCl (1 M) (2 × 5 mL), with NaHCO3 (s.s., 5 mL), and brine (5 mL). The organic phase was dried over Na2SO4 and evaporated under reduced pressure to obtain the title compound with no further purification (quantitative yield) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 12.60 (s, 1H, C2′OH), 7.65 (d, J = 8.8 Hz, 1H, C6′-H), 6.59 (d, J = 2.4 Hz, 1H, C3′-H), 6.55 (dd, J = 8.8, 2.4 Hz, 1H, C5′-H), 5.20 (s, 2H, OCH2OCH3), 3.47 (s, 3H, OCH2OCH3), 2.56 (s, 3H, COCH3). ESI-MS, m/z [M-H]− 195.
1-(2-Methoxy-4-(methoxymethoxy)phenyl)ethan-1-one (14b). To a solution of 1-(2-hydroxy-4-(methoxymethoxy)phenyl)ethan-1-one (14a, 1 eq., 1 mmol) in anhydrous DMF (6 mL) under N2 atmosphere, K2CO3 (1.5 eq., 1.5 mmol) was added, followed by CH3I (1.5 eq., 1.5 mmol). The mixture was stirred at 25 °C for 12 h. Then the reaction was quenched with NH4Cl (s.s., 5 mL) and extracted with EtOAc (3 × 5 mL). The combined organic phases were washed with NaHCO3 (s.s., 5 mL) and brine (5 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography (Pet. Et./EtOAc 4:1) to afford the title compound (37% yield) as a transparent oil. 1H NMR (300 MHz, CDCl3) δ 7.77 (d, J = 8.7 Hz, 1H, C6′-H), 6.62 (dd, J = 8.7, 2.2 Hz, 1H, C5′-H), 6.58 (d, J = 2.1 Hz, 1H, C3′-H), 5.18 (s, 2H, OCH2OCH3), 3.86 (s, 3H, C2′OCH3), 3.45 (s, 3H, OCH2OCH3), 2.54 (s, 3H, COCH3). ESI-MS, m/z [M + Na]+ 233.
2-Phenylbenzofuran-5-carbaldehyde (20a). Following the General Procedure A, starting from 3-iodo-4-hydroxybenzaldehyde (5b) and ethynylbenzene (19a), the title compound was obtained as a yellow solid (65% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 20:1). 1H NMR (300 MHz, CDCl3) δ 10.05 (s, 1H, CHO), 8.10 (s, 1H, C4-H), 7.86 (m, 3H, C6-H, C2-ArH), 7.62 (d, J = 8.5 Hz, 1H, C7-H), 7.54–7.36 (m, 3H, C2-ArH), 7.09 (s, 1H, C3-H). ESI-MS, m/z [M+H]+ 223.
2-(4-Methoxyphenyl)benzofuran-5-carbaldehyde (20b). Following the General Procedure A, starting from 3-iodo-4-hydroxybenzaldehyde (5b) and 1-ethynyl-4-methoxybenzene (19b), the title compound was obtained as an off-white solid (67% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 7:1). 1H NMR (300 MHz, CDCl3) δ 10.05 (s, 1H, CHO), 8.08 (s, 1H, C4-H), 7.92–7.68 (m, 3H, C6-H, C3′-H and C5′-H), 7.59 (d, J = 8.5 Hz, 1H, C7-H), 6.99 (d, J = 8.8 Hz, 2H, C2′-H and C6′-H), 6.96 (s, 1H, C3-H), 3.87 (s, 3H, ArOCH3). ESI-MS, m/z [M+H]+ 253.
(E)-1-(2-Hydroxy-4-(methoxymethoxy)phenyl)-3-(2-(4-(methoxymethoxy)phenyl)benzofuran-5-yl)prop-2-en-1-one (15a). Following the General Procedure B, starting from 1-(2-hydroxy-4-(methoxymethoxy)phenyl)ethan-1-one (14a) and 2-(4-(methoxymethoxy)phenyl)benzofuran-5-carbaldehyde (13), the title compound was obtained as an orange solid (56% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 4:1). 1H NMR (300 MHz, CDCl3) δ 13.39 (s, 1H, C2′OH), 8.00 (d, J = 15.4 Hz, 1H, CαH=CβH), 7.89 (d, J = 9.0 Hz, 1H, C6′-H), 7.86–7.74 (m, 3H, Ar-H and CαH=CβH), 7.69–7.49 (m, 3H, Ar-H), 7.13 (d, J = 8.8 Hz, 2H, C3″-H and C5″-H), 6.92 (s, 1H, C3-H), 6.66 (d, J = 2.4 Hz, 1H, C3′-H), 6.60 (dd, J = 8.8, 2.4 Hz, 1H, C5′-H), 5.23 (s, 4H), 3.51 (s, 6H). ESI-MS, m/z [M-H]− 459.
(E)-1-(2-Methoxy-4-(methoxymethoxy)phenyl)-3-(2-(4-(methoxymethoxy)phenyl)benzofuran-5-yl)prop-2-en-1-one (15b). Following the General Procedure B, starting from 1-(2-methoxy-4-(methoxymethoxy)phenyl)ethan-1-one (14b) and 2-(4-(methoxymethoxy)phenyl)benzofuran-5-carbaldehyde (13), the title compound was obtained as a yellow solid (63% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 2:1). 1H NMR (300 MHz, CDCl3) δ 7.76 (m, 5H Ar-H and CαH=CβH), 7.50 (m, 3H, Ar-H, and CαH=CβH), 7.10 (d, J = 8.6 Hz, 2H, C3″-H, C5″-H), 6.88 (s, 1H, C3-H), 6.71 (d, J = 8.6 Hz, 1H, C5′-H), 6.65 (s, 1H, C3′-H), 5.22 (s, 2H, OCH2OCH3), 5.21 (s, 2H, OCH2OCH3), 3.91 (s, 3H, C2′OCH3), 3.50 (s, 3H, OCH2OCH3), 3.49 (s, 3H, OCH2OCH3). ESI-MS, m/z [M+Na]+ 497.
(E)-3-(2-(4-(Methoxymethoxy)phenyl)benzofuran-5-yl)-1-phenylprop-2-en-1-one (15c). Following the General Procedure B, starting from acetophenone (14c) and 2-(4-(methoxymethoxy)phenyl)benzofuran-5-carbaldehyde (13), the title compound was obtained as an orange solid (48% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 4:1).1H NMR (300 MHz, CDCl3) δ 8.05 (d, J = 7.0 Hz, 2H, C2′-H and C6′-H), 7.94 (d, J = 15.7 Hz, 1H, CαH=CβH), 7.83 (s, 1H, Ar-H), 7.80 (d, J = 8.8 Hz, 2H, C2″-H, C6″-H), 7.64–7.56 (m, 4H, Ar-H and CαH=CβH), 7.53 (d, J = 7.0 Hz, 2H, C3′-H and C5′-H), 7.13 (d, J = 8.7 Hz, 2H, C3″-H, C5″-H), 6.93 (s, 1H, C3-H), 5.24 (s, 2H, OCH2OCH3), 3.51 (s, 3H, OCH2OCH3). ESI-MS, m/z [M+H]+ 385.
(E)-1-(2-Hydroxyphenyl)-3-(2-(4-(methoxymethoxy)phenyl)benzofuran-5-yl)prop-2-en-1-one (15d). Following the General Procedure B, starting from 1-(2-hydroxyphenyl)ethan-1-one (14d) and 2-(4-(methoxymethoxy)phenyl)benzofuran-5-carbaldehyde (13), the title compound was obtained as an orange solid (47% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 4:1). 1H NMR (300 MHz, CDCl3) δ 12.95 (s, 1H, C2′OH), 8.00 (m, 1H, Ar-H), 7.84 (s, 1H, Ar-H), 7.80 (d, J = 8.2 Hz, 2H, C2″-H, C6″-H), 7.68 (m, 1H, Ar-H), 7.62 (m, 1H, Ar-H), 7.57–7.40 (m, 3H, Ar-H), 7.13 (d, J = 8.1 Hz, 2H, C3″-H, C5″-H), 7.04 (m, 1H, Ar-H), 6.99 (m, 1H, Ar-H), 6.92 (s, 1H, C3-H), 5.23 (s, 2H, OCH2OCH3), 3.51 (s, 3H, OCH2OCH3). ESI-MS, m/z [M-H]− 399.
1-Methyl-1H-indole-5-carbaldehyde (23a). To a solution of 1H-indole-5-carbaldehyde (22, 1 eq., 1 mmol) in anhydrous DMF (5 mL), cooled to 0 °C NaH (60% w/w, 1.5 eq., 1.5 mmol) was added under N2 atmosphere. The suspension was stirred at the same temperature for 30 min, then CH3I (1.5 eq., 1.5 mmol) was added dropwise. The ice bath was removed, and the reaction mixture was stirred at 25 °C for 12 h. The reaction was quenched with NH4Cl (s.s., 5 mL) and extracted with EtOAc (3 × 5 mL). The combined organic phases were washed with NaHCO3 (s.s., 5 mL) and brine (5 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography (Pet. Et./EtOAc 4:1) to afford the title compound (91% yield) as a white solid. 1H NMR (300 MHz, CDCl3) δ 10.03 (s, 1H, CHO), 8.15 (s, 1H, C4-H), 7.80 (d, J = 8.9 Hz, 1H, C6-H), 7.40 (d, J = 8.6 Hz, 1H, C7-H), 7.15 (d, J = 3.2 Hz, 1H, C2-H), 6.65 (d, J = 3.1 Hz, 1H, C3-H), 3.85 (s, 3H, NCH3). ESI-MS, m/z [M+H]+ 160.
1-Benzyl-1H-indole-5-carbaldehyde (23b). Starting from 1H-indole-5-carbaldehyde 22, the title compounds were obtained as a yellow oil in 84% yield after purification by silica gel column chromatography (Pet. Et./EtOAc 8:1) following the procedure described for the preparation of 23a. 1H NMR (300 MHz, CDCl3) δ 10.03 (s, 1H, CHO), 8.18 (s, 1H, C4-H), 7.75 (d, J = 8.6 Hz, 1H, C6-H), 7.37 (d, J = 8.6 Hz, 1H, C7-H), 7.32 (m, 3H, NCH2Ph), 7.23 (d, J = 3.2 Hz, 1H, C3-H), 7.11 (m, 2H, NCH2Ph), 6.72 (d, J = 3.2 Hz, 1H, C2-H), 5.37 (s, 2H, NCH2Ph). ESI-MS, m/z [M+H]+ 236.
2-(4-(Methoxymethoxy)phenyl)-1-methyl-1H-indole-5-carbaldehyde (27a). A suspension of Pd(OAc)2, 1-iodo-4-(methoxymethoxy)benzene (26), 2-nitrobenzoic acid, Ag2O, and 1-methyl-1H-indole-5-carbaldehyde (23a) in anhydrous DMF under N2 atmosphere was stirred at 25 °C for 24 h. Then, the reaction mixture was filtered through Celite® using EtOAc as eluent and washed with NH4Cl (s.s., 5 mL), NaHCO3 (s.s., 5 mL), and brine (5 mL). The organic phase was dried over Na2SO4 and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography (Pet. Et./EtOAc 2:1) to afford the title compound (71% yield) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 10.03 (s, 1H, CHO), 8.13 (s, 1H, C4-H), 7.79 (d, J = 8.6 Hz, 1H, C6-H), 7.42 (m, 3H, C7-H, C3′-H and C5′-H), 7.16 (d, J = 8.7 Hz, 2H, C2′-H and C6′-H), 6.64 (s, 1H, C3-H), 5.25 (s, 2H, OCH2OCH3), 3.75 (s, 3H, NCH3), 3.53 (s, 3H, OCH2OCH3). ESI-MS, m/z [M+H]+ 296.
1-Benzyl-2-(4-(methoxymethoxy)phenyl)-1H-indole-5-carbaldehyde (27b). Starting from 1-iodo-4-(methoxymethoxy)benzene (26) and -benzyl-1H-indole-5-carbaldehyde (23b), the title compounds were obtained as a yellow oil in 80% yield after purification by silica gel column chromatography (Pet. Et./EtOAc 5:1) following the procedure described for the preparation of 27a. 1H NMR (300 MHz, CDCl3) δ 9.97 (s, 1H, CHO), 8.13 (s, 1H, C4-H), 7.66 (d, J = 8.5 Hz, 1H, C6-H), 7.32 (d, J = 8.7 Hz, 2H, C3′-H and C5′-H), 7.21 (m, 4H, C7-H and NCH2Ph), 7.04 (d, J = 8.7 Hz, 2H, C2′-H and C6′-H), 6.95 (m, 2H, NCH2Ph), 6.69 (s, 1H, C3-H), 5.34 (s, 2H, NCH2Ph), 5.16 (s, 2H, OCH2OCH3), 3.45 (s, 3H, OCH2OCH3). ESI-MS, m/z [M+H]+ 372.
(E)-1-(2-Hydroxy-4-(methoxymethoxy)phenyl)-3-(1H-indol-5-yl)prop-2-en-1-one (24a). Following the General Procedure B, starting from 1-(2-hydroxy-4-(methoxymethoxy)phenyl)ethan-1-one (14a) and 1H-indole-5-carbaldehyde (22), the title compound was obtained as a yellow solid (53% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 4:1). 1H NMR (300 MHz, CDCl3) δ 13.52 (s, 1H, C2′OH), 8.44 (br, 1H, NH), 8.07 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.95 (s, 1H, C4-H), 7.90 (d, J = 9.0 Hz, 1H, C6′-H), 7.62–7.52 (m, 2H, CαH=CβH and C6-H), 7.43 (d, J = 8.5 Hz, 1H, C7-H), 7.27 (m, 1H, C2-H), 6.68–6.54 (m, 3H, Ar′-H and C3-H), 5.23 (s, 2H, OCH2OCH3), 3.49 (s, 3H, OCH2OCH3). ESI-MS, m/z [M-H]− 322.
(E)-1-(2-Hydroxy-4-(methoxymethoxy)phenyl)-3-(1-methyl-1H-indol-5-yl)prop-2-en-1-one (24b). Following the General Procedure B, starting from 1-(2-hydroxy-4-(methoxymethoxy)phenyl)ethan-1-one (14a) and 1-methyl-1H-indole-5-carbaldehyde (23a), the title compound was obtained as a yellow oil (60% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 4:1). 1H NMR (300 MHz, CDCl3) δ 13.53 (s, 1H, C2′OH), 8.08 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.98–7.86 (m, 2H, C4-H and C6′-H), 7.58 (d, J = 15.4 Hz, 1H, CαH=CβH), 7.58 (m, 1H, C6-H), 7.36 (d, J = 8.6 Hz, 1H, C7-H), 7.10 (d, J = 3.1 Hz, 1H, C2-H), 6.65 (d, J = 2.4 Hz, 1H, C3′-H), 6.60 (dd, J = 8.9, 2.4 Hz, 1H, C5′-H), 6.56 (d, J = 3.1 Hz, 1H, C3-H), 5.23 (s, 2H, OCH2OCH3), 3.83 (s, 3H, NCH3), 3.50 (s, 3H, OCH2OCH3). ESI-MS, m/z [M-H]− 336.
(E)-1-(2,4-Dihydroxyphenyl)-3-(2-(4-hydroxyphenyl)benzofuran-5-yl)prop-2-en-1-one (1). Following the General Procedure C, starting from (E)-1-(2-hydroxy-4-(methoxymethoxy)phenyl)-3-(2-(4-(methoxymethoxy)phenyl)benzofuran-5-yl)prop-2-en-1-one (15a), the title compound was obtained as an orange solid (69% yield) after purification by silica gel column chromatography (DCM/MeOH 20:1). Spectroscopic data (1H NMR, 13C NMR, ESI-MS) were in agreement with the previously reported for the same compound.
(E)-1-(4-Hydroxy-2-methoxyphenyl)-3-(2-(4-hydroxyphenyl)benzofuran-5-yl)prop-2-en-1-one (16). Following the General Procedure C, starting from (E)-1-(2-methoxy-4-(methoxymethoxy)phenyl)-3-(2-(4-(methoxymethoxy)phenyl)benzofuran-5-yl)prop-2-en-1-one (15b) the title compound was obtained as an orange solid (69% yield) after purification by silica gel column chromatography (DCM/MeOH 100:1). 1H NMR (300 MHz, Acetone-d6) δ 7.93 (s, 1H, C4-H), 7.81 (d, J = 8.7 Hz, 2H, C2″-H and C6″-H), 7.73 (d, J = 15.7 Hz, 1H, CαH=CβH), 7.69–7.61 (m, 3H, CαH=CβH, C6-H and C7-H), 7.57 (d, J = 8.6 Hz, 1H, C6′-H), 7.13 (s, 1H, C3-H), 6.99 (d, J = 8.7 Hz, 2H, C3″-H and C5″-H), 6.61 (d, J = 2.0 Hz, 1H, C3′-H), 6.55 (dd, J = 8.5, 2.1 Hz, 1H, C5′-H), 3.95 (s, 3H). 13C NMR (75 MHz, Acetone-d6) δ 188.83, 162.79, 160.97, 158.56, 157.50, 155.52, 141.29, 132.66, 130.91, 130.37, 126.60 (2C), 126.34, 124.10, 121.53, 121.03, 120.95, 115.84 (2C), 111.22, 107.86, 99.27, 99.15, 55.23. ESI-MS, m/z [M-H]− 385.
(E)-3-(2-(4-Hydroxyphenyl)benzofuran-5-yl)-1-phenylprop-2-en-1-one (17). Following the General Procedure C, starting from (E)-3-(2-(4-(methoxymethoxy)phenyl)benzofuran-5-yl)-1-phenylprop-2-en-1-one (15c) the title compound was obtained as an orange solid (81% yield) after purification by silica gel column chromatography (DCM/MeOH 100:1). 1H NMR (300 MHz, Acetone-d6) δ 8.87 (br, 1H, C4″OH), 8.18 (m, 2H, Ar-H), 8.08 (s, 1H, Ar-H), 7.94 (d, J = 15.7 Hz, 1H, CαH=CβH), 7.88 (d, J = 15.7 Hz, 1H, CαH=CβH), 7.87–7.77 (m, 3H, C2″-H, C6″-H and Ar-H), 7.67–7.51 (m, 4H, Ar-H), 7.15 (s, 1H, C3-H), 6.99 (d, J = 8.7 Hz, 2H, C3″-H, C5″-H). ESI-MS, m/z [M-H]− 339.
(E)-1-(2-Hydroxyphenyl)-3-(2-(4-hydroxyphenyl)benzofuran-5-yl)prop-2-en-1-one (18). Following the General Procedure C, starting from (E)-1-(2-hydroxyphenyl)-3-(2-(4-(methoxymethoxy)phenyl)benzofuran-5-yl)prop-2-en-1-one (15d) the title compound was obtained as an orange solid (74% yield) after purification by silica gel column chromatography (DCM/MeOH 100:1). 1H NMR (300 MHz, Acetone-d6) δ 13.02 (s, 1H, C2′OH), 8.33 (d, J = 8.0 Hz, 1H, C6′-H), 8.14 (s, 1H, C4-H), 8.08 (s, 2H, CαH=CβH), 7.87 (dd, J = 8.6, 1.6 Hz, 1H, C6-H), 7.82 (d, J = 8.7 Hz, 2H, C2″-H and C6″-H), 7.67–7.53 (m, 2H, C7-H and C4′-H), 7.16 (s, C3-H), 7.05–6.93 (m, 2H, C3′-H and C5′-H), 7.00 (d, J = 8.7 Hz, 2H, C3″-H and C5″-H). 13C NMR (75 MHz, Acetone-d6) δ 193.97, 163.63, 163.29, 158.56, 157.82, 156.12, 146.09, 136.38, 130.49, 130.14, 126.67 (2C), 125.06, 122.03, 121.42, 120.03, 119.20, 118.94, 117.92, 115.79 (2C), 111.37, 99.22. ESI-MS, m/z [M-H]− 355.
(E)-1-(2,4-Dihydroxyphenyl)-3-(2-phenylbenzofuran-5-yl)prop-2-en-1-one (21a). Following the General Procedure B, starting from 1-(2-hydroxy-4-(methoxymethoxy)phenyl)ethan-1-one (14a) and 2-phenylbenzofuran-5-carbaldehyde (20a), (E)-1-(2-hydroxy-4-(methoxymethoxy)phenyl)-3-(2-phenylbenzofuran-5-yl)prop-2-en-1-one was obtained as a yellow solid (58% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 4:1). 1H NMR (300 MHz, CDCl3) δ 13.58 (s, 1H, C2′OH), δ 7.86 (m, 2H, Ar-H), 7.82–7.73 (m, 2H, Ar-H and CαH=CβH), 7.70 (d, J = 8.6 Hz, 1H, C6′-H), 7.59–7.42 (m, 5H, Ar-H and CαH=CβH), 7.38 (m, 1H, Ar-H), 7.02 (s, 1H, C3-H), 6.88 (d, J = 2.2 Hz, 1H, C3′-H), 6.79 (dd, J = 8.6, 2.2 Hz, 1H, C5′-H), 5.27 (s, 2H), 5.23 (s, 2H, OCH2OCH3), 3.51 (s, 3H, OCH2OCH3). ESI-MS, m/z [M-H]− 399. The abovementioned compound was then submitted to the General Procedure C, thus obtaining the title compound as a yellow solid (93% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 1:1). 1H NMR (300 MHz, Acetone-d6) δ 13.58 (s, 1H, C2′OH), 9.57 (br, 1H, C4′OH), 8.21 (d, J = 8.9 Hz, 1H, C6′-H), 8.16 (s, 1H, C4-H), 7.99 (m, 4H, CαH=CβH, Ar-H), 7.90 (d, J = 8.6, 1H, C6-H), 7.67 (d, J = 8.5 Hz, 1H, C7-H), 7.53 (m, 2H, Ar-H), 7.48–7.35 (m, 2H, Ar-H), 6.50 (dd, J = 8.8, 2.4 Hz, 1H, C5′-H), 6.39 (d, J = 2.3 Hz, 1H, C3′-H). 13C NMR (75 MHz, Acetone-d6) δ 191.88, 166.79, 164.88, 157.06, 156.13, 144.45, 132.66, 130.56, 130.04, 129.90, 129.07, 128.98 (2C), 125.44, 124.89 (2C), 122.29, 119.69, 113.61, 111.58, 107.91, 102.84, 101.66. ESI-MS, m/z [M-H]− 355.
(E)-1-(2-Hydroxy-4-(methoxymethoxy)phenyl)-3-(2-(4-methoxyphenyl)benzofuran-5-yl)prop-2-en-1-one (21b). Following the General Procedure B, starting from 1-(2-hydroxy-4-(methoxymethoxy)phenyl)ethan-1-one (14a) and 2-(4-methoxyphenyl)benzofuran-5-carbaldehyde (20b), (E)-1-(2-hydroxy-4-(methoxymethoxy)phenyl)-3-(2-(4-methoxyphenyl)benzofuran-5-yl)prop-2-en-1-one was obtained as a yellow solid (44% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 2:1). 1H NMR (300 MHz, CDCl3) δ 13.57 (s, 1H, C2′OH), δ 7.78 (m, 4H, Ar-H), 7.69 (d, J = 8.6 Hz, 1H, C6′-H), 7.49 (m, 3H, Ar-H), 6.98 (m, 2H, Ar-H), 6.88 (m, 2H, Ar-H), 6.79 (d, J = 8.5 Hz, 1H, C5′-H), 5.27 (s, 2H, OCH2OCH3), 3.85 (s, 3H, C4″OCH3), 3.51 (s, 3H, OCH2OCH3). ESI-MS, m/z [M-H]− 429. The abovementioned compound was then submitted to the General Procedure C, thus obtaining the title compound as a yellow solid (91% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 1:1). 1H NMR (300 MHz, d-DMSO) δ 13.54 (s, 1H, C2′OH), 8.23 (d, J = 8.9 Hz, 1H), 8.16 (s, 1H, C4-H), 7.99 (d, J = 15.4 Hz, 1H, CαH=CβH), 7.92 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.88 (d, J = 8.7 Hz, 2H, C2″-H and C6″-H), 7.85 (m, 1H, C6-H), 7.67 (d, J = 8.6 Hz, 1H, C7-H), 7.33 (s, 1H, C3-H), 7.07 (d, J = 8.7 Hz, 2H, C3″-H and C5″-H), 6.42 (dd, J = 8.9, 1.7 Hz, 1H, C5′-H), 6.28 (d, J = 1.7 Hz, 1H, C3′-H), 3.81 (s, 3H, C4″OCH3). 13C NMR (75 MHz, d-DMSO) δ 191.54, 166.62, 166.43, 160.49, 157.09, 155.82, 144.51, 133.48, 130.60, 130.25, 126.92 (2C), 125.80, 122.45, 120.55, 115.02 (2C), 113.10, 112.29, 111.91, 109.12, 103.13, 100.67, 55.75. ESI-MS, m/z [M-H]− 385.
(E)-1-(2,4-Dihydroxyphenyl)-3-(1H-indol-5-yl)prop-2-en-1-one (25a, CAS registry number 2412944-18-2). Following the General Procedure C, starting from (E)-1-(2-hydroxy-4-(methoxymethoxy)phenyl)-3-(1H-indol-5-yl)prop-2-en-1-one (24a) the title compound was obtained as a yellow solid (83% yield) after purification by silica gel column chromatography (DCM/MeOH 100:1). 1H NMR (300 MHz, CD3OD) δ 7.98 (m, 3H, CαH=CβH, C6′-H and C4-H), 7.72 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.60 (d, J = 8.6 Hz, 1H, C6-H), 7.43 (d, J = 8.5 Hz, 1H, C7-H), 7.28 (d, J = 2.9 Hz, 1H, C2-H), 6.53 (d, J = 3.0 Hz, 1H, C3-H), 6.43 (dd, J = 8.6, 1.8 Hz, 1H, C5′-H), 6.30 (d, J = 2.0 Hz, 1H, C3′-H). 13C NMR (75 MHz, CD3OD) δ 192.19, 166.09, 164.87, 146.64, 137.91, 131.93, 128.43, 126.16, 125.59, 122.89, 121.07, 116.42, 113.32, 111.46, 107.66, 102.34, 102.07. ESI-MS, m/z [M-H]− 278.
(E)-1-(2,4-Dihydroxyphenyl)-3-(1-methyl-1H-indol-5-yl)prop-2-en-1-one (25b). Following the General Procedure C, starting from (E)-1-(2-hydroxy-4-(methoxymethoxy)phenyl)-3-(1-methyl-1H-indol-5-yl)prop-2-en-1-one (24b) the title compound was obtained as a yellow solid (77% yield) after purification by silica gel column chromatography (DCM/MeOH 100:1). 1H NMR (300 MHz, Acetone-d6) δ 13.74 (s, 1H, C2′OH), 9.48 (s, 1H, C4′OH), 8.19 (d, J = 8.9 Hz, 1H, C6′-H), 8.07 (s, 1H, C4-H), 8.05 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.89 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.76 (d, J = 8.6 Hz, 1H, C6-H), 7.49 (d, J = 8.6 Hz, 1H, C7-H), 7.31 (d, J = 3.1 Hz, 1H, C2-H), 6.54 (d, J = 3.1 Hz, 1H, C3-H), 6.49 (dd, J = 8.8, 2.2 Hz, 1H, C5′-H), 6.38 (d, J = 2.2 Hz, 1H, C3′-H), 3.88 (s, 3H, NCH3). 13C NMR (75 MHz, Acetone-d6) δ 191.98, 166.71, 164.55, 146.44, 138.31, 132.42, 130.63, 128.97, 126.30, 123.39, 121.63, 117.04, 113.68, 110.07, 107.72, 102.82, 101.78, 32.21. ESI-MS, m/z [M-H]− 292.
(E)-1-(2,4-Dihydroxyphenyl)-3-(2-(4-hydroxyphenyl)-1-methyl-1H-indol-5-yl)prop-2-en-1-one (28a). Following the General Procedure B, starting from 1-(2-hydroxy-4-(methoxymethoxy)phenyl)ethan-1-one (14a) and 2-(4-(methoxymethoxy)phenyl)-1-methyl-1H-indole-5-carbaldehyde (27a), (E)-1-(2-hydroxy-4-(methoxymethoxy)phenyl)-3-(2-(4-(methoxymethoxy)phenyl)-1-methyl-1H-indol-5-yl)prop-2-en-1-one was obtained as a yellow oil (67% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 2:1). 1H NMR (300 MHz, CDCl3) δ 13.47 (s, 1H, C2′OH), δ 7.83 (s, 1H, C4-H), 7.82 (d, J = 15.6 Hz, 1H, CαH=CβH), 7.66 (d, J = 8.6 Hz, 1H, C6′-H), 7.53 (d, J = 8.6 Hz, 1H, C6-H), 7.43 (d, J = 15.7 Hz, 1H, CαH=CβH), 7.43 (d, J = 8.7 Hz, 2H, C2″-H and C6″-H), 7.34 (d, J = 8.6 Hz, 1H, C7-H), 7.15 (d, J = 8.7 Hz, 2H, C3″-H and C5″-H), 6.87 (d, J = 2.1 Hz, 1H, C3′-H), 6.78 (dd, J = 8.6, 2.2 Hz, 1H, C5′-H), 6.53 (s, 1H, C3-H), 5.23 (s, 4H, 2xOCH2OCH3), 3.74 (s, 3H, NCH3), 3.51 (s, 6H, 2xOCH2OCH3). ESI-MS, m/z [M-H]− 472. The abovementioned compound was then submitted to the General Procedure C, thus obtaining the title compound as a yellow solid (63% yield) after purification by silica gel column chromatography (DCM/MeOH 100:1). 1H NMR (300 MHz, Acetone-d6) δ 13.75 (s, 1H, C2′OH), 8.20 (d, J = 8.9 Hz, 1H, C6′-H), 8.06 (d, J = 15.2 Hz, 1H, CαH=CβH), 8.05 (s, 1H, C4-H), 7.91 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.76 (d, J = 8.6 Hz, 1HC6-H), 7.52 (d, J = 8.6 Hz, 1H, C7-H), 7.45 (d, J = 8.5 Hz, 2H, C2″-H and C6″-H), 7.01 (d, J = 8.5 Hz, 2H, C3″-H and C5″-H), 6.55 (s, 1H, C3-H), 6.50 (dd, J = 8.8, 2.2 Hz, 1H, C5′-H), 6.38 (d, J = 2.2 Hz, 1H, C3′-H), 3.80 (s, 3H, NCH3). 13C NMR (75 MHz, Acetone-d6) δ 191.98, 166.72, 164.56, 157.71, 146.45, 143.18, 139.77, 132.42, 130.65 (2C), 128.50, 126.72, 123.40, 122.53, 121.70, 117.04, 115.45 (2C), 113.69, 110.36, 107.72, 102.86, 101.29, 30.70. ESI-MS, m/z [M-H]− 384.
(E)-3-(1-Benzyl-2-(4-hydroxyphenyl)-1H-indol-5-yl)-1-(2,4-dihydroxyphenyl)prop-2-en-1-one (28b). Following the General Procedure B, starting from 1-(2-hydroxy-4-(methoxymethoxy)phenyl)ethan-1-one (14a) and 1-benzyl-2-(4-(methoxymethoxy)phenyl)-1H-indole-5-carbaldehyde (27b), (E)-3-(1-benzyl-2-(4-(methoxymethoxy)phenyl)-1H-indol-5-yl)-1-(2-hydroxy-4-(methoxymethoxy)phenyl)prop-2-en-1-one was obtained as a yellow oil (65% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 2:1). 1H NMR (300 MHz, Acetone-d6) δ 13.64 (s, 1H, C2′OH), 8.25 (d, J = 8.9 Hz, 1H, C6′-H), 8.13 (s, 1H, C4-H), 8.08 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.92 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.78 (d, J = 8.8 Hz, 1H, C6-H), 7.46 (d, J = 8.6 Hz, 2H, C2″-H and C6″-H), 7.40 (d, J = 8.6 Hz, 1H, C7-H), 7.32–7.20 (m, 3H, NCH2Ph), 7.12 (d, J = 8.6 Hz, 2H, C3″-H and C5″-), 7.01 (d, J = 7.0 Hz, 2H, NCH2Ph), 6.70 (s, 1H, C3-H), 6.63 (dd, J = 9.0, 2.2 Hz, 1H, C5′-H), 6.58 (d, J = 2.1 Hz, 1H, C3′-H), 5.53 (s, 2H, NCH2Ph), 5.30 (s, 2H, OCH2OCH3), 5.25 (s, 2H, OCH2OCH3), 3.46 (s, 6H, 2xOCH2OCH3). ESI-MS, m/z [M-H]− 548. The abovementioned compound was then submitted to the General Procedure C, thus obtaining the title compound as a yellow oil (61% yield) after purification by silica gel column chromatography (Pet. Et./EtOAc 1:1). 1H NMR (300 MHz, Acetone-d6) δ 13.71 (s, 1H, C2′OH), 8.17 (d, J = 8.9 Hz, 1H, C6′-H), 8.09 (s, 1H, C4-H), 8.04 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.88 (d, J = 15.3 Hz, 1H, CαH=CβH), 7.67 (d, J = 7.6 Hz, 1H, C6-H), 7.37 (m, 3H, C7-H, C2″-H and C6″), 7.32–7.20 (m, 3H, NCH2Ph), 7.01 (d, J = 6.9 Hz, 2H, NCH2Ph), 6.93 (d, J = 8.4 Hz, 2H, C3″-H and C5″-H), 6.65 (s, 1H, C3-H), 6.48 (dd, J = 8.8, 2.2 Hz, 1H, C5′-H),), 6.38 (d, J = 2.2 Hz, 1H, C3′-H),), 5.51 (s, 2H, NCH2Ph). 13C NMR (75 MHz, Acetone-d6) δ 192.41, 167.15, 165.02, 158.29, 146.65, 143.78, 139.73, 138.73, 132.84, 131.03, 129.32, 129.01 (2C), 127.58 (2C), 126.44 (2C), 123.77, 123.03, 122.35, 117.84, 115.94 (2C), 114.15, 111.60, 108.21, 103.33, 102.69, 102.66, 47.59. ESI-MS, m/z [M-H]− 460


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











