
Identification of an Orally Bioavailable, Brain-Penetrant Compound with Selectivity for the Cannabinoid Type 2 Receptor

 , ,
, ,  , , and
, , and 
Abstract
:
1. Introduction
2. Material and Methods
General Experimental Procedures
3. Experimental Methods
3.1. General Method for Synthesis of (S,E)-11-[2-(Arylmethylene)hydrazono]-pyrrolo [2,1-c] [1,4] Benzodiazepines (4a–4q)
3.2. (S,E)-11-[(1H-Pyrrol-2-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4a)
3.3. (S,E)-11-[(1-Methyl-1H-imidazol-2-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4b)
3.4. (S,E)-11-[(Thiazol-5-ylmethylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4c)
3.5. (S,E)-11-[(2-Methylthiazol-5-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4d)
3.6. (S,E)-11-[(4-Methylthiazol-5-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4e)
3.7. (S,E)-11-[(2-Aminothiazol-5-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4f)
3.8. (S,E)-11-[(2,4-Dichlorothiazol-5-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4g)
3.9. (S,E)-11-[2-(Benzo[b]thiophenylmethylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4h)
3.10. (S,E)-11-[(Benzo[b]thiophen-2-ylmethylene)hydrazono]-7-bromo-pyrrolo[2,1-c][1,4] Benzodiazepine (4i)
3.11. (S,E)-11-[(Benzo[b]thiophen-3-ylmethylene)hydrazono]-7-bromo-pyrrolo[2,1-c][1,4] Benzodiazepine (4j)
3.12. (S,E)-11-[(Benzo[b]thiophen-2-ylmethylene)hydrazono]-8-chloro-pyrrolo[2,1-c][1,4] Benzodiazepine (4k)
3.13. (S,E)-11-[(3-Methylbenzo[b]thiophen-2-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4l)
3.14. (S,E)-11-[(3-Chlorobenzo[b]thiophen-2-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4m)
3.15. (S,E)-11-[(Benzo[d]thiazol-2-ylmethylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4n)
3.16. (S,E)-11-[(1-Methyl-1H-indol-3-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4o)
3.17. (S,E)-11-[(5-Methoxy-1H-indol-3-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4p)
3.18. (S,E)-11-[(Adamantan-1-yl)methylene)hydrazono]-pyrrolo[2,1-c][1,4] Benzodiazepine (4q)
4. Cannabinoid Receptor Binding Assay
5. PK Evaluation in CD-1 Mice
5.1. Preparation of Stock Solutions, Calibration Standards, Quality Control Samples and Internal Standard Solution
5.2. Instrument and Analytical Conditions
5.3. Sample Preparation
5.4. In Vivo Studies in CD-1 Mice
5.5. Subjects
5.6. Tissue Preparation
5.7. Pharmacokinetic Assessments
6. Results and Discussion
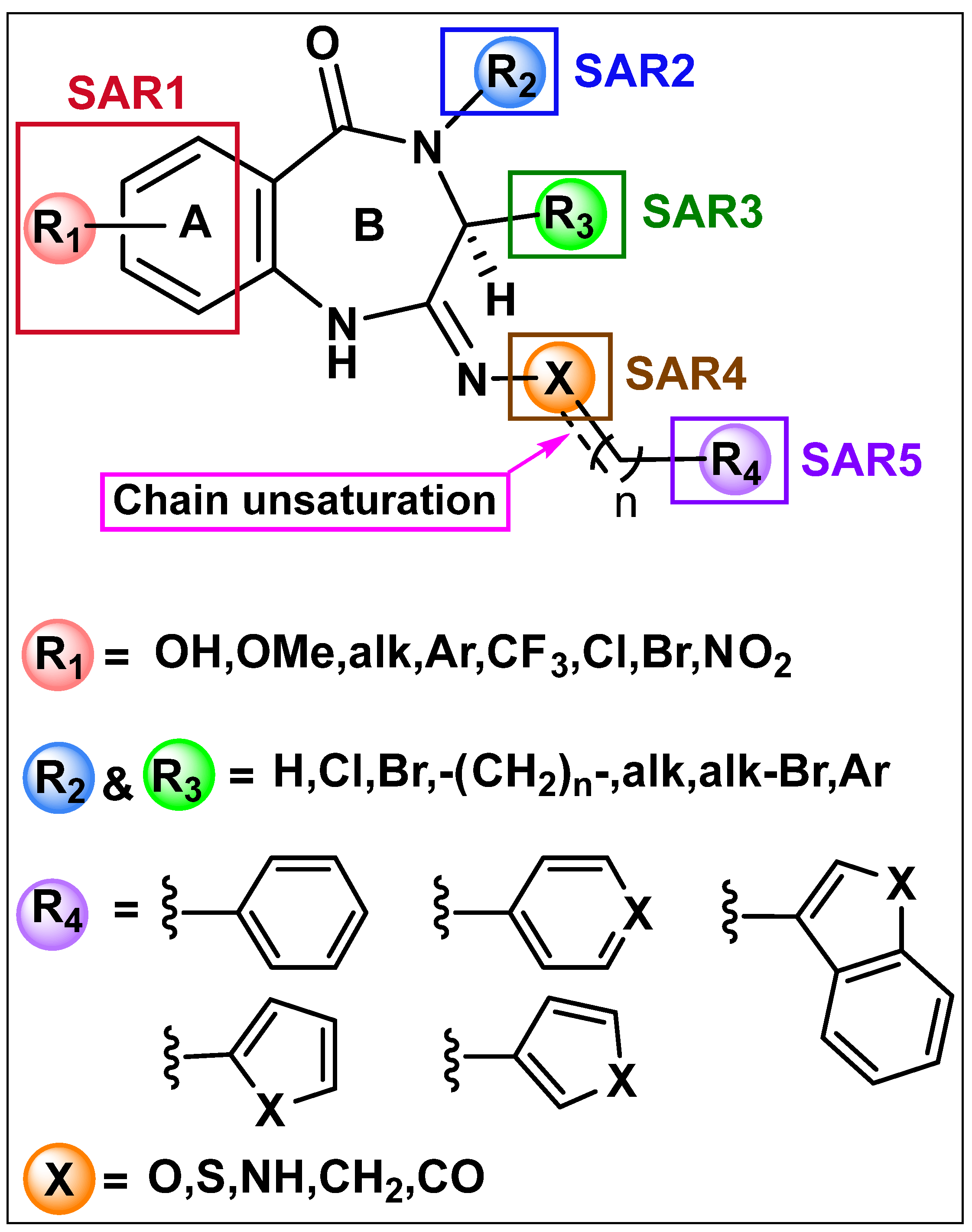
6.1. Chemistry
6.2. Biological Evaluation of the Synthesized Compounds
6.2.1. Cannabinoid Receptors Displacement Assay
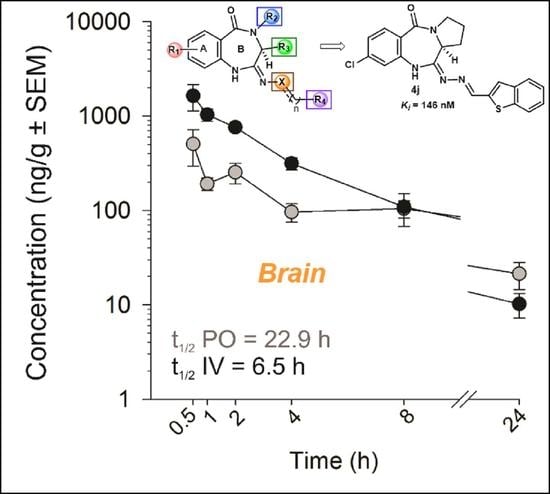
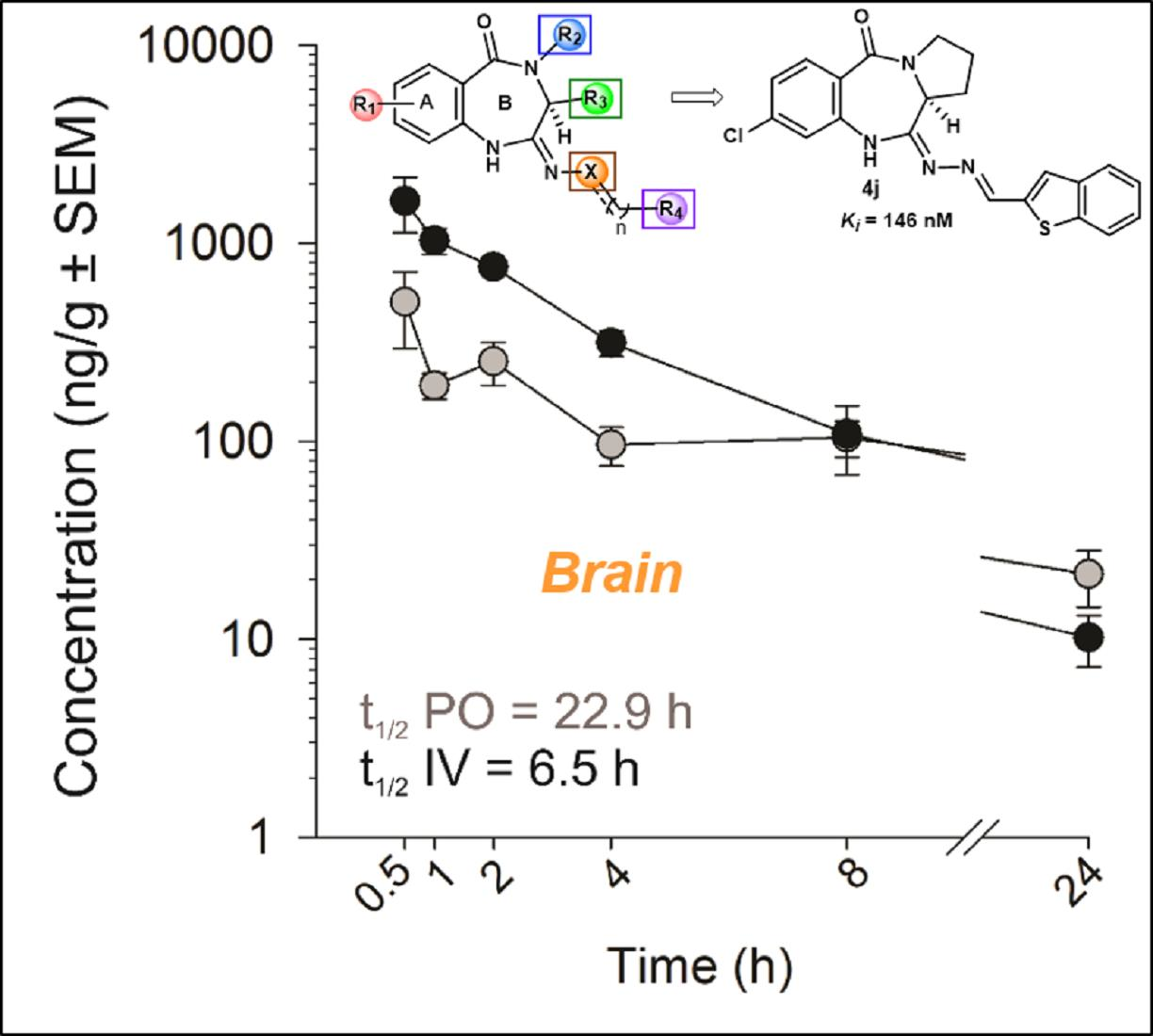
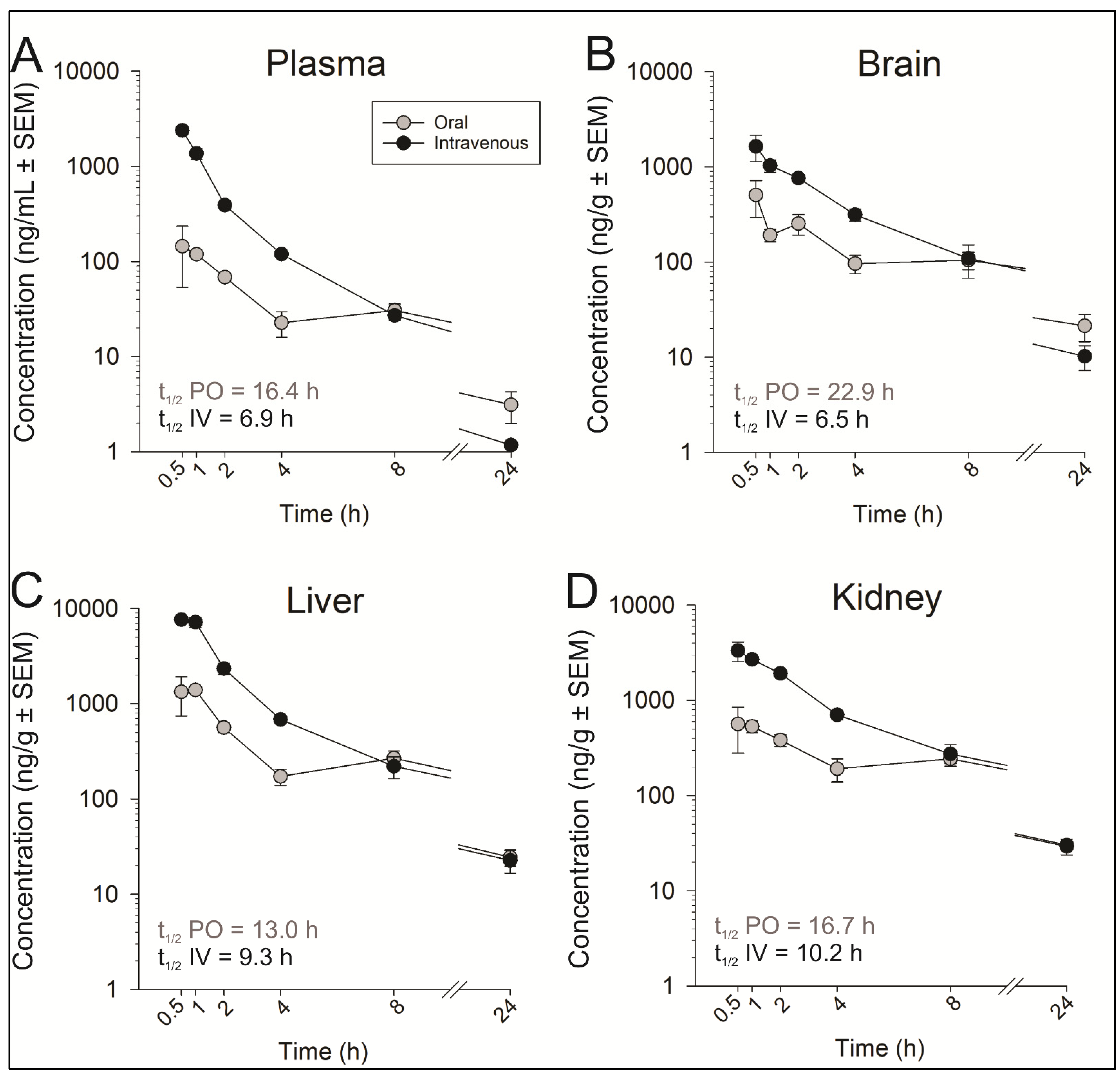
6.2.2. In Vivo PK Studies of the Lead Analog 4k
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| CB2 | Cannabinoid Receptor Subtype 2 |
| PBD | Pyrrolo[2,1-c][1,4]benzodiazepines |
| PK | Pharmacokinetics |
References
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettschneider, J.; Tredici, K.D.; Lee, V.M.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef] [PubMed]
- McPartland, J.M. Phylogenomic and chemotaxonomic analysis of the endocannabinoid system. Brain Res. Rev. 2004, 45, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, M.; Bernardini, N.; Bertorelli, R.; Campanella, M.; Nicolussi, E.; Fredduzzi, S.; Reggiani, A. CB2 receptor-mediated antihyperalgesia: Possible direct involvement of neural mechanisms. Eur. J. Neurosci. 2006, 23, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Das, S.; Williams, E.A.; Moore, D.; Jones, J.D.; Zahm, D.S.; Ndengele, M.M.; Lechner, A.J.; Howlett, A.C. Lypopolysaccharide and cyclic AMP regulation of CB2 cannabinoid receptor levels in rat brain and mouse RAW 264.7 macrophages. J. Neuroimmunol. 2006, 181, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.W. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat. Rev. Immunol. 2005, 5, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Chiba, K. Role of microglial m1/m2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals 2014, 7, 1028–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Bie, B.; Yang, H.; Xu, J.J.; Brown, D.L.; Naguib, M. Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol. Aging 2013, 34, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Shilabin, A.G.; Nieger, M. Syntheses and tautomerization of amino-substituted and pyrimidine-annulated pyrrolobenzodiazepines. Heterocycles 2005, 65, 625–632. [Google Scholar] [CrossRef]
- Schmidt, A.; Shilabin, A.G.; Namyslo, J.C.; Nieger, M.; Hemmen, S. Pyrimidine-annulated Pyrrolobenzodiazepines. A New Ring System Related to Aspergillus Alkaloids. Eur. J. Org. Chem. 2005, 1781–1789. [Google Scholar] [CrossRef]
- Cipolla, L.; Araújo, A.C.; Airoldi, C.; Bini, D. Pyrrolo[2,1-c][1,4]benzodiazepine as a scaffold for the design and synthesis of anti-tumour drugs. Anticancer Agents Med. Chem. 2009, 9, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Mingle, D.; Ospanov, M.; Radwan, M.O.; Ashpole, N.; Otsuka, M.; Ross, S.A.; Walker, L.; Shilabin, A.G.; Ibrahim, M.A. First In Class (S,E)-11-[2-(Arylmethylene)Hydrazono]-PBD Analogs As Selective CB2 Modulators Targeting Neurodegenerative Disorders. Med. Chem. Res. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dean, B.; Sundram, S.; Bradbury, R.; Scarr, E.; Copolov, D. Studies on [3H]CP-55940 binding in the human central nervous system: Regional specific changes in density of cannabinoid-1 receptors associated with schizophrenia and cannabis use. Neuroscience 2001, 103, 9–15. [Google Scholar] [CrossRef]
- Stempel, A.V.; Stumpf, A.; Zhang, H.Y.; Ozdogan, T.; Pannasch, U.; Theis, A.K.; Otte, D.M.; Wojtalla, A.; Racz, I.; Ponomarenko, A.; et al. Cannabinoid type 2 receptors mediate a cell type-specific plasticity in the hippocampus. Neuron 2016, 90, 795–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayant, S.; Sharma, B.M.; Bansal, R.; Sharma, B. Pharmacological benefits of selective modulation of cannabinoid receptor type 2 (CB2) in experimental Alzheimer’s disease. Pharmacol. Biochem. Behav. 2016, 140, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Qu, L.; et al. Crystal structure of the human cannabinoid receptor CB2. Cell 2019, 176, 459–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CB1(%) | CB2(%) | Compound | CB1(%) | CB2(%) |
|---|---|---|---|---|---|
| 4a | −3.09 | −12.50 | 4j | 66.81 | 65.24 |
| 4b | −13.99 | −14.48 | 4k | 41.26 | 84.60 |
| 4c | 0.25 | 3.06 | 4l | 77.17 | 51.73 |
| 4d | −27.99 | −33.65 | 4m | 5.22 | 50.05 |
| 4e | 21.88 | 19.86 | 4n | 44.14 | 43.34 |
| 4f | 1.75 | 3.72 | 4o | 5.09 | 1.01 |
| 4g | 24.19 | 29.27 | 4p | −27.20 | −29.47 |
| 4h | 36.88 | 61.05 | 4q | 55.27 | 95.58 |
| 4i | 49.82 | 36.39 |
| Compound | Structure | Ki (nM) |
|---|---|---|
| 4k |  | 146 |
| 4q |  | 137 |
| PK Parameters | Intravenous | Oral | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Plasma | Brain | Kidney | Liver | Plasma | Brain | Kidney | Liver | ||
| t1/2 | h | 6.91 | 6.54 | 10.2 | 9.26 | 16.4 | 22.9 | 16.7 | 13.0 |
| Cmax or C0 | ng/mL or ng/g | 4345 | 2260 | 4725 | 13497 | 198 | 602 | 795 | 1891 |
| Tmax | h | - | - | - | - | 0.88 | 1.25 | 0.75 | 0.75 |
| AUC0–∞ | ng × h/mL or ng × h/g | 4015 | 5685 | 13272 | 19991 | 739 | 3069 | 5223 | 6423 |
| CL | mL/min/kg | 23.5 | 15.7 | 6.38 | 4.28 | - | - | - | - |
| Vd | L/kg | 14.3 | - | - | - | - | - | - | - |
| F | % | - | - | - | - | 18 | 43 | 37 | 32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ospanov, M.; Sulochana, S.P.; Paris, J.J.; Rimoldi, J.M.; Ashpole, N.; Walker, L.; Ross, S.A.; Shilabin, A.G.; Ibrahim, M.A. Identification of an Orally Bioavailable, Brain-Penetrant Compound with Selectivity for the Cannabinoid Type 2 Receptor. Molecules 2022, 27, 509. https://doi.org/10.3390/molecules27020509
Ospanov M, Sulochana SP, Paris JJ, Rimoldi JM, Ashpole N, Walker L, Ross SA, Shilabin AG, Ibrahim MA. Identification of an Orally Bioavailable, Brain-Penetrant Compound with Selectivity for the Cannabinoid Type 2 Receptor. Molecules. 2022; 27(2):509. https://doi.org/10.3390/molecules27020509
Chicago/Turabian StyleOspanov, Meirambek, Suresh P. Sulochana, Jason J. Paris, John M. Rimoldi, Nicole Ashpole, Larry Walker, Samir A. Ross, Abbas G. Shilabin, and Mohamed A. Ibrahim. 2022. "Identification of an Orally Bioavailable, Brain-Penetrant Compound with Selectivity for the Cannabinoid Type 2 Receptor" Molecules 27, no. 2: 509. https://doi.org/10.3390/molecules27020509
APA StyleOspanov, M., Sulochana, S. P., Paris, J. J., Rimoldi, J. M., Ashpole, N., Walker, L., Ross, S. A., Shilabin, A. G., & Ibrahim, M. A. (2022). Identification of an Orally Bioavailable, Brain-Penetrant Compound with Selectivity for the Cannabinoid Type 2 Receptor. Molecules, 27(2), 509. https://doi.org/10.3390/molecules27020509