
Family Tree for Aqueous Organic Redox Couples for Redox Flow Battery Electrolytes: A Conceptual Review



Abstract
:1. Introduction
Redox Flow Batteries
2. Requirements for Organic Redox Flow Battery Electrolytes
2.1. Solubility
2.2. Solubility and Strategies for Its Improvement
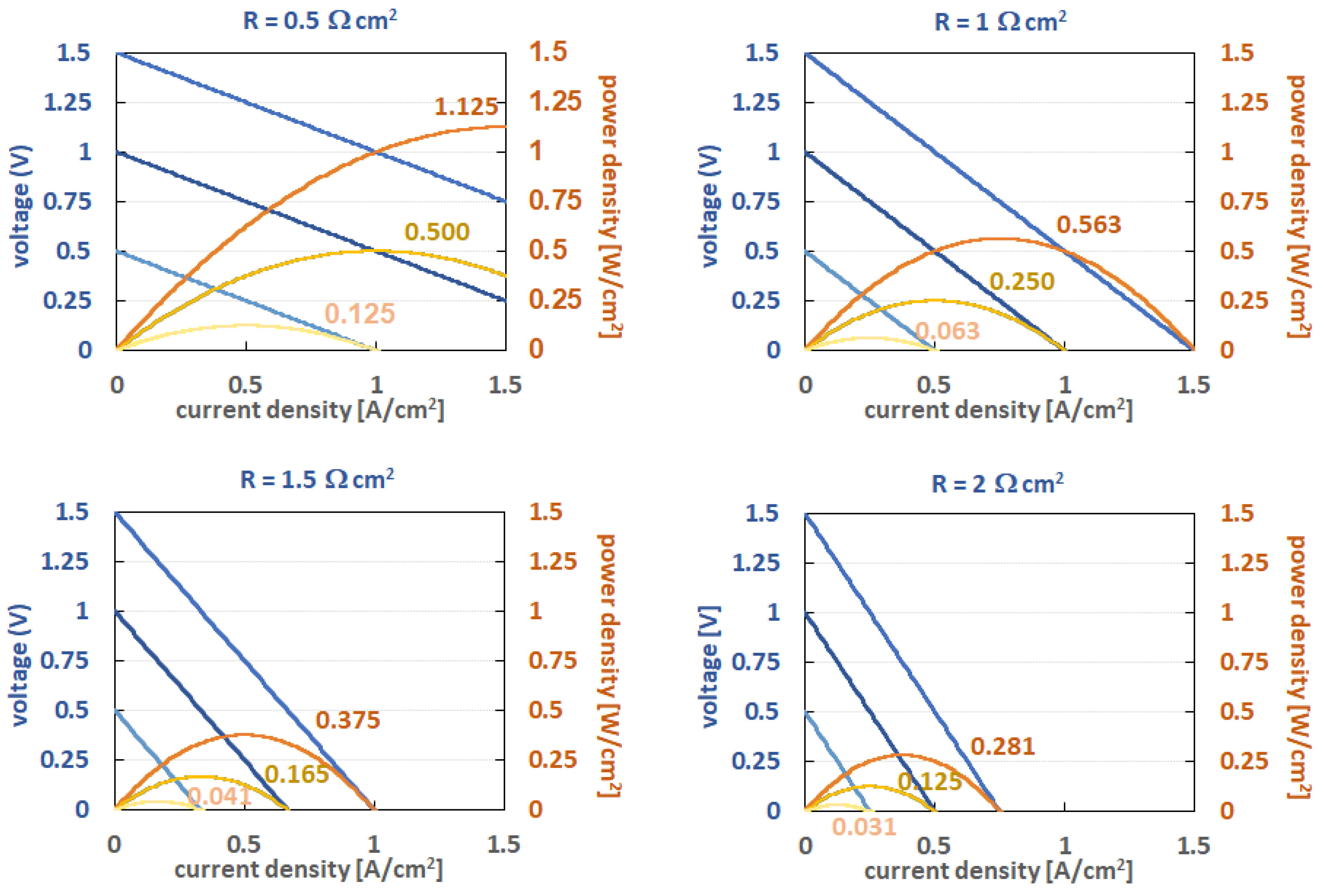
2.3. Redox Potential
2.4. Redox Kinetics
2.5. Chemical and Electrochemical Stability
2.5.1. Stability of the Supporting Electrolyte–Electrolyte–Electrode Interphase
2.5.2. Electrochemical Stability of Active Species
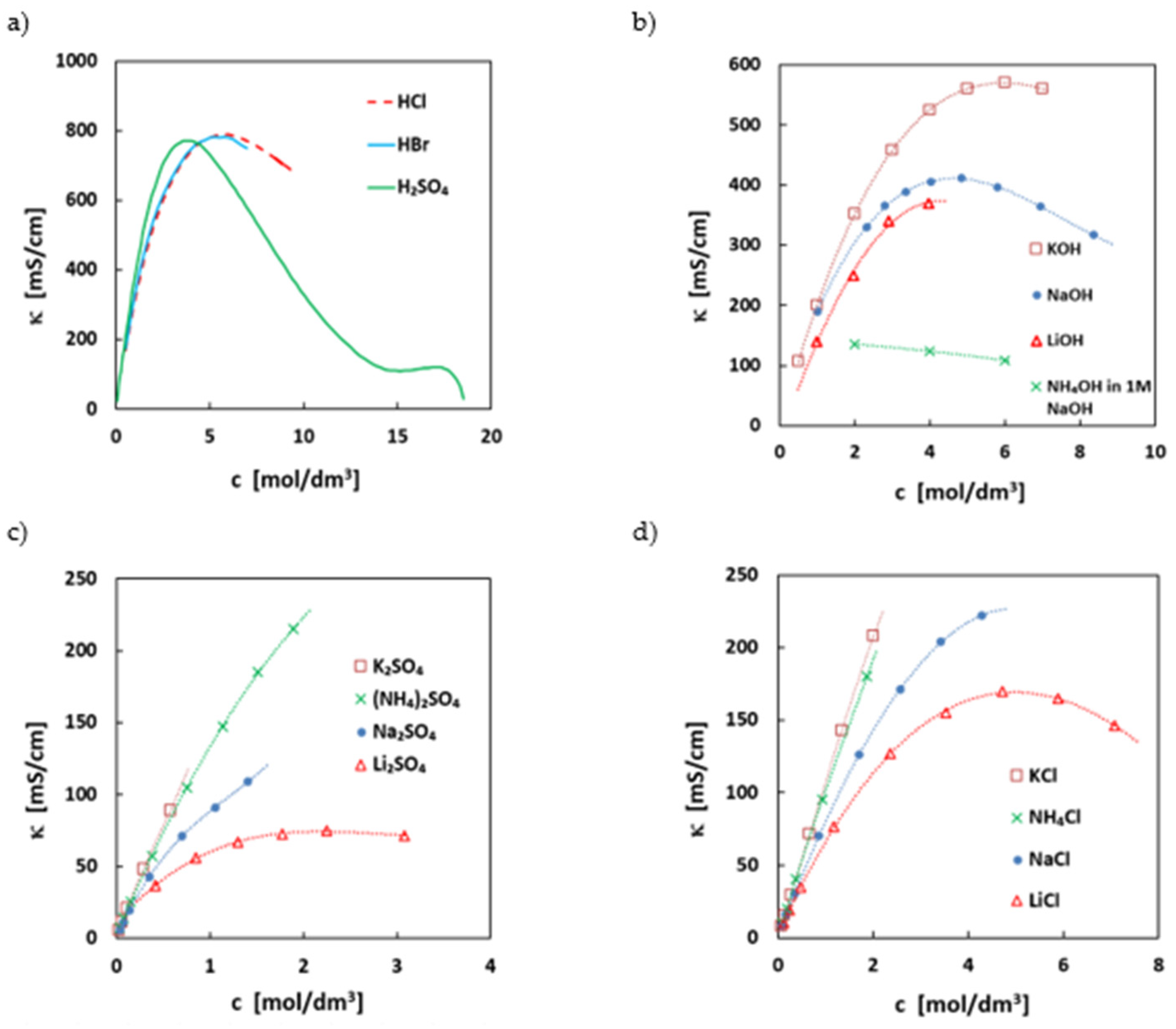
2.6. Ionic Conductivity
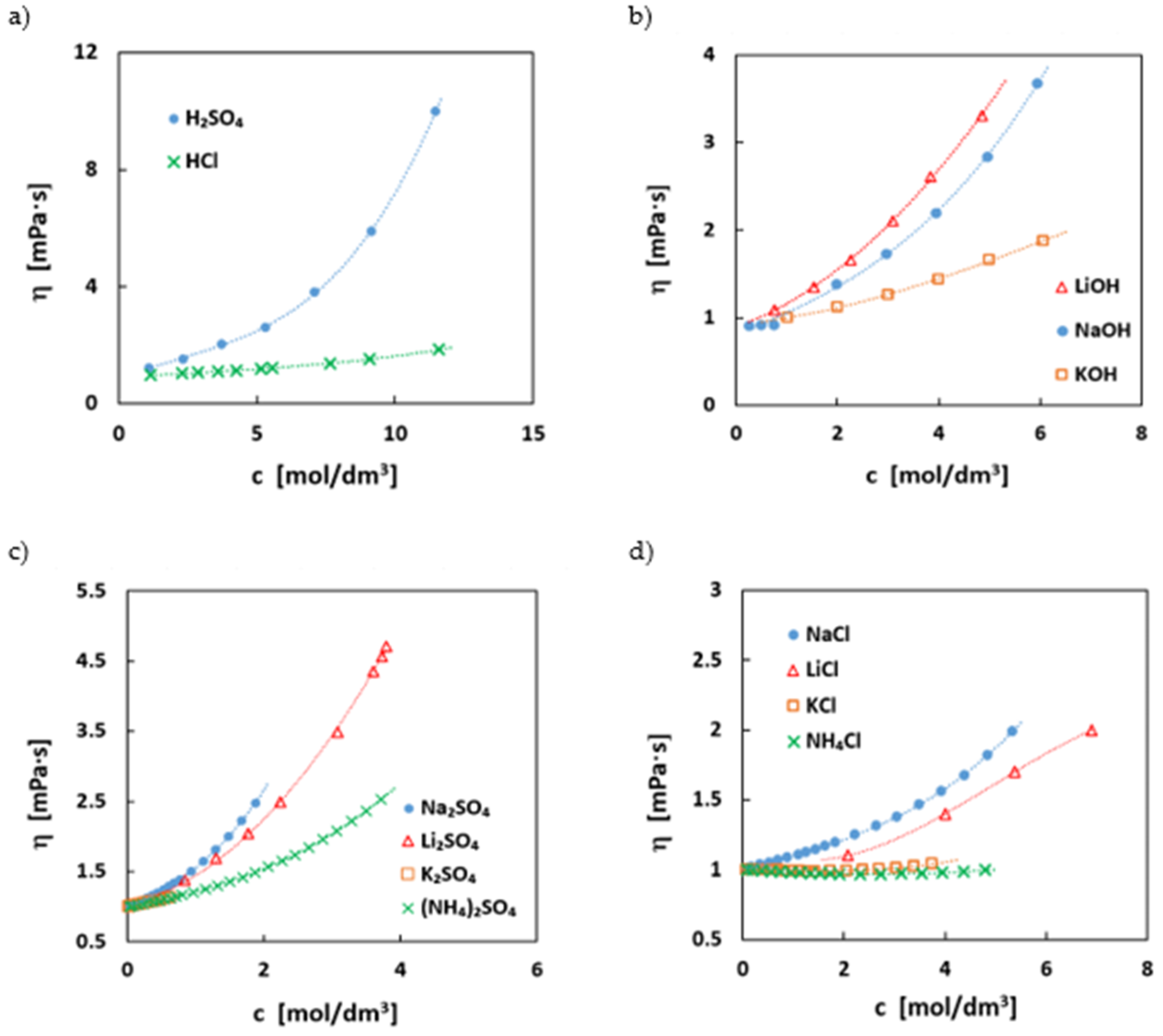
2.7. Dynamic Viscosity
2.8. Permeation
2.9. Effect of Temperature
2.10. Aquatic and Human Toxicity
2.11. Abundance of Materials
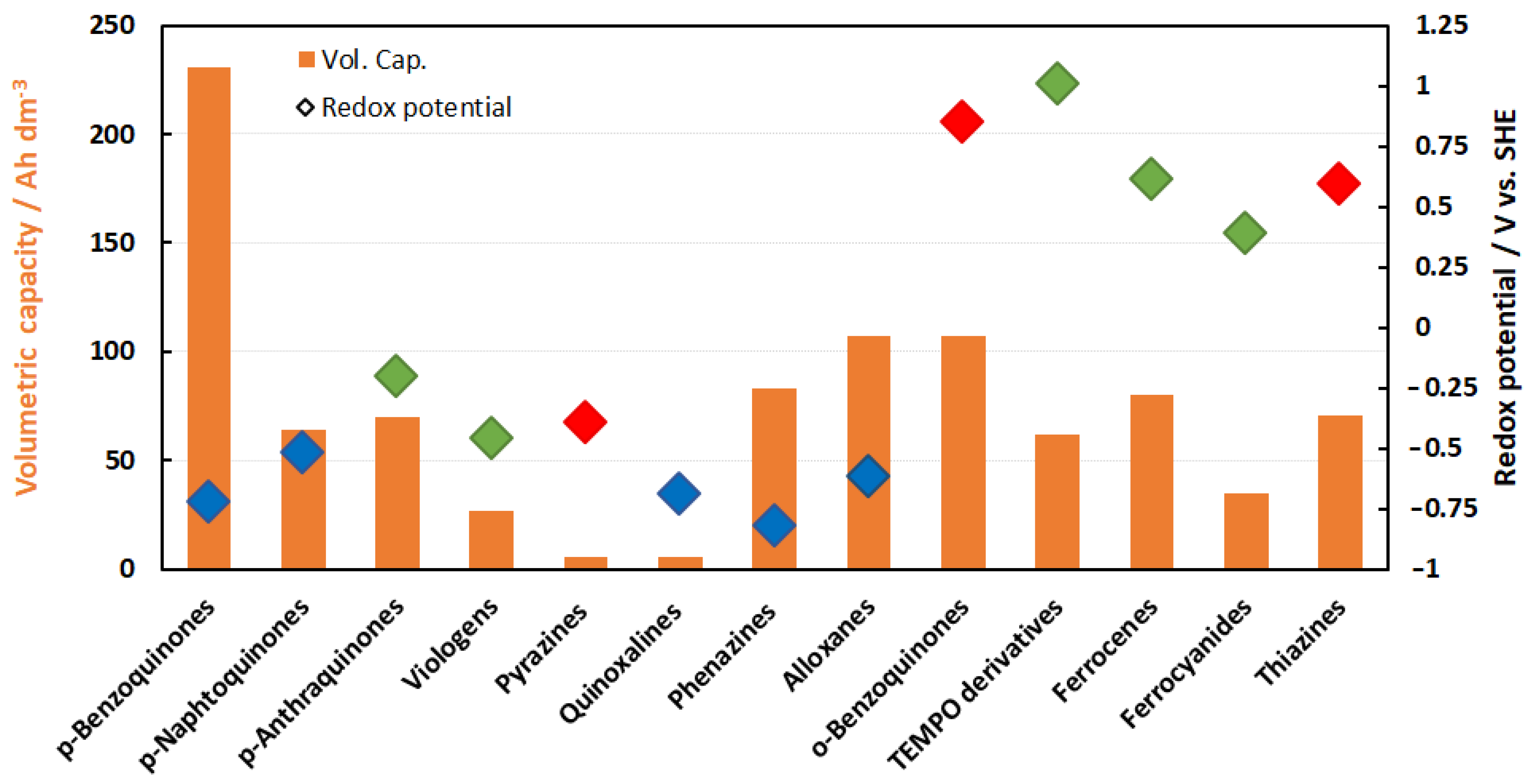
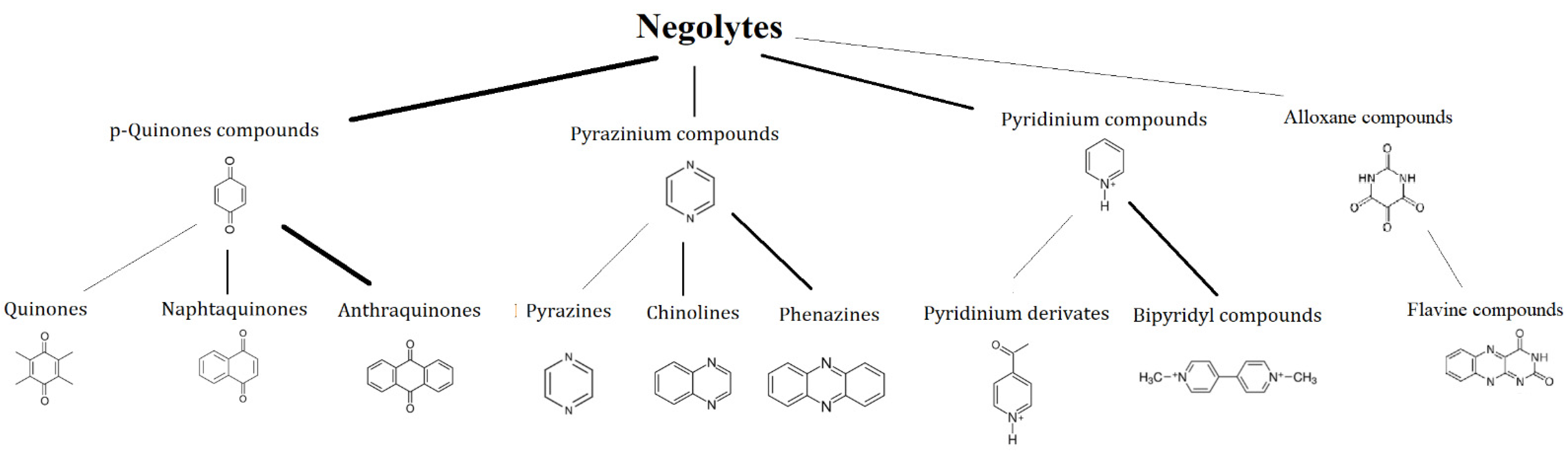
3. Family Tree
- (1)
- Energy density can be limited by decreased solubility of the redox compound in the charged state (e.g., reduction of pyridiniums resulting in loss of its ionic nature).
- (2)
- No mixed electrolytes were considered. If this is considered, the energy density is divided by a factor of two.
- (3)
- In a mixed electrolyte, it is unlikely that solubility is not affected by the other redox species. This usually leads to lower solubilities of the active species and an increase in viscosity.
- (4)
- Solubility of equal amounts of conducting salt for each redox equivalent plus an excess to ensure base conductivity are required, further reducing the solubility of the active material.
3.1. Negolytes
3.1.1. p-Quinones
Quinones (One Ring)
Naphthoquinone (Two Rings)
Anthraquinones (Three Rings)
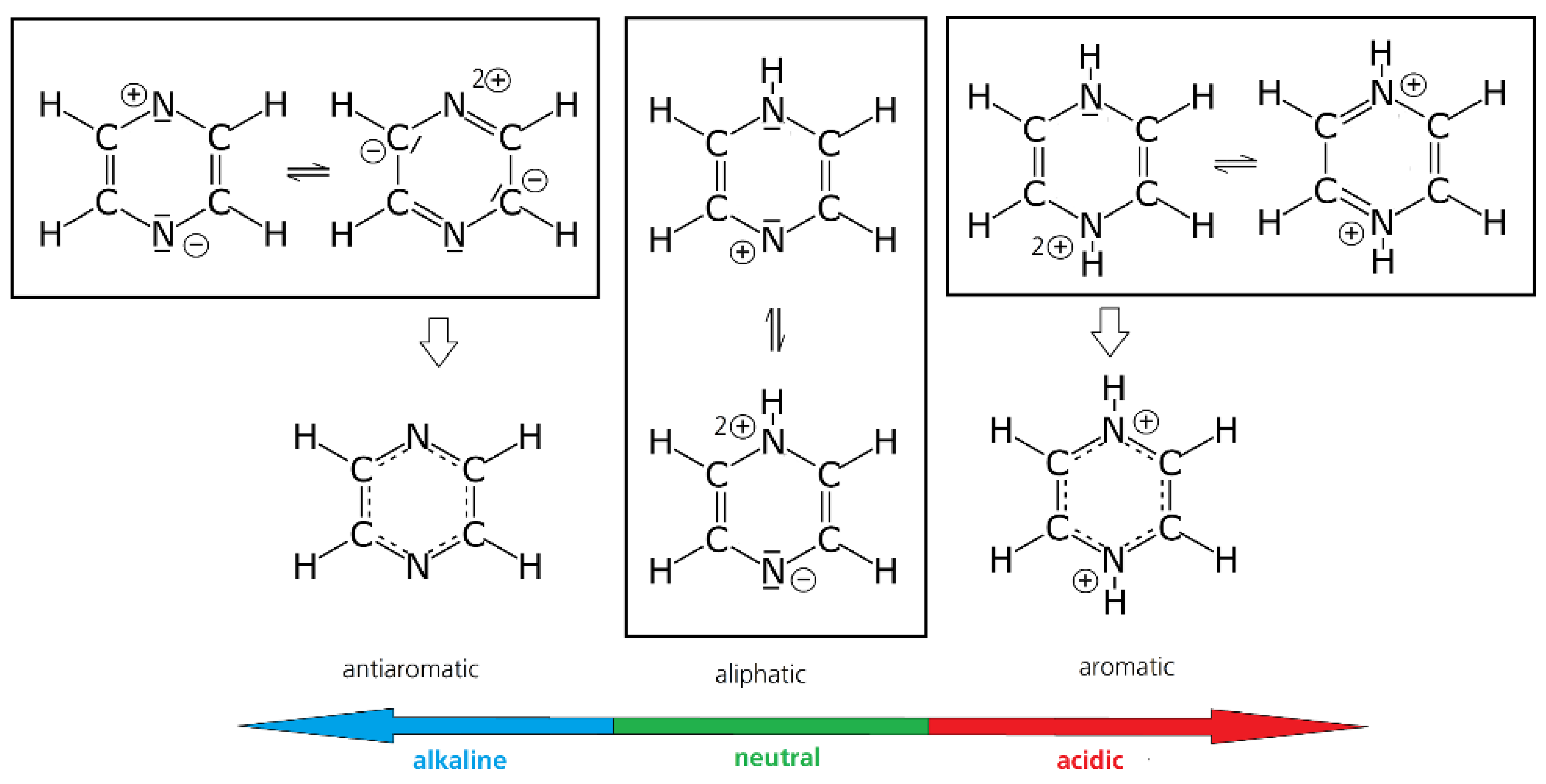
3.1.2. Pyridinium Compounds
3.1.3. Pyrazine Compounds
Pyrazines (One Ring)
Quinoxalines (Two Rings)
Phenazines (Three Rings)
3.1.4. Alloxan Compounds
3.1.5. Thiazine Compounds
Thiazines (One Ring)
Naphthathiazines (Two Rings)
Phenothiazines (Three Rings)
3.2. Posilytes
3.2.1. o-Quinones (One Ring)
3.2.2. p-Quinone (One Ring)
3.2.3. N-O∙ Radicals
3.2.4. Metal Complex–Low Spin
3.2.5. Metal Complex–High-Spin (Ferrocenes)
4. Customization of Components Used in Non-Aqueous Systems
4.1. Sulfur Compounds
4.2. N-heterocycles
4.3. Stable Organic Radicals
5. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhang, H.; Sun, C. Cost-effective iron-based aqueous redox flow batteries for large-scale energy storage application: A review. J. Power Sources 2021, 493, 229445. [Google Scholar] [CrossRef]
- Sun, C.; Negro, E.; Vezzù, K.; Pagot, G.; Cavinato, G.; Nale, A.; Herve Bang, Y.; Di Noto, V. Hybrid inorganic-organic proton-conducting membranes based on SPEEK doped with WO3 nanoparticles for application in vanadium redox flow batteries. Electrochim. Acta 2019, 309, 311–325. [Google Scholar] [CrossRef]
- Yuan, Z.; Yin, Y.; Xie, C.; Zhang, H.; Yao, Y.; Li, X.; Yuan, Z.Z.; Yin, Y.B.; Xie, C.X.; Zhang, H.M.; et al. Advanced Materials for Zinc-Based Flow Battery: Development and Challenge. Adv. Mater. 2019, 31, 1902025. [Google Scholar] [CrossRef] [PubMed]
- Brushett, F.R.; Aziz, M.J.; Rodby, K.E. On Lifetime and Cost of Redox-Active Organics for Aqueous Flow Batteries. ACS Energy Lett. 2020, 5, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Leung, P.; Aili, D.; Xu, Q.; Rodchanarowan, A.; Shah, A.A. Rechargeable organic-air redox flow batteries. Sustain. Energy Fuels 2018, 2, 2252–2259. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Díez, E.; Ventosa, E.; Guarnieri, M.; Trovò, A.; Flox, C.; Marcilla, R.; Soavi, F.; Mazur, P.; Aranzabe, E.; Ferret, R. Redox flow batteries: Status and perspective towards sustainable stationary energy storage. J. Power Sources 2021, 481, 228804. [Google Scholar] [CrossRef]
- Noack, J.; Roznyatovskaya, N.; Herr, T.; Fischer, P. The Chemistry of Redox-Flow Batteries. Angew. Chem. Int. Ed. 2015, 54, 9776–9809. [Google Scholar] [CrossRef] [PubMed]
- Izutsu, K. Electrochemistry in Nonaqueous Solutions; John Wiley & Sons: Hoboken, NJ, USA, 2002. [Google Scholar] [CrossRef]
- Barker, B.J.; Rosenfarb, J.; Caruso, J.A. Harnstoffe als Lösungsmittel in der chemischen Forschung. Angew. Chem. 1979, 91, 560–564. [Google Scholar] [CrossRef]
- Stroka, J.; Herfort, I.; Schneider, H. Dimethylpropyleneurea-water mixtures: 1. Physical properties. J. Solut. Chem. 1990, 19, 743–753. [Google Scholar] [CrossRef]
- Taniewska-Osińska, S.; Jóźwiak, M. Densimetric and viscosimetric investigations of NaI in hexamethylphosphoramide–water mixtures at 293.15, 298.15 and 303.15 K. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1989, 85, 2141–2147. [Google Scholar] [CrossRef]
- Merck Safety Data Sheets. Available online: https://www.sigmaaldrich.com/PL/pl/search/646-06-0?focus=products&page=1&perPage=30&sort=relevance&term=646-06-0&type=cas_number# (accessed on 10 March 2021).
- Agmon, N. The Grotthuss mechanism. Chem. Phys. Lett. 1995, 244, 456–462. [Google Scholar] [CrossRef]
- US9929425B2—High Solubility Iron Hexacyanides—Google Patents. Available online: https://patents.google.com/patent/US9929425B2/en (accessed on 30 November 2021).
- Luo, J.; Hu, B.; Debruler, C.; Bi, Y.; Zhao, Y.; Yuan, B.; Hu, M.; Wu, W.; Liu, T.L. Unprecedented Capacity and Stability of Ammonium Ferrocyanide Catholyte in pH Neutral Aqueous Redox Flow Batteries. Joule 2019, 3, 149–163. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Ruan, W.; Chen, Q. Understanding the Aqueous Solubility of Anthraquinone Sulfonate Salts: The Quest for High Capacity Electrolytes of Redox Flow Batteries. J. Electrochem. Soc. 2020, 167, 070522. [Google Scholar] [CrossRef]
- Wedege, K.; Dražević, E.; Konya, D.; Bentien, A. Organic Redox Species in Aqueous Flow Batteries: Redox Potentials, Chemical Stability and Solubility. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Niu, Z.; Peng, S.; Ding, Y.; Zhang, L.; Guo, X.; Zhao, Y.; Yu, G. Phenothiazine-Based Organic Catholyte for High-Capacity and Long-Life Aqueous Redox Flow Batteries. Adv. Mater. 2019, 31, 1901052. [Google Scholar] [CrossRef]
- Boobier, S.; Hose, D.R.J.; Blacker, A.J.; Nguyen, B.N. Machine learning with physicochemical relationships: Solubility prediction in organic solvents and water. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Delaney, J.S. Predicting aqueous solubility from structure. Drug Discov. Today 2005, 10, 289–295. [Google Scholar] [CrossRef]
- Bard, A.; Parsons, R.; Jordan, J. Standard Potentials in Aqueous Solution; M. Dekker: New York, NY, USA, 1985; ISBN 13-978-0824772918. [Google Scholar]
- De La Cruz, C.; Molina, A.; Patil, N.; Ventosa, E.; Marcilla, R.; Mavrandonakis, A. New insights into phenazine-based organic redox flow batteries by using high-throughput DFT modelling. Sustain. Energy Fuels 2020, 4, 5513–5521. [Google Scholar] [CrossRef]
- Gencten, M.; Sahin, Y. A critical review on progress of the electrode materials of vanadium redox flow battery. Int. J. Energy Res. 2020, 44, 7903–7923. [Google Scholar] [CrossRef]
- Tichter, T.; Andrae, D.; Mayer, J.; Schneider, J.; Gebhard, M.; Roth, C. Theory of cyclic voltammetry in random arrays of cylindrical microelectrodes applied to carbon felt electrodes for vanadium redox flow batteries. Phys. Chem. Chem. Phys. 2019, 21, 9061–9068. [Google Scholar] [CrossRef]
- Zhang, J.; Anson, F.C. Voltammetry and in-situ Fourier transform IR spectroscopy of two anthraquinone disulfonates adsorbed on graphite electrodes. J. Electroanal. Chem. 1992, 331, 945–957. [Google Scholar] [CrossRef]
- Rak, K.; Klikar, M.; Burešová, Z.; Mazúr, P.; Mikešová, M.; Kvíčala, J.; Bureš, F. Electrochemical investigation of structurally varied azinium scaffolds. Org. Biomol. Chem. 2021, 19, 8830–8839. [Google Scholar] [CrossRef]
- Armand, J.; Chekir, K.; Pinson, J. Electrochemical Reduction of Pyrazines into Dihydropyrazines. Preparation of Dihydropyrazines. Can. J. Chem. 2011, 52, 3971–3980. [Google Scholar] [CrossRef]
- Miao, S.; Brombosz, S.M.; Schleyer, P.V.R.; Wu, J.I.; Barlow, S.; Marder, S.R.; Hardcastle, K.I.; Bunz, U.H.F. Are N,N-dihydrodiazatetracene derivatives antiaromatic? J. Am. Chem. Soc. 2008, 130, 7339–7344. [Google Scholar] [CrossRef]
- Kwabi, D.G.; Ji, Y.; Aziz, M.J. Electrolyte Lifetime in Aqueous Organic Redox Flow Batteries: A Critical Review. Chem. Rev. 2020, 120, 6467–6489. [Google Scholar] [CrossRef]
- Goulet, M.-A.; Aziz, M.J. Flow Battery Molecular Reactant Stability Determined by Symmetric Cell Cycling Methods. J. Electrochem. Soc. 2018, 165, A1466–A1477. [Google Scholar] [CrossRef] [Green Version]
- Tabor, D.P.; Gómez-Bombarelli, R.; Tong, L.; Gordon, R.G.; Aziz, M.J.; Aspuru-Guzik, A. Mapping the frontiers of quinone stability in aqueous media: Implications for organic aqueous redox flow batteries. J. Mater. Chem. A 2019, 7, 12833–12841. [Google Scholar] [CrossRef]
- Luo, J.; Sam, A.; Hu, B.; DeBruler, C.; Wei, X.; Wang, W.; Liu, T.L. Unraveling pH dependent cycling stability of ferricyanide/ferrocyanide in redox flow batteries. Nano Energy 2017, 42, 215–221. [Google Scholar] [CrossRef]
- Beck, F. Design and Materials for Metal-Free Rechargeable Batteries. In New Promising Electrochemical Systems for Rechargeable Batteries; Springer: Amsterdam, The Netherlands, 1996; pp. 393–417. [Google Scholar]
- Kosydar, R.; Drelinkiewicz, A.; Ganhy, J.P. Degradation reactions in anthraquinone process of hydrogen peroxide synthesis. Catal. Lett. 2010, 139, 105–113. [Google Scholar] [CrossRef]
- Goulet, M.A.; Tong, L.; Pollack, D.A.; Tabor, D.P.; Odom, S.A.; Aspuru-Guzik, A.; Kwan, E.E.; Gordon, R.G.; Aziz, M.J. Extending the lifetime of organic flow batteries via redox state management. J. Am. Chem. Soc. 2020, 141, 8014–8019. [Google Scholar] [CrossRef] [PubMed]
- DeBruler, C.; Hu, B.; Moss, J.; Liu, X.; Luo, J.; Sun, Y.; Liu, T.L. Designer Two-Electron Storage Viologen Anolyte Materials for Neutral Aqueous Organic Redox Flow Batteries. Chem 2017, 3, 961–978. [Google Scholar] [CrossRef] [Green Version]
- Bird, C.L.; Kuhn, A.T. Electrochemistry of the viologens. Chem. Soc. Rev. 1981, 10, 49–82. [Google Scholar] [CrossRef]
- Beh, E.S.; De Porcellinis, D.; Gracia, R.L.; Xia, K.T.; Gordon, R.G.; Aziz, M.J. A neutral pH aqueous organic-organometallic redox flow battery with extremely high capacity retention. ACS Energy Lett. 2017, 2, 639–644. [Google Scholar] [CrossRef]
- Burešová, Z.; Klikar, M.; Mazúr, P.; Mikešová, M.; Kvíčala, J.; Bystron, T.; Bureš, F. Redox Property Tuning in Bipyridinium Salts. Front. Chem. 2021, 8, 1289. [Google Scholar] [CrossRef]
- Choe, Y.K.; Tsuchida, E.; Ikeshoji, T. First-principles molecular dynamics study on aqueous sulfuric acid solutions. J. Chem. Phys. 2007, 126, 154510. [Google Scholar] [CrossRef]
- Hamer, W.J.; DeWane, H.J. Electrolytic Conductance and the Conductances of the Halogen Acids in Water; U.S. National Bureau of Standards: Washington, DC, USA, 1970. Available online: https://www.worldcat.org/title/electrolytic-conductance-and-the-conductances-of-the-halogen-acids-in-water/oclc/114176 (accessed on 19 March 2021).
- Ling-Wei Woo, A. Conductance Studies of Concentrated Solutions of Sodium Hydroxide and Potassium Hydroxide Electrolytes Recommended Citation. Master’s Thesis, South Dakota State University, Brookings, SD, USA, 1968. [Google Scholar]
- Gilliam, R.J.; Graydon, J.W.; Kirk, D.W.; Thorpe, S.J. A review of specific conductivities of potassium hydroxide solutions for various concentrations and temperatures. Int. J. Hydrog. Energy 2007, 32, 359–364. [Google Scholar] [CrossRef]
- Gierszewski, P.J.; Finn, P.A.; Kirk, D.W. Properties of LiOH and LiNO3 aqueous solutions. Fusion Eng. Des. 1990, 13, 59–71. [Google Scholar] [CrossRef]
- Kuznetsova, O.G.; Levin, A.M.; Sevost’yanov, M.A.; Tsybin, O.I.; Bol’shikh, A.O. Electrical Conductivity of Ammonia–Alkali Solutions and Its Activation Energy. Russ. Metall. 2019, 2019, 93–97. [Google Scholar] [CrossRef]
- Lide, D.R.; Baysinger, G.; Berger, L.I.; Goldberg, R.N.; Kehiaian, H.V.; Kuchitsu, K.; Roth, D.L.; Zwillinger, D. CRC Handbook of Chemistry and Physics Editor-in-Chief, 71st ed.; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Cartón, A.; Sobrón, F.; Bolado, S.; Gerbolés, J.I. Density, Viscosity, and Electrical Conductivity of Aqueous Solutions of Lithium Sulfate. J. Chem. Eng. Data 1995, 40, 987–991. [Google Scholar] [CrossRef]
- Charvát, J.; Mazúr, P.; Dundálek, J.; Pocedič, J.; Vrána, J.; Mrlík, J.; Kosek, J.; Dinter, S. Performance enhancement of vanadium redox flow battery by optimized electrode compression and operational conditions. J. Energy Storage 2020, 30, 101468. [Google Scholar] [CrossRef]
- Liler, M. Properties of Sulphuric Acid/Water Mixtures as Solvents; Elsevier: Amsterdam, The Netherlands, 1971; Volume 23, pp. 1–25. [Google Scholar]
- Nishikata, E.; Ishii, T.; Ohta, T. Viscosities of Aqueous Hydrochloric Acid Solutions, and Densities and Viscosities of Aqueous Hydroiodic Acid Solutions. J. Chem. Eng. Data 1981, 26, 254–256. [Google Scholar] [CrossRef]
- Sipos, P.M.; Hefter, G.; May, P.M. Viscosities and densities of highly concentrated aqueous MOH solutions (M+ = Na+, K+, Li+, Cs+, (CH3)4N+) at 25.0 °C. J. Chem. Eng. Data 2000, 45, 613–617. [Google Scholar] [CrossRef]
- Wahab, A.; Mahiuddin, S. Isentropic compressibility and viscosity of aqueous and methanolic lithium chloride solutions. Can. J. Chem. 2002, 80, 175–182. [Google Scholar] [CrossRef]
- Mähler, J.; Persson, I. A study of the hydration of the alkali metal ions in aqueous solution. Inorg. Chem. 2012, 51, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Vrána, J.; Charvát, J.; Mazúr, P.; Bělský, P.; Dundálek, J.; Pocedič, J.; Kosek, J. Commercial perfluorosulfonic acid membranes for vanadium redox flow battery: Effect of ion-exchange capacity and membrane internal structure. J. Memb. Sci. 2018, 552, 202–212. [Google Scholar] [CrossRef]
- Teng, X.; Guo, Y.; Liu, D.; Li, G.; Yu, C.; Dai, J. A polydopamine-coated polyamide thin film composite membrane with enhanced selectivity and stability for vanadium redox flow battery. J. Memb. Sci. 2020, 601, 117906. [Google Scholar] [CrossRef]
- Qian, P.; Jiang, Y.; Wang, H.; Zhang, L.; Shi, H. Improved proton conductivity and structure stability of an SPEEK/SPPS blend membrane for vanadium redox flow batteries. Mater. Chem. Front. 2021, 5, 8171–8182. [Google Scholar] [CrossRef]
- Sun, C.; Negro, E.; Nale, A.; Pagot, G.; Vezzù, K.; Zawodzinski, T.A.; Meda, L.; Gambaro, C.; Di Noto, V. An efficient barrier toward vanadium crossover in redox flow batteries: The bilayer [Nafion/(WO3)x] hybrid inorganic-organic membrane. Electrochim. Acta 2021, 378, 138133. [Google Scholar] [CrossRef]
- Shi, Y.; Eze, C.; Xiong, B.; He, W.; Zhang, H.; Lim, T.M.; Ukil, A.; Zhao, J. Recent development of membrane for vanadium redox flow battery applications: A review. Appl. Energy 2019, 238, 202–224. [Google Scholar] [CrossRef]
- Janoschka, T.; Martin, N.; Martin, U.; Friebe, C.; Morgenstern, S.; Hiller, H.; Hager, M.D.; Schubert, U.S. An aqueous, polymer-based redox-flow battery using non-corrosive, safe, and low-cost materials. Nature 2015, 527, 78–81. [Google Scholar] [CrossRef]
- Janoschka, T.; Morgenstern, S.; Hiller, H.; Friebe, C.; Wolkersdörfer, K.; Häupler, B.; Hager, M.D.; Schubert, U.S. Synthesis and characterization of TEMPO- and viologen-polymers for water-based redox-flow batteries. Polym. Chem. 2015, 6, 7801–7811. [Google Scholar] [CrossRef]
- Hagemann, T.; Winsberg, J.; Grube, M.; Nischang, I.; Janoschka, T.; Martin, N.; Hager, M.D.; Schubert, U.S. An aqueous all-organic redox-flow battery employing a (2,2,6,6-tetramethylpiperidin-1-yl)oxyl-containing polymer as catholyte and dimethyl viologen dichloride as anolyte. J. Power Sources 2018, 378, 546–554. [Google Scholar] [CrossRef]
- Lai, Y.Y.; Li, X.; Liu, K.; Tung, W.Y.; Cheng, C.F.; Zhu, Y. Stable Low-Cost Organic Dye Anolyte for Aqueous Organic Redox Flow Battery. ACS Appl. Energy Mater. 2020, 3, 2290–2295. [Google Scholar] [CrossRef]
- Luo, J.; Hu, B.; Hu, M.; Zhao, Y.; Liu, T.L. Status and Prospects of Organic Redox Flow Batteries toward Sustainable Energy Storage. ACS Energy Lett. 2019, 4, 2220–2240. [Google Scholar] [CrossRef]
- Potash, R.A.; McKone, J.R.; Conte, S.; Abruña, H.D. On the Benefits of a Symmetric Redox Flow Battery. J. Electrochem. Soc. 2016, 163, A338–A344. [Google Scholar] [CrossRef]
- Fornari, R.P.; Mesta, M.; Hjelm, J.; Vegge, T.; De Silva, P. Molecular Engineering Strategies for Symmetric Aqueous Organic Redox Flow Batteries. ACS Mater. Lett. 2020, 2, 239–246. [Google Scholar] [CrossRef]
- Carretero-González, J.; Castillo-Martínez, E.; Armand, M. Highly water-soluble three-redox state organic dyes as bifunctional analytes. Energy Environ. Sci. 2016, 9, 3521–3530. [Google Scholar] [CrossRef]
- Tong, L.; Jing, Y.; Gordon, R.G.; Aziz, M.J. Symmetric All-Quinone Aqueous Battery. ACS Appl. Energy Mater. 2019, 2, 4016–4021. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, F.; Niu, Z.; Wu, H.; He, Y.; Zhu, H.; Ye, J.; Zhao, Y.; Zhang, X. Enhanced cyclability of organic redox flow batteries enabled by an artificial bipolar molecule in neutral aqueous electrolyte. J. Power Sources 2019, 417, 83–89. [Google Scholar] [CrossRef]
- Janoschka, T.; Friebe, C.; Hager, M.D.; Martin, N.; Schubert, U.S. An Approach Toward Replacing Vanadium: A Single Organic Molecule for the Anode and Cathode of an Aqueous Redox-Flow Battery. ChemistryOpen 2017, 6, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Winsberg, J.; Stolze, C.; Muench, S.; Liedl, F.; Hager, M.D.; Schubert, U.S. TEMPO/Phenazine Combi-Molecule: A Redox-Active Material for Symmetric Aqueous Redox-Flow Batteries. ACS Energy Lett. 2016, 1, 976–980. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, M.; Ma, T.; Liu, J.; Zhang, Q.; Tao, Z.; Liang, J. A symmetric aqueous redox flow battery based on viologen derivative. Chin. Chem. Lett. 2020, 31, 1690–1693. [Google Scholar] [CrossRef]
- Liu, B.; Tang, C.W.; Jiang, H.; Jia, G.; Zhao, T. An aqueous organic redox flow battery employing a trifunctional electroactive compound as anolyte, catholyte and supporting electrolyte. J. Power Sources 2020, 477, 228985. [Google Scholar] [CrossRef]
- Bhattarai, A.; Ghimire, P.C.; Whitehead, A.; Schweiss, R.; Scherer, G.G.; Wai, N.; Hng, H.H. Novel Approaches for Solving the Capacity Fade Problem during Operation of a Vanadium Redox Flow Battery. Batteries 2018, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Mazúr, P.; Charvát, J.; Mrlík, J.; Pocedič, J.; Akrman, J.; Kubáč, L.; Řeháková, B.; Kosek, J. Evaluation of Electrochemical Stability of Sulfonated Anthraquinone-Based Acidic Electrolyte for Redox Flow Battery Application. Molecules 2021, 26, 2484. [Google Scholar] [CrossRef]
- Egorova, K.S.; Ananikov, V.P. Toxicity of Metal Compounds: Knowledge and Myths. Organometallics 2017, 36, 4071–4090. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.; Sun, B.; Prakash, J.; Nirmalakhandan, N.N. Toxicity of Organic Chemicals and Their Mixtures to Activated Sludge Microorganisms. J. Environ. Eng. 1996, 122, 424–429. [Google Scholar] [CrossRef]
- Puzyn, T.; Leszczynski, J.; Cronin, M.T. Recent Advances in QSAR Studies: Methods and Applications; (Challenges and Advances in Computational Chemistry and Physics, 8); Springer: Dordrecht Library, Dordrecht, The Netherlands, 2010. [Google Scholar]
- Melnikov, F.; Kostal, J.; Voutchkova-Kostal, A.; Zimmerman, J.B.; Anastas, P.T. Assessment of predictive models for estimating the acute aquatic toxicity of organic chemicals. Green Chem. 2016, 18, 4432–4445. [Google Scholar] [CrossRef] [Green Version]
- Bolton, J.L.; Dunlap, T. Formation and biological targets of quinones: Cytotoxic versus cytoprotective effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef]
- Monks, T.J.; Hanzlik, R.P.; Cohen, G.M.; Ross, D.; Graham, D.G. Quinone Chemistry and Toxicity. Toxicol. Appl. Pharmacol. 1992, 112, 2–16. [Google Scholar] [CrossRef]
- Bakhite, E.A.; Abd-Ella, A.A.; El-Sayed, M.E.A.; Abdel-Raheem, S.A.A. Pyridine derivatives as insecticides. part 1: Synthesis and Toxicity of some pyridine derivatives against cowpea aphid, Aphis craccivora koch (homoptera: Aphididae). J. Agric. Food Chem. 2014, 62, 9982–9986. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.T.; Stewart, M.S.; Levitt, T.; Widdop, B. Paraquat poisoning: Significance of plasma-paraquat concentrations. Lancet 1979, 314, 330–332. [Google Scholar] [CrossRef]
- Rychen, G.; Aquilina, G.; Azimonti, G.; Bampidis, V.; de Lourdes Bastos, M.; Bories, G.; Cocconcelli, P.S.; Flachowsky, G.; Gropp, J.; Kolar, B.; et al. Safety and efficacy of pyrazine derivatives including saturated ones belonging to chemical group 24 when used as flavourings for all animal species. EFSA J. 2017, 15, 4671. [Google Scholar] [CrossRef] [Green Version]
- Jaidann, M.; Roy, S.; Abou-Rachid, H.; Lussier, L.S. A DFT theoretical study of heats of formation and detonation properties of nitrogen-rich explosives. J. Hazard. Mater. 2010, 176, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, C.; Wu, H.; Feng, P. Activation and Delivery of Tetrazine-Responsive Bioorthogonal Prodrugs. Molecules 2020, 25, 5640. [Google Scholar] [CrossRef]
- Asif, M. Chemical and Pharmacological Potential of Various Substituted Thiazine Derivatives. J. Pharm. Appl. Chem. 2015, 1, 49. [Google Scholar] [CrossRef]
- Lange, F.T.; Furrer, R.; Brauch, H.J. Polar Aromatic Sulfonates and Their Relevance to Waterworks; RIWA Association of River Water Works: Utrecht, The Netherlands, 2000. [Google Scholar]
- Lichtfouse, E.; Schwarzbauer, J.; Robert, D. Environmental Chemistry: Green Chemistry and Pollutants in Ecosystems; Springer: Berlin/Heidelberg, Germany, 2005; ISBN 97-835-40228608. [Google Scholar]
- Ferber, K.H. Toxicology of indigo. A review. J. Environ. Pathol. Toxicol. Oncol. 1987, 7, 73–83. [Google Scholar] [PubMed]
- Henschler, D. Toxicity of Chlorinated Organic Compounds: Effects of the Introduction of Chlorine in Organic Molecules. Angew. Chem. Int. Ed. Engl. 1994, 33, 1920–1935. [Google Scholar] [CrossRef]
- Salkinoja-Salonen, M.; Uotila, J.; Jokela, J.; Laine, M.; Saski, E. Organic Halogens in the Environment: Studies of Environmental Biodegradability and Human Exposure. Environ. Health Perspect. 1995, 103, 63–69. [Google Scholar]
- Schlemmer, W.; Nothdurft, P.; Petzold, A.; Riess, G.; Frühwirt, P.; Schmallegger, M.; Gescheidt-Demner, G.; Fischer, R.; Freunberger, S.A.; Kern, W.; et al. 2-Methoxyhydroquinone from Vanillin for Aqueous Redox-Flow Batteries. Angew. Chem. Int. Ed. 2020, 59, 22943–22946. [Google Scholar] [CrossRef]
- Yang, Z.; Tong, L.; Tabor, D.P.; Beh, E.S.; Goulet, M.-A.; De Porcellinis, D.; Aspuru-Guzik, A.; Gordon, R.G.; Aziz, M.J. Alkaline Benzoquinone Aqueous Flow Battery for Large-Scale Storage of Electrical Energy. Adv. Energy Mater. 2018, 8, 1702056. [Google Scholar] [CrossRef]
- Hu, P.; Lan, H.; Wang, X.; Yang, Y.; Liu, X.; Wang, H.; Guo, L. Renewable-lawsone-based sustainable and high-voltage aqueous flow battery. Energy Storage Mater. 2019, 19, 62–68. [Google Scholar] [CrossRef]
- Wang, C.; Yang, Z.; Wang, Y.; Zhao, P.; Yan, W.; Zhu, G.; Ma, L.; Yu, B.; Wang, L.; Li, G.; et al. High-Performance Alkaline Organic Redox Flow Batteries Based on 2-Hydroxy-3-carboxy-1,4-naphthoquinone. ACS Energy Lett. 2018, 3, 2404–2409. [Google Scholar] [CrossRef]
- Tong, L.; Goulet, M.A.; Tabor, D.P.; Kerr, E.F.; De Porcellinis, D.; Fell, E.M.; Aspuru-Guzik, A.; Gordon, R.G.; Aziz, M.J. Molecular Engineering of an Alkaline Naphthoquinone Flow Battery. ACS Energy Lett. 2019, 4, 1880–1887. [Google Scholar] [CrossRef]
- Mirle, C.; Medabalmi, V.; Ramanujam, K. Crossover-free hydroxy-substituted quinone anolyte and potassium ferrocyanide catholyte for aqueous alkaline organic redox flow battery. Catal. Today 2021, 370, 173–180. [Google Scholar] [CrossRef]
- Huskinson, B.; Marshak, M.P.; Suh, C.; Er, S.; Gerhardt, M.R.; Galvin, C.J.; Chen, X.; Aspuru-Guzik, A.; Gordon, R.G.; Aziz, M.J. A metal-free organic-inorganic aqueous flow battery. Nature 2014, 505, 195–198. [Google Scholar] [CrossRef]
- Gerhardt, M.R.; Tong, L.; Gómez-Bombarelli, R.; Chen, Q.; Marshak, M.P.; Galvin, C.J.; Aspuru-Guzik, A.; Gordon, R.G.; Aziz, M.J.; Gerhardt, M.R.; et al. Anthraquinone Derivatives in Aqueous Flow Batteries. Adv. Energy Mater. 2016, 1601488. [Google Scholar] [CrossRef]
- Lin, K.; Chen, Q.; Gerhardt, M.R.; Tong, L.; Kim, S.B.; Eisenach, L.; Valle, A.W.; Hardee, D.; Gordon, R.G.; Aziz, M.J.; et al. Alkaline quinone flow battery. Science 2015, 349, 1529–1532. [Google Scholar] [CrossRef] [Green Version]
- Kwabi, D.G.; Lin, K.; Ji, Y.; Aspuru-Guzik, A.; Gordon, R.G.; Aziz, M.J.; Kerr, E.F.; Goulet, M.-A.; De Porcellinis, D.; Tabor, D.P.; et al. Alkaline Quinone Flow Battery with Long Lifetime at pH 12. Joule 2018, 2, 1894–1906. [Google Scholar] [CrossRef]
- Er, S.; Suh, C.; Marshak, M.P.; Aspuru-Guzik, A. Computational design of molecules for an all-quinone redox flow battery. Chem. Sci. 2015, 6, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Eisenach, L.A.; Valle, A.W.; Gerhardt, M.R.; Wong, A.A.; Beh, E.S.; Aziz, M.J. Molecular Transport through Ion-Selective Membranes in Quinone-Based Redox Flow Batterie. ECS Meet. Abstr. 2016, MA2016-01, 385. [Google Scholar] [CrossRef]
- Gerhardt, M.R.; Beh, E.S.; Tong, L.; Gordon, R.G.; Aziz, M.J. Comparison of Capacity Retention Rates during Cycling of Quinone-Bromide Flow Batteries. MRS Adv. 2017, 2, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Murali, A.; Nirmalchandar, A.; Jayathilake, B.; Prakash, G.K.S.; Narayanan, S.R. A Durable, Inexpensive and Scalable Redox Flow Battery Based on Iron Sulfate and Anthraquinone Disulfonic Acid. J. Electrochem. Soc. 2020, 167, 060520. [Google Scholar] [CrossRef] [Green Version]
- Charvát, J.; Mazúr, P.; Pocedič, J.; Richtr, P.; Mrlík, J.; Kosek, J.; Akrman, J.; Kubáč, L. New organic-air flow fuel cell and electrolyser for stationary energy storage. J. Power Sources 2022, 520, 230811. [Google Scholar] [CrossRef]
- Khataee, A.; Wedege, K.; Dražević, E.; Bentien, A. Differential pH as a method for increasing cell potential in organic aqueous flow batteries. J. Mater. Chem. A 2017, 5, 21875–21882. [Google Scholar] [CrossRef]
- Lee, W.; Permatasari, A.; Kwon, Y. Neutral pH aqueous redox flow batteries using an anthraquinone-ferrocyanide redox couple. J. Mater. Chem. C 2020, 8, 5727–5731. [Google Scholar] [CrossRef]
- Hu, B.; Luo, J.; Hu, M.; Yuan, B.; Liu, T.L. A pH-Neutral, Metal-Free Aqueous Organic Redox Flow Battery Employing an Ammonium Anthraquinone Anolyte. Angew. Chem. 2019, 131, 16782–16789. [Google Scholar] [CrossRef]
- Jin, S.; Jing, Y.; Kwabi, D.G.; Ji, Y.; Tong, L.; De Porcellinis, D.; Goulet, M.A.; Pollack, D.A.; Gordon, R.G.; Aziz, M.J. A Water-Miscible Quinone Flow Battery with High Volumetric Capacity and Energy Density. ACS Energy Lett. 2019, 4, 1342–1348. [Google Scholar] [CrossRef]
- Schwarz, W.M.; Kosower, E.M.; Shain, I. Formation of stable free radicals on electroreduction of N-alkylpyridinium salts. J. Am. Chem. Soc. 1961, 83, 3164–3165. [Google Scholar] [CrossRef]
- Rusling, J.F. Electrodimerization of the pyridiniumcarboxylic acids homarine and trigonelline. J. Electroanal. Chem. 1981, 125, 447–458. [Google Scholar] [CrossRef]
- Sevov, C.S.; Hendriks, K.H.; Sanford, M.S. Low-potential pyridinium anolyte for aqueous redox flow batteries. J. Phys. Chem. C 2017, 121. [Google Scholar] [CrossRef]
- Janoschka, T.; Martin, N.; Hager, M.D.; Schubert, U.S. Wasserbasierte Redox-Flow-Batterie mit hoher Kapazität und Leistung: Das TEMPTMA/MV-System. Angew. Chem. 2016, 128, 14639–14643. [Google Scholar] [CrossRef]
- Liu, T.; Wei, X.; Nie, Z.; Sprenkle, V.; Wang, W. A Total Organic Aqueous Redox Flow Battery Employing a Low Cost and Sustainable Methyl Viologen Anolyte and 4-HO-TEMPO Catholyte. Adv. Energy Mater. 2016, 6, 1501449. [Google Scholar] [CrossRef]
- Hu, B.; Liu, T.L. Two electron utilization of methyl viologen anolyte in nonaqueous organic redox flow battery. J. Energy Chem. 2018, 27, 1326–1332. [Google Scholar] [CrossRef] [Green Version]
- Debruler, C.; Hu, B.; Moss, J.; Luo, J.; Liu, T.L. A Sulfonate-Functionalized Viologen Enabling Neutral Cation Exchange, Aqueous Organic Redox Flow Batteries toward Renewable Energy Storage. ACS Energy Lett. 2018, 3, 663–668. [Google Scholar] [CrossRef]
- Liu, Q.; Li, X.; Yan, C.; Tang, A. A dopamine-based high redox potential catholyte for aqueous organic redox flow battery. J. Power Sources 2020, 460, 228124. [Google Scholar] [CrossRef]
- Jin, C.; Shijian, E.M.; Fell, L.; Vina-Lopez, Y.; Jing, P.W.; Michalak, R.G.; Gordon, M.J.A. Near Neutral pH Redox Flow Battery with Low Permeability and Long Lifetime Phosphonated Viologen Active Species. Adv. Energy Mater. 2020, 10, 2000100. [Google Scholar] [CrossRef]
- Ohira, A.; Funaki, T.; Ishida, E.; Kim, J.D.; Sato, Y. Redox-Flow Battery Operating in Neutral and Acidic Environments with Multielectron-Transfer-Type Viologen Molecular Assembly. ACS Appl. Energy Mater. 2020, 3, 4377–4383. [Google Scholar] [CrossRef]
- Inzelt, G.; Puskás, Z. Adsorption and precipitation during the redox transformations of phenazine. Electrochim. Acta 2004, 49, 1969–1980. [Google Scholar] [CrossRef]
- Klatt, L.N.; Rouseff, R.L. Electrochemical Reduction of Pyrazine in Aqueous Media. J. Am. Chem. Soc. 1972, 94, 7295–7304. [Google Scholar] [CrossRef]
- Butkus, A.M.; Fink, W.H. Ab initio model calculations for pyridine, pyrazine, and their protonated forms. J. Chem. Phys. 1979, 71, 5329–5335. [Google Scholar] [CrossRef]
- Pasadakis-Kavounis, A.; Baj, V.; Hjelm, J. Electrochemical Characterization of Aromatic Molecules with 1,4-Diaza Groups for Flow Battery Applications. Molecules 2021, 26, 2227. [Google Scholar] [CrossRef]
- Milshtein, J.D.; Su, L.; Liou, C.; Badel, A.F.; Brushett, F.R. Voltammetry study of quinoxaline in aqueous electrolytes. Electrochim. Acta 2015, 180, 695–704. [Google Scholar] [CrossRef]
- Hong, J.; Lee, M.; Lee, B.; Seo, D.H.; Park, C.B.; Kang, K. Biologically inspired pteridine redox centres for rechargeable batteries. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollas, A.; Wei, X.; Murugesan, V.; Nie, Z.; Li, B.; Reed, D.; Liu, J.; Sprenkle, V.; Wang, W. A biomimetic high-capacity phenazine-based anolyte for aqueous organic redox flow batteries. Nat. Energy 2018, 3, 508–514. [Google Scholar] [CrossRef]
- Wang, C.; Li, X.; Yu, B.; Wang, Y.; Yang, Z.; Wang, H.; Lin, H.; Ma, J.; Li, G.; Jin, Z. Molecular Design of Fused-Ring Phenazine Derivatives for Long-Cycling Alkaline Redox Flow Batteries. ACS Energy Lett. 2020, 6, 411–417. [Google Scholar] [CrossRef]
- Xu, J.; Pang, S.; Wang, X.; Wang, P.; Ji, Y. Ultrastable aqueous phenazine flow batteries with high capacity operated at elevated temperatures. Joule 2021, 5, 2437–2449. [Google Scholar] [CrossRef]
- Kakkar, R.; Bhandari, M. Theoretical investigation of the alloxan-dialuric acid redox cycle. Int. J. Quantum Chem. 2013, 113, 2060–2069. [Google Scholar] [CrossRef]
- Mohran, H.S. An electrochemical investigation of the redox properties of murexide in aqueous and non-aqueous media. Am. J. Appl. Sci. 2009, 6, 964–969. [Google Scholar] [CrossRef]
- Kosswattaarachchi, A.M.; Cook, T.R. Repurposing the Industrial Dye Methylene Blue as an Active Component for Redox Flow Batteries. ChemElectroChem 2018, 5, 3437–3442. [Google Scholar] [CrossRef]
- Lee, W.; Kwon, Y. Performance evaluation of aqueous organic redox flow battery using methylene blue and vanadium redox couple. Korean Chem. Eng. Res. 2018, 56, 890–894. [Google Scholar] [CrossRef]
- Nawar, S.; Huskinson, B.; Aziz, M. Benzoquinone-hydroquinone couple for flow battery. MRS Proc. 2013, 1491, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.; Han, Y.K. Computational screening of organic molecules as redox active species in redox flow batteries. Curr. Appl. Phys. 2016, 16, 939–943. [Google Scholar] [CrossRef]
- Yang, B.; Hoober-Burkhardt, L.; Krishnamoorthy, S.; Murali, A.; Prakash, G.K.S.; Narayanan, S.R. High-Performance Aqueous Organic Flow Battery with Quinone-Based Redox Couples at Both Electrodes. J. Electrochem. Soc. 2016, 163, A1442–A1449. [Google Scholar] [CrossRef]
- Yang, B.; Hoober-Burkhardt, L.; Wang, F.; Surya Prakash, G.K.; Narayanan, S.R. An Inexpensive Aqueous Flow Battery for Large-Scale Electrical Energy Storage Based on Water-Soluble Organic Redox Couples. J. Electrochem. Soc. 2014, 161, A1371–A1380. [Google Scholar] [CrossRef] [Green Version]
- Hoober-Burkhardt, L.; Krishnamoorthy, S.; Yang, B.; Murali, A.; Nirmalchandar, A.; Prakash, G.K.S.; Narayanan, S.R. A New Michael-Reaction-Resistant Benzoquinone for Aqueous Organic Redox Flow Batteries. J. Electrochem. Soc. 2017, 164, A600–A607. [Google Scholar] [CrossRef]
- Rubio-Garcia, J.; Kucernak, A.; Parra-Puerto, A.; Liu, R.; Chakrabarti, B. Hydrogen/functionalized benzoquinone for a high-performance regenerative fuel cell as a potential large-scale energy storage platform. J. Mater. Chem. A 2020, 8, 3933–3941. [Google Scholar] [CrossRef]
- Ojani, R.; Raoof, J.-B.; Zamani, S. Electrochemical Behavior of Chloranil Chemically Modified Carbon Paste Electrode. Application to the Electrocatalytic Determination of Ascorbic Acid. Electroanalysis 2005, 17, 1740–1745. [Google Scholar] [CrossRef]
- Gerken, J.B.; Anson, C.W.; Preger, Y.; Symons, P.G.; Genders, J.D.; Qiu, Y.; Li, W.; Root, T.W.; Stahl, S.S. Comparison of Quinone-Based Catholytes for Aqueous Redox Flow Batteries and Demonstration of Long-Term Stability with Tetrasubstituted Quinones. Adv. Energy Mater. 2020, 10, 2000340. [Google Scholar] [CrossRef]
- Drazevic, E.; Szabo, C.; Konya, D.; Lund, T.; Wedege, K.; Bentien, A. Investigation of Tetramorpholinohydroquinone as a Potential Catholyte in a Flow Battery. ACS Appl. Energy Mater. 2019, 2, 4745–4754. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Garcia, S.N.; Janoschka, T.; Kónya, D.; Hager, M.D.; Schubert, U.S. Novel, Stable Catholyte for Aqueous Organic Redox Flow Batteries: Symmetric Cell Study of Hydroquinones with High Accessible Capacity. Molecules 2021, 26, 3823. [Google Scholar] [CrossRef]
- Orita, A.; Verde, M.G.; Sakai, M.; Meng, Y.S. The impact of pH on side reactions for aqueous redox flow batteries based on nitroxyl radical compounds. J. Power Sources 2016, 321, 126–134. [Google Scholar] [CrossRef]
- Chang, Z.; Henkensmeier, D.; Chen, R. One-Step Cationic Grafting of 4-Hydroxy-TEMPO and its Application in a Hybrid Redox Flow Battery with a Crosslinked PBI Membrane. ChemSusChem 2017, 10, 3193–3197. [Google Scholar] [CrossRef]
- Liu, Y.; Goulet, M.A.; Tong, L.; Liu, Y.; Ji, Y.; Wu, L.; Gordon, R.G.; Aziz, M.J.; Yang, Z.; Xu, T. A Long-Lifetime All-Organic Aqueous Flow Battery Utilizing TMAP-TEMPO Radical. Chem 2019, 5, 1861–1870. [Google Scholar] [CrossRef]
- Chang, Z.; Henkensmeier, D.; Chen, R. Shifting redox potential of nitroxyl radical by introducing an imidazolium substituent and its use in aqueous flow batteries. J. Power Sources 2019, 418, 11–16. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, W.; Qin, M.; Chen, Z.; Xu, J.; Cao, J.; Li, J. Fundamental properties of TEMPO-based catholytes for aqueous redox flow batteries: Effects of substituent groups and electrolytes on electrochemical properties, solubilities and battery performance. RSC Adv. 2020, 10, 21839–21844. [Google Scholar] [CrossRef]
- Winsberg, J.; Stolze, C.; Schwenke, A.; Muench, S.; Hager, M.D.; Schubert, U.S. Aqueous 2,2,6,6-Tetramethylpiperidine-N-oxyl Catholytes for a High-Capacity and High Current Density Oxygen-Insensitive Hybrid-Flow Battery. ACS Energy Lett. 2017, 2, 411–416. [Google Scholar] [CrossRef]
- Koshino, N.; Saha, B.; Espenson, J.H. Kinetic Study of the Phthalimide N-Oxyl Radical in Acetic Acid. Hydrogen Abstraction from Substituted Toluenes, Benzaldehydes, and Benzyl Alcohols. J. Org. Chem. 2003, 68, 9364–9370. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wu, K.H.; Cao, L.; Saputera, W.H.; Amal, R.; Wang, D.W. Unlocking high-potential non-persistent radical chemistry for semi-aqueous redox batteries. Chem. Commun. 2019, 55, 2154–2157. [Google Scholar] [CrossRef]
- Fischer, P. Photoreduction of aqueous ferricyanide solutions—Raman measurements. unpublished data. 2005. [Google Scholar]
- Kim, H.S.; Yoon, T.; Jang, J.; Mun, J.; Park, H.; Ryu, J.H.; Oh, S.M. A tetradentate Ni(II) complex cation as a single redox couple for non-aqueous flow batteries. J. Power Sources 2015, 283, 300–304. [Google Scholar] [CrossRef]
- Kim, H.S.; Hwang, S.; Mun, J.; Park, H.; Ryu, J.H.; Oh, S.M. Counter anion effects on the energy density of Ni(II)-chelated tetradentate azamacrocyclic complex cation as single redox couple for non-aqueous flow batteries. Electrochim. Acta 2019, 308, 227–230. [Google Scholar] [CrossRef]
- Froehlich, J.D.; Kubiak, C.P. Homogeneous CO2 Reduction by Ni(cyclam) at a Glassy Carbon Electrode. Inorg. Chem. 2012, 51, 3932–3934. [Google Scholar] [CrossRef] [PubMed]
- Goeltz, J.; Millard, M. Processes for Forming Titanium Catechol Complex, 2017. EP3275887A1, 31 January 2018. [Google Scholar]
- Bae, C.; Roberts, E.P.L.; Chakrabarti, M.H.; Saleem, M. All-Chromium Redox Flow Battery for Renewable Energy Storage. Int. J. Green Energy 2011, 8, 248–264. [Google Scholar] [CrossRef]
- Bae, C.H.; Roberts, E.P.L.; Dryfe, R.A.W. Chromium redox couples for application to redox flow batteries. Electrochim. Acta 2002, 48, 279–287. [Google Scholar] [CrossRef]
- Freeman, M.B.; Wang, L.; Jones, D.S.; Bejger, C.M. A cobalt sulfide cluster-based catholyte for aqueous flow battery applications. J. Mater. Chem. A 2018, 6, 21927–21932. [Google Scholar] [CrossRef]
- Wen, Y.H.; Zhang, H.M.; Qian, P.; Zhou, H.T.; Zhao, P.; Yi, B.L.; Yang, Y.S. A study of the Fe(III)/Fe(II)-triethanolamine complex redox couple for redox flow battery application. Electrochim. Acta 2006, 51, 3769–3775. [Google Scholar] [CrossRef]
- Hu, B.; Debruler, C.; Rhodes, Z.; Liu, T.L. Long-Cycling aqueous organic Redox flow battery (AORFB) toward sustainable and safe energy storage. J. Am. Chem. Soc. 2017, 139, 1207–1214. [Google Scholar] [CrossRef]
- Chen, Q.; Li, Y.; Liu, Y.; Sun, P.; Yang, Z.; Xu, T. Designer Ferrocene Catholyte for Aqueous Organic Flow Batteries. ChemSusChem 2020, 14, 1295–1301. [Google Scholar] [CrossRef]
- Yu, J.; Salla, M.; Zhang, H.; Ji, Y.; Zhang, F.; Zhou, M.; Wang, Q. A robust anionic sulfonated ferrocene derivative for pH-neutral aqueous flow battery. Energy Storage Mater. 2020, 29, 216–222. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, B.; Schrage, B.R.; Ziegler, C.J.; Boika, A. Investigations into aqueous redox flow batteries based on ferrocene bisulfonate. ACS Appl. Energy Mater. 2020, 3, 10270–10277. [Google Scholar] [CrossRef]
- Páez, T.; Martínez-Cuezva, A.; Palma, J.; Ventosa, E. Revisiting the cycling stability of ferrocyanide in alkaline media for redox flow batteries. J. Power Sources 2020, 471, 228453. [Google Scholar] [CrossRef]
- Páez, T.; Martínez-Cuezva, A.; Palma, J.; Ventosa, E. Mediated alkaline flow batteries: From fundamentals to application. ACS Appl. Energy Mater. 2019, 2, 8328–8336. [Google Scholar] [CrossRef]
- Hawthorne, K.L.; Wainright, J.S.; Savinell, R.F. Studies of Iron-Ligand Complexes for an All-Iron Flow Battery Application. J. Electrochem. Soc. 2014, 161, A1662–A1671. [Google Scholar] [CrossRef]
- Tsitovich, P.B.; Kosswattaarachchi, A.M.; Crawley, M.R.; Tittiris, T.Y.; Cook, T.R.; Morrow, J.R. An Fe III Azamacrocyclic Complex as a pH-Tunable Catholyte and Anolyte for Redox-Flow Battery Applications. Chem. A Eur. J. 2017, 23, 15327–15331. [Google Scholar] [CrossRef]
- Gong, K.; Xu, F.; Grunewald, J.B.; Ma, X.; Zhao, Y.; Gu, S.; Yan, Y. All-Soluble All-Iron Aqueous Redox-Flow Battery. ACS Energy Lett. 2016, 1, 89–93. [Google Scholar] [CrossRef]
- Shin, M.; Noh, C.; Chung, Y.; Kwon, Y. All iron aqueous redox flow batteries using organometallic complexes consisting of iron and 3-[bis (2-hydroxyethyl)amino]-2-hydroxypropanesulfonic acid ligand and ferrocyanide as redox couple. Chem. Eng. J. 2020, 398, 125631. [Google Scholar] [CrossRef]
- Modrzynski, C.; Burger, P. Energy storage inspired by nature—Ionic liquid iron–sulfur clusters as electrolytes for redox flow batteries. Dalt. Trans. 2019, 48, 1941–1946. [Google Scholar] [CrossRef] [Green Version]
- Schröder, H.V.; Schalley, C.A. Tetrathiafulvalene—A redox-switchable building block to control motion in mechanically interlocked molecules. Beilstein J. Org. Chem. 2018, 14, 2163–2185. [Google Scholar] [CrossRef] [PubMed]
- Bendikov, M.; Wudl, F.; Perepichka, D.F. Tetrathiafulvalenes, oligoacenenes, and their buckminsterfullerene derivatives: The brick and mortar of organic electronics. Chem. Rev. 2004, 104, 4891–4945. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q. Synthesis of New Tetrazines Functionalized with Photoactive and Electroactive Groups. Ph.D. Thesis, ENS Cachan, Paris, France, 2012. [Google Scholar]
- Stone, E.W.; Maki, A.H. ESR study of polyazine anions. J. Chem. Phys. 1963, 39, 1635–1642. [Google Scholar] [CrossRef]
- Fritea, L.; Audebert, P.; Galmiche, L.; Gorgy, K.; Le Goff, A.; Villalonga, R.; Săndulescu, R.; Cosnier, S. First Occurrence of Tetrazines in Aqueous Solution: Electrochemistry and Fluorescence. ChemPhysChem 2015, 16, 3695–3699. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.D.; Schmalisch, S.; Schwan, S.; Hong, L.; Wegner, H.A.; Mollenhauer, D.; Janek, J.; Schröder, D. Tailoring Dihydroxyphthalazines to Enable Their Stable and Efficient Use in the Catholyte of Aqueous Redox Flow Batteries. Chem. Mater. 2020, 32, 3427–3438. [Google Scholar] [CrossRef]
- Honarmand, E.; Mostaanzadeh, H.; Aalaiy, M. Computational assisted electrochemical studies for 1,4-diazabicyclo[2,2,2]octane determination at multiwalled carbon nanotube paste electrode. Russ. J. Electrochem. 2017, 53, 479–485. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical, Chemical and Biological Properties of Electrolytes | |||||
|---|---|---|---|---|---|
| 1 | Solubility | a (b, f) | 6 | Dynamic viscosity | e (b) |
| 2 | Redox potential | a (b, f) | 7 | Permeation | a (c) |
| 3 | Redox kinetic | b (e) | 8 | Effect of temperature | c, e |
| 4 | Chemical/electrochemical stability | c | 9 | Aquatic and human toxicity | d |
| 5 | Ionic conductivity | b (e) | 10 | Abundance of materials | f |
| ε at 20 °C | η at 20 °C [cP] | DN [kcal/mol] | AN [kcal/mol] | π* | ρ at 20 °C [g/cm3] | |
|---|---|---|---|---|---|---|
| Formamide (FA) | 109 | 3.75 | 24 | 39.8 | 97 | 1.13 |
| Water | 80.1 | 1.002 | 18 | 54.8 | 109 | 0.998 |
| Propylenecarbonate (PC) | 64.9 | 2.53 | 15.1 | 18.3 | 83 | 1.21 |
| Dimethylsulfoxide (DMSO) | 46.5 | 1.99 | 29.8 | 19.3 | 100 | 1.10 |
| g-Butyrolactone (GBL) | 39.1 | 1.73 | 18 | 17.3 | 87 | 1.13 |
| Dimethylformamide (DMF) | 36.7 | 0.80 | 26.6 | 16.0 | 88 | 0.94 |
| N, N′-Dimethylpropyleneurea (DMPU) | 36.1 | 2.93 | 15 | 42.0 | - | 1.06 |
| Acetonitrile (AN) | 35.9 | 0.34 | 14.1 | 18.9 | 75 | 0.78 |
| Hexamethylphosphoramide (HMPA) | 30.0 | 3.53 | 38.8 | 10.6 | - | 1.02 |
| 1,3-Dioxalane (DO) | 7.3 | 0.60 | - | - | 69 | 1.06 |
| Dimethoxyethane (DMO) | 7.2 | 0.46 | 23.9 | 10.2 | 53 | 1.38 |
| Methylacetate (MA) | 6.7 | 0.36 | 16.5 | 10.7 | 60 | 0.94 |
| Ethylacetate (EA) | 6.0 | 0.42 | 17.1 | 9.3 | 55 | 0.90 |
| Critical Raw Materials | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 01 | Antimony | 07 | Cobalt | 13 | HREEs | 19 | Natural rubber | 25 | Silicon metal |
| 02 | Baryte | 08 | Coking coal | 14 | Indium | 20 | Niobium | 26 | Tantalum |
| 03 | Bauxite | 09 | Fluorspar | 15 | Lithium | 21 | PMGs | 27 | Titanium |
| 04 | Beryllium | 10 | Gallium | 16 | LREEs | 22 | Phosphate rock | 28 | Vanadium |
| 05 | Bismuth | 11 | Germanium | 17 | Magnesium | 23 | Phosphorus | 29 | Tungsten |
| 06 | Borates | 12 | Hafnium | 18 | Naturalgraphite | 24 | Scandium | 30 | Strontium |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, P.; Mazúr, P.; Krakowiak, J. Family Tree for Aqueous Organic Redox Couples for Redox Flow Battery Electrolytes: A Conceptual Review. Molecules 2022, 27, 560. https://doi.org/10.3390/molecules27020560
Fischer P, Mazúr P, Krakowiak J. Family Tree for Aqueous Organic Redox Couples for Redox Flow Battery Electrolytes: A Conceptual Review. Molecules. 2022; 27(2):560. https://doi.org/10.3390/molecules27020560
Chicago/Turabian StyleFischer, Peter, Petr Mazúr, and Joanna Krakowiak. 2022. "Family Tree for Aqueous Organic Redox Couples for Redox Flow Battery Electrolytes: A Conceptual Review" Molecules 27, no. 2: 560. https://doi.org/10.3390/molecules27020560
APA StyleFischer, P., Mazúr, P., & Krakowiak, J. (2022). Family Tree for Aqueous Organic Redox Couples for Redox Flow Battery Electrolytes: A Conceptual Review. Molecules, 27(2), 560. https://doi.org/10.3390/molecules27020560