All chemical reagents were purchased and utilized without further purification. Solvents were used directly or treated with standard methods before use. Melting points (m.p.) were determined on an X-6a digital melting point apparatus (Gongyi Tech Instrument Co., Ltd., Gongyi, China) and were uncorrected. Infrared spectra (IR) were recorded on a Bruker TENSOR 27 spectrometer. Proton nuclear magnetic resonance spectra (1H NMR) and carbon nuclear magnetic resonance spectra (13C NMR) were recorded in CDCl3 on a Bruker Avance 400, 500, or 600 MHz instruments using tetramethylsilane (TMS) as the internal standard. High-resolution mass spectra (HRMS) were carried out with IonSpec 4.7 Tesla FTMS instrument. The purities of all the title compounds were determined on an UltiMate 3000 (Dionex, Sunnyvale, CA, USA) HPLC system and were of > 95% purity.
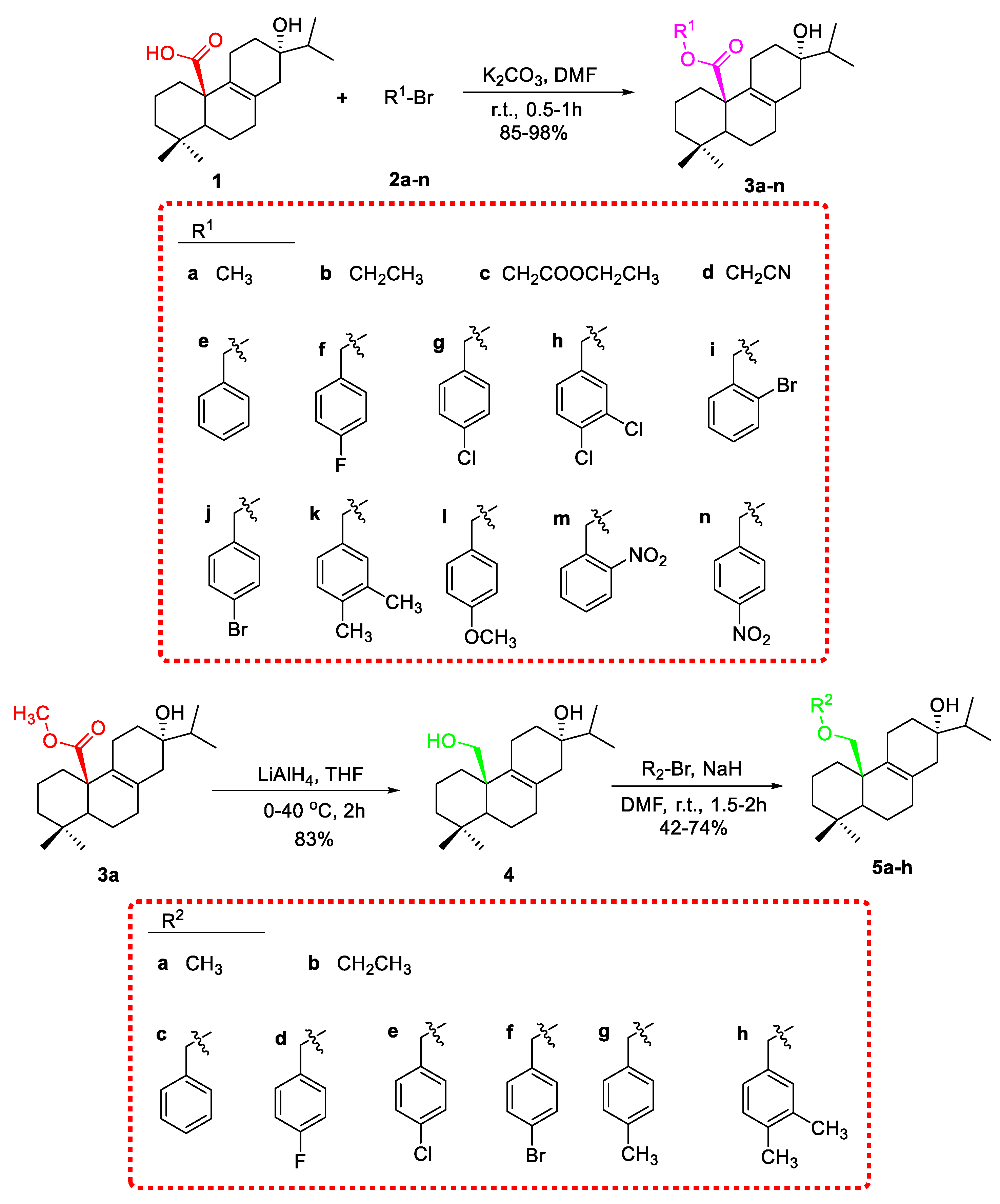
3.1.1. General Procedure for the Synthesis of Compound 3a–n
Lophanic acid (1, 100 mg, 0.31 mmol) and potassium carbonate (86 mg, 0.62 mmol) were dissolved in N, N-Dimethylformamide (DMF, 5 mL), and the solution was stirred at room temperature. Then a solution of substituent haloalkane (0.62 mmol) in DMF (2 mL) was added dropwise for 10 min. When the reaction was complete, checked by thin-layer chromatography (TLC) analysis, pure water (30 mL) was added to the reaction, which was extracted with ethyl acetate (3 × 30 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography eluting with petroleum ether/ethyl acetate to afford compound 3a-n in 85–98%.
Data for
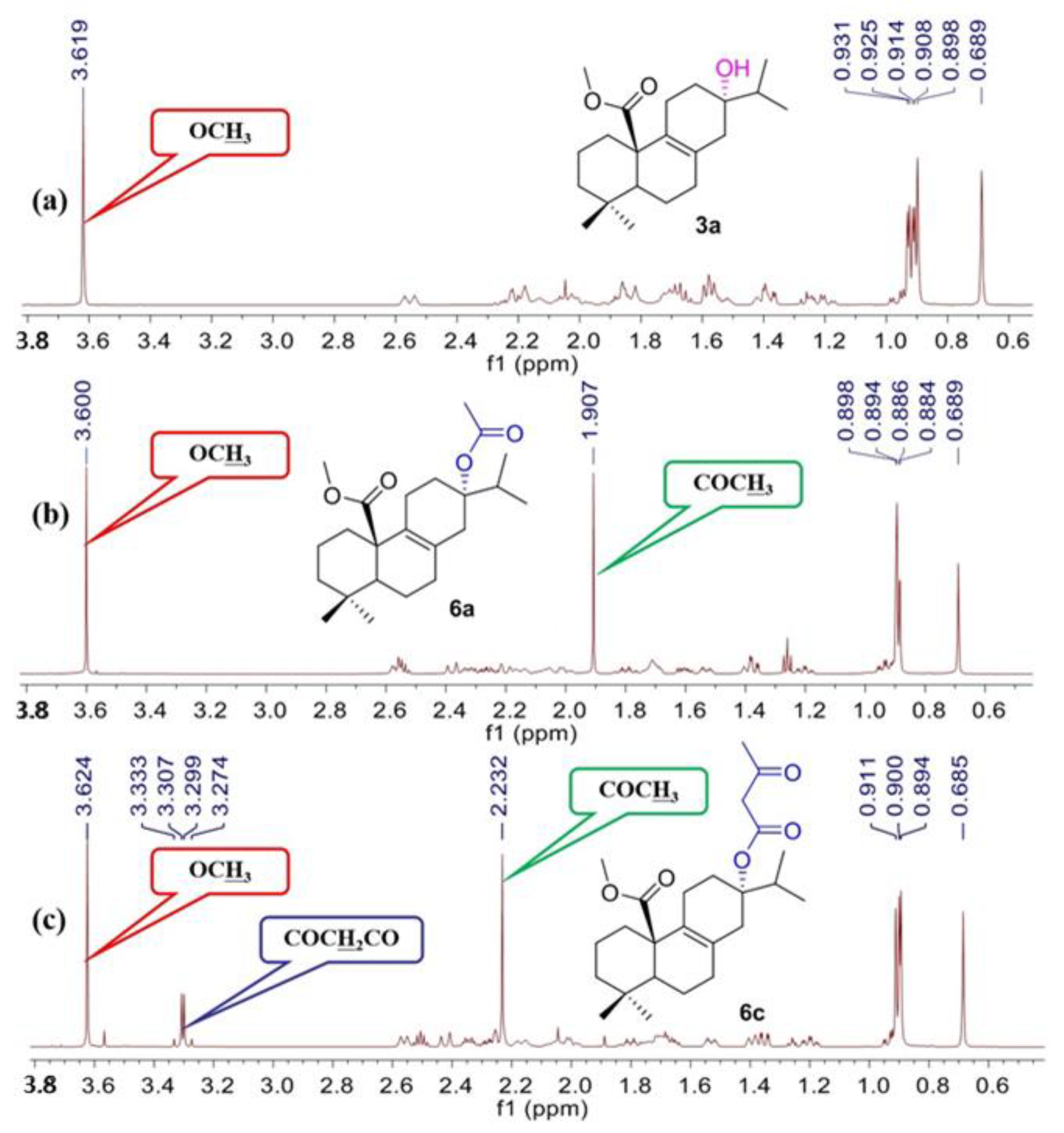
3a: White solid, yield: 98%, m.p. 99–101 °C; IR cm
−1: 3527, 1709, 1213, 1141;
1H NMR (400 MHz, CDCl
3):
δ 3.61 (s, 3H, OC
H3), 2.57 (d,
J = 12.6 Hz, 1H), 2.27–2.13 (m, 3H), 2.07–1.98 (m, 2H), 1.89–1.81 (m, 3H), 1.72–1.65 (m, 3H), 1.59–1.51 (m, 3H), 1.42–1.36 (m, 2H), 1.26–1.20 (m, 1H), 0.98–0.94 (m, 1H), 0.92–0.91 (m, 6H, H-16, 17), 0.89 (s, 3H, H-19), 0.68 (s, 3H, H-18);
13C NMR (100 MHz, CDCl
3):
δ 176.2 (C-20), 130.1 (C-9), 129.6 (C-8), 72.0 (C-13), 52.4 (O
CH
3), 51.4 (C-5), 48.7 (C-10), 41.8 (C-3), 41.2 (C-14), 34.5 (C-1), 34.4 (C-4), 33.7 (C-15), 32.2 (C-12), 32.0 (C-7), 31.6 (C-18), 21.5 (C-11), 20.1 (C-2), 19.8 (C-19), 18.1 (C-6), 16.7 (C-16), 16.7 (C-17); HRMS
m/z calcd for C
21H
34O
3Na ([M + Na]
+) 357.2400, found 357.2401 (
Figures S1–S3).
Data for
3b: White solid, yield: 95%, m.p. 52–54 °C; IR cm
−1: 3540, 1709, 1214, 1145;
1H NMR (600 MHz, CDCl
3):
δ 4.13–4.03 (m, 2H, C
H2CH
3), 2.56 (d,
J = 12.0 Hz, 1H), 2.29–2.05 (m, 4H), 2.02–1.97 (m, 1H), 1.92–1.81 (m, 3H), 1.71–1.64 (m, 3H), 1.58–1.51 (m, 3H), 1.41–1.36 (m, 2H), 1.25–1.18 (m, 4H), 0.96–0.93 (m, 1H), 0.92–0.89 (m, 6H, H-16, 17), 0.90 (s, 3H, H-19), 0.72 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.6 (C-20), 130.3 (C-9), 129.5 (C-8), 72.0 (C-13), 60.2 (
CH
2CH
3), 52.4 (C-5), 48.7 (C-10), 41.9 (C-3), 41.2 (C-14), 34.5 (C-1), 34.5 (C-4), 33.8 (C-15), 32.2 (C-12), 32.0 (C-7), 31.6 (C-18), 21.5 (C-11), 20.1 (C-2), 20.0 (C-19), 18.2 (C-6), 16.8 (C-16), 16.8 (C-17), 14.2 (CH
2CH
3); HRMS
m/z calcd for C
22H
36O
3Na ([M + Na]
+) 371.2557, found 371.2558 (
Figures S4–S6).
Data for
3c: colorless oil, yield: 89%; IR cm
−1: 3660, 1735, 1726, 1381, 1130;
1H NMR (600 MHz, CDCl
3):
δ 4.74 (d,
J = 15.6 Hz, 1H, C
H2COOCH
2CH
3), 4.32 (d,
J = 15.6 Hz, 1H, C
H2COOCH
2CH
3), 4.25–4.16 (m, 2H, CH
2COOC
H2CH
3), 2.63 (d,
J = 12.1 Hz, 1H), 2.26–2.12 (m, 4H), 2.06–2.00 (m, 2H), 1.85–1.78 (m, 2H), 1.76–1.64 (m, 3H), 1.56–1.51 (m, 3H), 1.47–1.39 (m, 2H), 1.27–1.20 (m, 4H), 1.04–0.99 (m, 1H), 0.96 (d,
J = 6.6 Hz, 3H, H-16), 0.93 (d,
J = 6.6 Hz, 3H, H-17), 0.90 (s, 3H, H-19), 0.71 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.1 (C-20), 168.2 (CH
2COOCH
2CH
3), 130.6 (C-9), 129.2 (C-8), 71.4 (C-13), 61.5 (
CH
2COOCH
2CH
3), 60.2 (CH
2COO
CH
2CH
3), 51.9 (C-5), 48.5 (C-10), 41.9 (C-3), 40.3 (C-14), 36.1 (C-1), 34.7 (C-4), 33.9 (C-15), 32.0 (C-12), 31.9 (C-7), 31.4 (C-18), 20.5 (C-11), 20.0 (C-2), 19.9 (C-19), 18.2 (C-6), 17.1 (C-16), 16.9 (C-17), 14.1 (CH
2COOCH
2CH
3); HRMS
m/z calcd for C
24H
38O
5Na ([M + Na]
+) 429.2611, found 429.2613 (
Figures S7–S9).
Data for
3d: colorless oil, yield: 92%; IR cm
−1: 1732, 1457, 1115;
1H NMR (600 MHz, CDCl
3):
δ 4.68 (s, 2H, C
H2CN), 2.59 (d,
J = 13.8 Hz, 1H), 2.21–2.13 (m, 3H), 2.09–2.01 (m, 2H), 1.87–1.74 (m, 4H), 1.67–1.56 (m, 5H), 1.45–1.42 (m, 2H), 1.25–1.19 (m, 1H), 1.03–0.98 (m, 1H), 0.89–0.91 (m, 9H, H-16, 17, 19), 0.71 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 174.3 (C-20), 131.3 (C-9), 128.9 (C-8), 114.5 (CH
2CN), 71.9 (C-13), 52.4 (C-5), 49.1 (
CH
2CN), 48.2 (C-10), 41.7 (C-3), 41.3 (C-14), 34.6 (C-1), 34.5 (C-4), 33.9 (C-15), 31.9 (C-12), 31.9 (C-7), 29.8 (C-18), 21.6 (C-11), 20.2 (C-2), 20.1 (C-19), 18.2 (C-6), 16.8 (C-16), 16.8 (C-17); HRMS
m/z calcd for C
22H
33O
3NNa ([M + Na]
+) 382.2352, found 382.2352 (
Figures S10–S12).
Data for
3e: colorless oil, yield: 97%; IR cm
−1: 1716, 1456, 1205, 1129;
1H NMR (600 MHz, CDCl
3):
δ 7.26–7.35 (m, 5H, H-Ph), 5.16 (d,
J = 12.6 Hz, 1H, PhC
H2), 5.00 (d,
J = 12.6 Hz, 1H, PhC
H2), 2.58 (d,
J = 13.2 Hz, 1H), 2.30–2.17 (m, 2H), 2.13–1.97 (m, 3H), 1.90–1.78 (m, 3H), 1.70–1.63 (m, 2H), 1.59–1.45 (m, 3H), 1.40–1.37 (m, 3H), 1.22–1.17 (m, 1H), 0.97–0.92 (m, 1H), 0.90 (s, 3H, H-16), 0.89 (s, 3H, H-17), 0.88 (s, 3H, H-19), 0.67 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.5 (C-20), 136.3 (C-Ph), 130.1 (C-9), 130.0 (C-Ph), 128.6 (C-8), 128.1 (C-Ph), 128.0 (C-Ph), 71.9 (C-13), 66.2 (Ph
CH
2), 52.5 (C-5), 48.9 (C-10), 42.0 (C-3), 41.5 (C-14), 34.6 (C-1), 34.0 (C-4), 33.9 (C-15), 32.1 (C-12), 32.0 (C-7), 32.0 (C-18), 21.8 (C-11), 20.2 (C-2), 20.2 (C-19), 18.3 (C-6), 16.8 (C-16), 16.8 (C-17); HRMS
m/z calcd for C
27H
38O
3Na ([M + Na]
+) 433.2713, found 433.2713 (
Figures S13–S15).
Data for
3f: colorless oil, yield: 85%; IR cm
−1: 1715, 1513, 1224;
1H NMR (600 MHz, CDCl
3):
δ 7.30–7.26 (m, 2H, H-Ph), 7.03–7.01 (m, 2H, H-Ph), 5.11 (d,
J = 12.0 Hz, 1H, PhC
H2), 4.97 (d,
J = 12.0 Hz, 1H, PhC
H2), 2.56 (d,
J = 13.8 Hz, 1H), 2.26–2.17 (m, 2H), 2.13–1.95 (m, 3H), 1.87–1.76 (m, 3H), 1.70–1.63 (m, 2H), 1.59–1.46 (m, 4H), 1.39–1.37 (m, 2H), 1.22–1.17 (m, 1H), 0.97–0.92 (m, 1H), 0.91 (s, 3H, H-16), 0.90 (s, 3H, H-17), 0.87 (s, 3H, H-19), 0.63 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.3 (C-20), 163.3 (C-Ph), 132.0 (C-Ph), 131.9 (C-9), 130.1 (C-Ph), 130.0 (C-8), 115.3 (C-Ph), 71.9 (C-13), 65.4 (Ph
CH
2), 52.3 (C-5), 48.8 (C-10), 41.8 (C-3), 41.3 (C-14), 34.4 (C-1), 34.0 (C-4), 33.8 (C-15), 31.9 (C-12), 31.9 (C-7), 31.8 (C-18), 21.6 (C-11), 20.1 (C-2), 20.0 (C-19), 18.1 (C-6), 16.7 (C-16), 16.7 (C-17); HRMS
m/z calcd for C
27H
37O
3FNa ([M + Na]
+) 451.2619, found 451.2617 (
Figures S16–S18).
Data for
3g: White solid, yield: 98%, m.p. 76–78 °C; IR cm
−1: 1720, 1456, 1201, 1146, 1128, 1089, 981, 806;
1H NMR (600 MHz, CDCl
3):
δ 7.31 (d,
J = 8.4 Hz, 2H, H-Ph), 7.25 (d,
J = 8.4 Hz, 2H, H-Ph), 5.11 (d,
J = 12.6 Hz, 1H, PhC
H2), 4.95 (d,
J = 12.6 Hz, 1H, PhC
H2), 2.56 (d,
J = 12.6 Hz, 1H), 2.26–2.17 (m, 2H), 2.13–1.96 (m, 3H), 1.85–1.76 (m, 3H), 1.70–1.46 (m, 6H), 1.40–1.37 (m, 2H), 1.22–1.17 (m, 1H), 0.97–0.92 (m, 1H), 0.91 (s, 3H, H-16), 0.90 (s, 3H, H-17), 0.88 (s, 3H, H-19), 0.64 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.4 (C-20), 134.8 (C-Ph), 133.9 (C-Ph), 130.1 (C-9), 129.9 (C-Ph), 129.5 (C-8), 128.8 (C-Ph), 71.9 (C-13), 65.4 (Ph
CH
2), 52.4 (C-5), 48.9 (C-10), 41.9 (C-3), 41.4 (C-14), 34.5 (C-1), 34.1 (C-4), 33.9 (C-15), 32.0 (C-12), 32.0 (C-7), 32.0 (C-18), 21.8 (C-11), 20.2 (C-2), 20.1 (C-19), 18.3 (C-6), 16.8 (C-16), 16.8 (C-17); HRMS
m/z calcd for C
27H
37O
3ClNa ([M + Na]
+) 467.2323, found 467.2326 (
Figures S19–S21).
Data for
3h: colorless oil, yield: 88%; IR cm
−1: 1716, 1472, 1205, 1128;
1H NMR (600 MHz, CDCl
3):
δ 7.41–7.40 (m, 2H, H-Ph), 7.14–7.13 (m, 1H, H-Ph), 5.14 (d,
J = 12.6 Hz, 1H, PhC
H2), 4.89 (d,
J = 12.6 Hz, 1H, PhC
H2), 2.57 (d,
J = 13.2 Hz, 1H), 2.22–2.16 (m, 2H), 2.14–1.98 (m, 3H), 1.88–1.77 (m, 3H), 1.73–1.48 (m, 6H), 1.42–1.39 (m, 2H), 1.23–1.18 (m, 1H), 0.99–0.94 (m, 1H), 0.92 (s, 3H, H-16), 0.90 (s, 3H, H-17), 0.89 (s, 3H, H-19), 0.66 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.4 (C-20), 136.7 (C-Ph), 132.7 (C-Ph), 132.1 (C-Ph), 130.6 (C-9), 130.2 (C-Ph), 129.9 (C-8), 129.8 (C-Ph), 127.2 (C-Ph), 72.0 (C-13), 64.6 (Ph
CH
2), 52.4 (C-5), 49.0 (C-10), 41.9 (C-3), 41.5 (C-14), 34.6 (C-1), 34.3 (C-4), 33.9 (C-15), 32.0 (C-12), 32.0 (C-7), 29.7 (C-18), 21.8 (C-11), 20.2 (C-2), 20.1 (C-19), 18.4 (C-6), 16.8 (C-16), 16.8 (C-17); HRMS
m/z calcd for C
27H
36O
3Cl
2Na ([M + Na]
+) 501.1934, found 501.1935 (
Figures S22–S24).
Data for
3i: White solid, yield: 96%, m.p. 65–67 °C; IR cm
−1: 1720, 1199, 1145, 1129, 1112, 746;
1H NMR (600 MHz, CDCl
3):
δ 7.55–7.52 (m, 1H, H-Ph), 7.37–7.28 (m, 2H, H-Ph), 7.17–7.15 (m, 1H, H-Ph), 5.28 (d,
J = 13.2 Hz, 1H, PhC
H2), 5.03 (d,
J = 13.2 Hz, 1H, PhC
H2), 2.59 (d,
J = 13.8 Hz, 1H), 2.30–2.17 (m, 2H), 2.11–1.98 (m, 3H), 1.88–1.76 (m, 3H), 1.71–1.57 (m, 4H), 1.47–1.38 (m, 4H), 1.23–1.18 (m, 1H), 0.97–0.92 (m, 1H), 0.90 (s, 3H, H-16), 0.89 (s, 3H, H-17), 0.88 (s, 3H, H-19), 0.67 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.3 (C-20), 135.6 (C-Ph), 132.8 (C-Ph), 132.6 (C-Ph), 130.2 (C-9), 129.8 (C-Ph), 129.7 (C-8), 127.7 (C-Ph), 123.7 (C-Ph), 71.9 (C-13), 65.7 (Ph
CH
2), 52.4 (C-5), 49.0 (C-10), 42.0 (C-3), 41.5 (C-14), 34.6 (C-1), 33.9 (C-4), 33.8 (C-15), 32.1 (C-12), 32.0 (C-7), 32.0 (C-18), 21.8 (C-11), 20.2 (C-2), 20.1 (C-19), 18.3 (C-6), 16.8 (C-16), 16.8 (C-17); HRMS
m/z calcd for C
27H
37O
3BrNa ([M + Na]
+): 511.1818, found 511.1822 (
Figures S25–S27).
Data for
3j: White solid, yield: 93%, m.p. 63–65 °C; IR cm
−1: 1719, 1455, 1200, 1146, 1127;
1H NMR (600 MHz, CDCl
3):
δ 7.47 (d,
J = 8.4 Hz, 2H, H-Ph), 7.19 (d,
J = 8.4 Hz, 2H, H-Ph), 5.10 (d,
J = 12.6 Hz, 1H, PhC
H2), 4.93 (d,
J = 12.6 Hz, 1H, PhC
H2), 2.57 (d,
J = 13.2 Hz, 1H), 2.25–2.17 (m, 2H), 2.13–1.96 (m, 3H), 1.87–1.77 (m, 3H), 1.71–1.47 (m, 6H), 1.40–1.37 (m, 2H), 1.22–1.17 (m, 1H), 0.98–0.92 (m, 1H), 0.91 (s, 3H, H-16), 0.90 (s, 3H, H-17), 0.88 (s, 3H, H-19), 0.65 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.4 (C-20), 135.3 (C-Ph), 131.7 (C-Ph), 130.1 (C-9), 130.0 (C-Ph), 129.9 (C-8), 122.1 (C-Ph), 72.0 (C-13), 65.5 (Ph
CH
2), 52.5 (C-5), 49.0 (C-10), 41.9 (C-3), 41.5 (C-14), 34.6 (C-1), 34.2 (C-4), 33.9 (C-15), 32.1 (C-12), 32.0 (C-7), 32.0 (C-18), 21.8 (C-11), 20.2 (C-2), 20.2 (C-19), 18.3 (C-6), 16.8 (C-16), 16.8 (C-17); HRMS
m/z calcd for C
27H
37O
3BrNa ([M + Na]
+) 511.1818, found 511.1822 (
Figures S28–S30).
Data for
3k: colorless oil, yield: 95%; IR cm
−1: 1714, 1455, 1202, 1126;
1H NMR (600 MHz, CDCl
3):
δ 7.09 (d,
J = 7.8 Hz, 1H, H-Ph), 7.06 (s, 1H, H-Ph), 7.03 (d,
J = 7.2 Hz, 1H, H-Ph), 5.09 (d,
J = 12.6 Hz, 1H, PhC
H2), 4.92 (d,
J = 12.0 Hz, 1H, PhC
H2), 2.57 (d,
J = 13.2 Hz, 1H), 2.30–2.17 (m, 8H), 2.13–1.96 (m, 3H), 1.91–1.78 (m, 3H), 1.69–1.63 (m, 2H), 1.59–1.48 (m, 3H), 1.39–1.38 (m, 3H), 1.22–1.17 (m, 1H), 0.96–0.91 (m, 1H), 0.90 (s, 3H, H-16), 0.89 (s, 3H, H-17), 0.88 (s, 3H, H-19), 0.68 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.5 (C-20), 136.7 (C-Ph), 136.4 (C-Ph), 133.7 (C-Ph), 130.2 (C-9), 129.8 (C-Ph), 129.7 (C-8), 129.4 (C-Ph), 125.5 (C-Ph), 71.9 (C-13), 66.2 (Ph
CH
2), 52.4 (C-5), 48.9 (C-10), 42.0(C-3), 41.5 (C-14), 34.5 (C-1), 34.0 (C-4), 33.9 (C-15), 32.1 (C-12), 32.1 (C-7), 32.0 (C-18), 21.8 (C-11), 20.2 (C-2), 20.2 (C-19), 19.8 (
CH
3Ph), 19.6 (
CH
3Ph), 18.3 (C-6), 16.8 (C-16), 16.8 (C-17); HRMS
m/z calcd for C
29H
42O
3Na([M + Na]
+) 461.3026, found 461.3025 (
Figures S31–S33).
Data for
3l: colorless oil, yield: 94%; IR cm
−1: 1713, 1515, 1249;
1H NMR (600 MHz, CDCl
3):
δ 7.25 (d,
J = 8.0 Hz, 2H, H-Ph), 6.86 (d,
J = 8.0 Hz, 2H, H-Ph), 5.06 (d,
J = 12.0 Hz, 1H, PhC
H2), 4.95 (d,
J = 12.0 Hz, 1H, PhC
H2), 3.79 (s, 3H, OC
H3), 2.55 (d,
J = 13.2 Hz, 1H), 2.28–2.16 (m, 2H), 2.12–1.96 (m, 3H), 1.88–1.76 (m, 3H), 1.68–1.63 (m, 2H), 1.58–1.45 (m, 4H), 1.39–1.35 (m, 2H), 1.21–1.16 (m, 1H), 0.95–0.92 (m, 1H), 0.90 (s, 3H, H-16), 0.89 (s, 3H, H-17), 0.87 (s, 3H, H-19), 0.65 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.5 (C-20), 159.5 (C-Ph), 130.1 (C-9), 129.9 (C-Ph), 129.8 (C-8), 128.3 (C-Ph), 113.9 (C-Ph), 71.9 (C-13), 66.0 (Ph
CH
2), 55.3 (O
CH
3), 52.4 (C-5), 48.9 (C-10), 41.9 (C-3), 41.4 (C-14), 34.5 (C-1), 34.1 (C-4), 33.9 (C-15), 32.1 (C-12), 32.0 (C-7), 31.9 (C-18), 21.7 (C-11), 20.2 (C-2), 20.1 (C-19), 18.2 (C-6), 16.8 (C-16), 16.8 (C-17); HRMS
m/z calcd for C
28H
40O
4Na ([M + Na]
+) 463.2819, found 463.2820 (
Figures S34–S36).
Data for
3m: White solid, yield: 91%, m.p. 163–165 °C; IR cm
−1: 1715, 1525, 1358, 1199, 736;
1H NMR (600 MHz, CDCl
3):
δ 8.06 (d,
J = 7.8 Hz, 1H, H-Ph), 7.60 (td,
J = 7.8, 1.2 Hz, 1H, H-Ph), 7.52 (d,
J = 9.0 Hz, 1H, H-Ph), 7.48 (t,
J = 7.2 Hz, 1H, H-Ph), 5.72 (d,
J = 14.4 Hz, 1H, PhC
H2), 5.21 (d,
J = 14.4 Hz, 1H, PhC
H2), 2.60 (d,
J = 13.2 Hz, 1H), 2.23–1.98 (m, 5H), 1.91–1.78 (m, 3H), 1.72–1.64 (m, 2H), 1.60–1.51 (m, 4H), 1.42–1.40 (m, 2H), 1.24–1.18 (m, 1H), 0.99–0.94 (m, 1H), 0.93 (s, 3H, H-16), 0.92 (s, 3H, H-17), 0.88 (s, 3H, H-19), 0.69 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.1 (C-20), 147.8 (C-Ph), 133.9 (C-Ph), 132.5 (C-Ph), 130.6 (C-9) 129.7 (C-Ph), 129.5 (C-8), 128.9 (C-Ph), 125.1 (C-Ph), 71.9 (C-13), 63.0 (Ph
CH
2), 52.3 (C-5), 49.0 (C-10), 42.0 (C-3), 41.5 (C-14), 34.8 (C-1), 34.5 (C-4), 34.0 (C-15), 32.2 (C-12), 32.1 (C-7), 31.9 (C-18), 21.5 (C-11), 20.1 (C-2), 20.1 (C-19), 18.4 (C-6), 16.9 (C-16), 16.8 (C-17); HRMS
m/z calcd for C
27H
37O
5NNa([M + Na]
+) 478.2564, found 478.2568 (
Figures S37–S39).
Data for
3n: White solid, yield: 95%, m.p. 99–101 °C; IR cm
−1: 1710, 1518, 1345, 1162, 1122;
1H NMR (600 MHz, CDCl
3):
δ 8.21 (d,
J = 8.4 Hz, 2H, H-Ph), 7.48 (d,
J = 8.4 Hz, 2H, H-Ph), 5.29 (d,
J = 13.2 Hz, 1H, PhC
H2), 5.05 (d,
J = 13.5 Hz, 1H, PhCH
2), 2.60 (d,
J = 13.2 Hz, 1H), 2.26–2.19 (m, 2H), 2.13–1.99 (m, 3H), 1.87–1.77 (m, 3H), 1.75–1.45 (m, 6H), 1.44–1.39 (m, 2H), 1.25–1.19 (m, 1H), 1.01–0.96 (m, 1H), 0.92 (s, 3H, H-16), 0.91 (s, 3H, H-17), 0.89 (s, 3H, H-19), 0.66 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.3 (C-20), 147.7 (C-Ph), 143.7 (C-Ph), 130.4 (C-9), 129.7 (C-8), 128.3 (C-Ph), 123.9 (C-Ph), 72.0 (C-13), 64.8 (Ph
CH
2), 52.4 (C-5), 49.0 (C-10), 41.8 (C-3), 41.4 (C-14), 34.6 (C-1), 34.6 (C-4), 33.9 (C-15), 32.0 (C-12), 32.0 (C-7), 32.0 (C-18), 21.7 (C-11), 20.2 (C-2), 20.1 (C-19), 18.3 (C-6), 16.8 (C-16), 16.8 (C-17); HRMS
m/z calcd for C
27H
37O
5NNa ([M + Na]
+) 478.2564, found 478.2568 (
Figures S40–S42).
3.1.3. General Procedure for the Synthesis of Compound 5a–i
To a solution of 4 (100 mg, 0.33 mmol) and Sodium hydride (26 mg, 1.0 mmol) in DMF (10 mL) was added substituent haloalkane (0.66 mmol) in DMF (2 mL) was added dropwise for 10 min. The resulting mixture was stirred at room temperature for 1.5–2 h until the starting materials were completely transformed. After termination by pure water (30 mL), the reaction was extracted with ethyl acetate (3 × 30 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography eluting with petroleum ether/ethyl acetate to afford compound 5a-i in 42–74%.
Data for
5a: colorless oil, yield: 43%; IR cm
−1: 2925, 2358, 988, 957;
1H NMR (600 MHz, CDCl
3):
δ 3.51–3.48 (m, 2H, H-20), 3.25 (s, 3H, OC
H3), 2.24–2.05 (m, 6H), 1.94–1.90 (m, 1H), 1.74–1.58 (m, 5H), 1.52–1.42 (m, 4H), 1.30–1.27 (m, 1H), 1.20–1.15 (m, 1H), 1.11–1.08 (m, 1H), 0.95–0.92 (m, 6H, H-16, 17), 0.89 (s, 3H, H-19), 0.86 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 134.9 (C-9), 126.2 (C-8), 76.8 (C-20), 72.3 (C-13), 59.3 (O
CH
3), 51.1 (C-5), 41.9 (C-10), 41.6 (C-3), 40.3 (C-14), 36.1 (C-1), 34.2 (C-4), 33.5 (C-15), 33.4 (C-12), 32.6 (C-7), 31.1 (C-18), 22.9 (C-11), 22.1 (C-2), 19.2 (C-19), 18.6 (C-6), 17.2 (C-16), 17.2 (C-17); HRMS
m/z calcd for C
21H
36O
2Na ([M + Na]
+): 343.2608, found 343.2607 (
Figures S46–S48).
Data for
5b: colorless oil, yield: 73%; IR cm
−1: 2925, 2358, 988, 957;
1H NMR (600 MHz, CDCl
3):
δ 3.53 (s, 2H, H-20), 3.37 (q,
J = 7.2 Hz, 2H, C
H2CH
3), 2.30–2.06 (m, 6H), 1.94–1.90 (m, 1H), 1.74–1.65 (m, 4H), 1.60–1.56 (m, 1H), 1.53–1.41 (m, 4H), 1.30–1.28 (m, 1H), 1.20–1.15 (m, 1H), 1.14 (t,
J = 7.2 Hz, 3H, CH
2C
H3), 1.10–1.06 (m, 1H), 0.95–0.92 (m, 6H, H-16, 17), 0.89 (s, 3H, H-19), 0.85 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 134.9 (C-9), 126.1 (C-8), 74.4 (C-20), 72.3 (C-13), 66.7 (
CH
2CH
3), 51.0 (C-5), 42.0 (C-10), 41.5 (C-3), 40.2 (C-14), 36.2 (C-1), 34.2 (C-4), 33.5 (C-15), 33.4 (C-12), 32.5 (C-7), 31.1 (C-18), 22.9 (C-11), 22.1 (C-2), 19.2 (C-19), 18.6 (C-6), 17.2 (C-16), 17.2 (C-17), 15.4 (CH
2CH
3); HRMS
m/z calcd for C
22H
38O
2Na ([M + Na]
+): 357.2764, found 357.2764 (
Figures S49–S51).
Data for
5c: colorless oil, yield: 43%; IR cm
−1: 2941, 1455, 1386, 1090;
1H NMR (600 MHz, CDCl
3):
δ 7.33–7.23(m, 5H, H-Ph), 4.47 (d,
J = 12.6 Hz, 1H, PhC
H2), 4.41 (d,
J = 12.6 Hz, 1H, PhC
H2), 3.59–3.55 (m, 2H, H-20), 2.34–2.02 (m, 6H), 1.95–1.91 (m, 1H), 1.76–1.58 (m, 5H), 1.51–1.37 (m, 4H), 1.28–1.25 (m, 1H), 1.17–1.12 (m, 1H), 1.07–1.02 (m, 1H), 0.94–0.92 (m, 6H, H-16, 17), 0.86 (s, 3H, H-19), 0.74 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 138.9 (C-Ph), 135.1 (C-Ph), 128.4 (C-9), 127.6 (C-8), 127.4 (C-Ph), 126.1 (C-Ph), 74.0 (C-20), 73.5 (Ph
CH
2), 72.3 (C-13), 51.2 (C-5), 41.9 (C-10), 41.5 (C-3), 40.4 (C-14), 35.8 (C-1), 34.0 (C-4), 33.5 (C-15), 33.5 (C-12), 32.4 (C-7), 31.4 (C-18), 22.8 (C-11), 22.0 (C-2), 19.1 (C-19), 18.6 (C-6), 17.2 (C-16), 17.1 (C-17); HRMS
m/z calcd for C
27H
40O
2Na ([M + Na]
+) 419.2921, found 419.2913 (
Figures S52–S54).
Data for
5d: colorless oil, yield: 46%; IR cm
−1: 2941, 1455, 1386, 1090;
1H NMR (600 MHz, CDCl
3):
δ 7.26–7.23 (m, 2H, H-Ph), 7.02–6.99 (m, 2H, H-Ph), 4.43 (d,
J = 12.0 Hz, 1H, PhC
H2), 4.36 (d,
J = 12.0 Hz, 1H, PhC
H2), 3.57–3.53 (m, 2H, H-20), 2.31–2.04 (m, 6H), 1.94–1.91 (m, 1H), 1.75–1.57 (m, 5H), 1.51–1.38 (m, 4H), 1.28–1.25 (m, 1H), 1.18–1.13 (m, 1H), 1.08–1.03 (m, 1H), 0.94–0.92 (m, 6H, H-16, 17), 0.87 (s, 3H, H-19), 0.75 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 163.1 (C-Ph), 135.0 (C-Ph), 129.4 (C-9), 129.3 (C-8), 126.2 (C-Ph), 115.3 (C-Ph), 74.05 (C-20), 72.8 (Ph
CH
2), 72.3 (C-13), 51.2 (C-5), 41.9 (C-10), 41.5 (C-3), 40.4 (C-14), 35.9 (C-1), 34.0 (C-4), 33.5 (C-15), 33.5 (C-12), 32.4 (C-7), 31.4 (C-18), 22.8 (C-11), 22.0 (C-2), 19.1 (C-19), 18.6 (C-6), 17.2 (C-16), 17.1 (C-17); HRMS
m/z calcd for C
27H
39O
2FNa ([M + Na]
+) 437.2828, found 437.2823 (
Figures S55–S57).
Data for
5e: colorless oil, yield: 53%; IR cm
−1: 2921, 1490, 1455, 1089, 1015, 806;
1H NMR (600 MHz, CDCl
3):
δ 7.29 (d,
J = 7.8 Hz, 2H, H-Ph), 7.22 (d,
J = 8.4 Hz, 2H, H-Ph), 4.43 (d,
J = 12.0 Hz, 1H, PhC
H2), 4.36 (d,
J = 12.0 Hz, 1H, PhC
H2), 3.59–3.54 (m, 2H, H-20), 2.31–2.04 (m, 6H), 1.95–1.91 (m, 1H), 1.76–1.57 (m, 5H), 1.51–1.39 (m, 4H), 1.29–1.26 (m, 1H), 1.18–1.13 (m, 1H), 1.09–1.03 (m, 1H), 0.94–0.92 (m, 6H, H-16, 17), 0.87 (s, 3H, H-19), 0.76 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 137.4 (C-Ph), 135.0 (C-Ph), 133.2 (C-Ph), 128.9 (C-9), 128.6 (C-8), 126.3 (C-Ph), 74.3 (C-20), 72.8 (Ph
CH
2), 72.3 (C-13), 51.2 (C-5), 41.8 (C-10), 41.5 (C-3), 40.4 (C-14), 35.9 (C-1), 34.0 (C-4), 33.5 (C-15), 32.4 (C-12), 31.4 (C-7), 31.4 (C-18), 22.8 (C-11), 22.0 (C-2), 19.1 (C-19), 18.6 (C-6), 17.1 (C-16), 17.1 (C-17); HRMS
m/z calcd for C
27H
39O
2ClNa ([M + Na]
+) 453.2531, found 453.2533 (
Figures S58–S60).
Data for
5f: white solid, yield: 53%, m.p. 63–65 °C; IR cm
−1: 2358, 1455, 1089, 1012, 800, 668;
1H NMR (600 MHz, CDCl
3):
δ 7.44 (d,
J = 8.4 Hz, 2H, H-Ph), 7.16 (d,
J = 8.4 Hz, 2H, H-Ph), 4.42 (d,
J = 12.6 Hz, 1H, PhC
H2), 4.35 (d,
J = 12.6 Hz, 1H, PhC
H2), 3.59–3.54 (m, 2H, H-20), 2.29–2.03 (m, 6H), 1.95–1.91 (m, 1H), 1.76–1.57 (m, 5H), 1.51–1.39 (m, 4H), 1.29–1.25 (m, 1H), 1.18–1.13 (m, 1H), 1.08–1.03 (m, 1H), 0.94–0.91 (m, 6H, H-16, 17), 0.87 (s, 3H, H-19), 0.76 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 137.9 (C-Ph), 135.0 (C-Ph), 131.5 (C-9), 129.2 (C-8), 126.2 (C-Ph), 121.3 (C-Ph), 74.3 (C-20), 72.8 (Ph
CH
2), 72.2 (C-13), 51.2 (C-5), 41.8 (C-10), 41.5 (C-3), 40.4 (C-14), 35.9 (C-1), 33.9 (C-4), 33.5 (C-15), 33.4 (C-12), 32.4 (C-7), 31.4 (C-18), 22.8 (C-11), 22.0 (C-2), 19.1 (C-19), 18.6 (C-6), 17.1 (C-16), 17.1 (C-17); HRMS
m/z calcd for C
27H
39O
2BrNa ([M + Na]
+) 497.2026, found 497.2028 (
Figures S61–S63).
Data for
5g: colorless oil, yield: 74%; IR cm
−1: 2928, 1466, 1091;
1H NMR (600 MHz, CDCl
3):
δ 7.16 (d,
J = 7.8 Hz, 2H, H-Ph), 7.12 (d,
J = 7.8 Hz, 2H, H-Ph), 4.43 (d,
J = 12.0 Hz, 1H, PhC
H2), 4.36 (d,
J = 12.0 Hz, 1H, PhC
H2), 3.57–3.54 (m, 2H, H-20), 2.32 (s, 3H, C
H3), 2.29–2.03 (m, 6H), 1.94–1.91 (m, 1H), 1.75–1.57 (m, 5H), 1.50–1.37 (m, 4H), 1.27–1.25 (m, 1H), 1.17–1.12 (m, 1H), 1.08–1.01 (m, 1H), 0.94–0.91 (m, 6H, H-16, 17), 0.86 (s, 3H, H-19), 0.76 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 137.0 (C-Ph), 135.1 (C-Ph), 129.1 (C-Ph), 129.0 (C-9), 128.0 (C-Ph), 127.6 (C-8), 73.9 (C-20), 73.3 (Ph
CH
2), 72.2 (C-13), 51.2 (C-5), 41.9 (C-10), 41.5 (C-3), 40.4 (C-14), 35.8 (C-1), 34.0 (C-4), 33.5 (C-15), 33.4 (C-12), 32.4 (C-7), 31.4 (C-18), 22.7 (C-11), 22.0 (C-2), 21.2 (
CH
3), 19.1 (C-19), 18.6 (C-6), 17.1 (C-16), 17.1 (C-17); HRMS
m/z calcd for C
28H
42O
2Na ([M + Na]
+) 433.3077, found 433.3076 (
Figures S64–S66).
Data for
5h: colorless oil, yield: 43%; IR cm
−1: 2941, 1455, 1386, 1090;
1H NMR (600 MHz, CDCl
3):
δ 7.07 (d,
J = 7.2 Hz, 1H, H-Ph), 7.04 (s, 1H, H-Ph), 7.00 (d,
J = 7.2 Hz, 1H, H-Ph), 4.41 (d,
J = 12.1 Hz, 1H, PhC
H2), 4.34 (d,
J = 12.0 Hz, 1H, PhC
H2), 3.57–3.54 (m, 2H, H-20), 2.34–2.27 (m, 1H), 2.24 (s, 3H, C
H3), 2.23 (s, 3H, C
H3), 2.18–2.04 (m, 5H), 1.95–1.91 (m, 1H), 1.76–1.58 (m, 5H), 1.51–1.38 (m, 4H), 1.28–1.25 (m, 1H), 1.17–1.12 (m, 1H), 1.07–1.02 (m, 1H), 0.95–0.92 (m, 6H, H-16, 17), 0.86 (s, 3H, H-19), 0.76 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 136.3 (C-Ph), 135.6 (C-Ph), 135.1 (C-Ph), 129.6 (C-9), 129.0 (C-Ph), 126.1 (C-Ph), 125.1 (C-8), 73.9 (C-20), 73.3 (Ph
CH
2), 72.2 (C-13), 51.2 (C-5), 41.9 (C-10), 41.5 (C-3), 40.5 (C-14), 35.8 (C-1), 34.1 (C-4), 33.5 (C-15), 33.5 (C-12), 32.5 (C-7), 31.4 (C-18), 22.8 (C-11), 22.0 (C-2), 19.8 (C-19), 19.6 (
CH
3), 19.1 (
CH
3), 18.6 (C-6), 17.2 (C-16), 17.1 (C-17); HRMS
m/z calcd for C
29H
44O
2Na ([M + Na]
+) 447.3234, found 447.3233 (
Figures S67–S69).
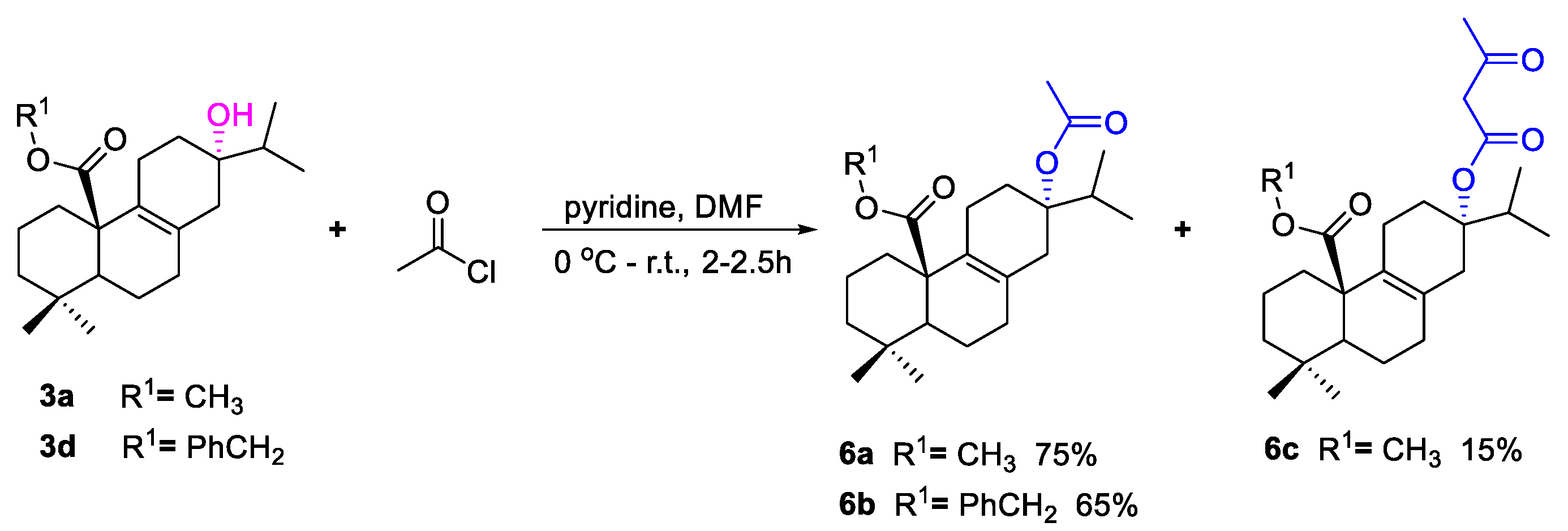
3.1.4. Synthesis of Compound 6a–c
To a solution of 3a or 3d (0.3 mmol) and pyridine (1.2 mmol) in DMF (10 mL) at 0 °C was added acetyl chloride (0.6 mmol). The mixture was warmed up to room temperature and stirred for 2–2.5 h. When the reaction was complete, pure water (30 mL) was added to the reaction, which was extracted with ethyl acetate (3 × 30 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography eluting with petroleum ether/ethyl acetate to afford compounds 6a (75%), 6b (65%), or 6c (15%).
Data for
6a: white solid, yield: 75%, m.p. 118–120 °C; IR cm
−1: 1726, 1361, 1272, 1226, 1203, 1130;
1H NMR (600 MHz, CDCl
3):
δ 3.59 (s, 3H, OC
H3), 2.57–2.52 (m, 2H), 2.39 (d,
J = 17.4 Hz, 1H), 2.34–2.30 (m, 1H), 2.28–2.24 (m, 1H), 2.21–1.97 (m, 4H), 1.90 (s, 3H, COC
H3), 1.84–1.76 (m, 1H), 1.74–1.68 (m, 2H), 1.62–1.57 (m, 1H), 1.55–1.51 (m, 1H), 1.40–1.35 (m, 2H), 1.22–1.17 (m, 1H), 0.95–0.90 (m, 1H), 0.89–0.88 (m, 9H, H-16, 17, 19), 0.68 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 176.1 (C-20), 170.7 (
COCH
3), 129.6 (C-9), 129.3 (C-8), 85.9 (C-13), 52.4 (O
CH
3), 51.3 (C-5), 48.6 (C-10), 42.0 (C-3), 36.2 (C-14), 34.8 (C-1), 33.9 (C-4), 32.1 (C-15), 32.1 (C-12), 32.1 (C-7), 31.8 (C-18), 22.3 (CO
CH
3), 21.7 (C-11), 20.3 (C-2), 20.1 (C-19), 18.3 (C-6), 17.5 (C-16), 17.2 (C-17); HRMS
m/z calcd for C
23H
36O
4Na ([M + Na]
+) 399.2506, found 399.2507 (
Figures S70–S72).
Data for
6b: colorless oil, yield: 65%; IR cm
−1: 2957, 1748, 1687, 1458, 1127, 906;
1H NMR (600 MHz, CDCl
3):
δ 7.36–7.28 (5H, H-Ph), 5.13 (d,
J = 12.6 Hz, 1H, PhC
H2), 4.98 (d,
J = 12.6 Hz, 1H, PhC
H2), 2.61 (d,
J = 16.8 Hz, 1H), 2.54–2.50 (m, 1H), 2.43 (d,
J = 12.6 Hz, 1H), 2.35–2.27 (m, 2H), 2.23 (d,
J = 16.8 Hz, 1H), 2.17–2.13 (m, 1H), 2.10–1.98 (m, 2H), 1.85–1.63 (m, 6H), 1.64–1.59 (m, 1H), 1.54–1.52 (m, 1H), 1.40–1.37 (m, 2H), 1.22–1.17 (m, 1H), 0.97–0.92 (m, 1H), 0.89–0.87 (m, 9H, H-16, 17, 19), 0.66 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 175.3 (C-20), 170.7 (
COCH
3), 136.3 (C-Ph), 129.8 (C-9), 129.6 (C-8), 128.6 × 2 (C-Ph), 128.0 (C-Ph), 127.8 (C-Ph) × 2, 85.9 (C-13), 66.2 (Ph
CH
2), 52.3 (C-5), 48.8 (C-10), 42.0 (C-3), 36.3 (C-14), 34.7 (C-1), 34.0 (C-4), 32.0 (C-15), 31.9 (C-12), 31.6 (C-7), 31.6 (C-18), 28.2 (CO
CH
3), 22.3 (C-11), 21.7 (C-2), 20.2 (C-19), 18.3 (C-6), 17.5 (C-16), 17.2 (C-17); HRMS
m/z calcd for C
29H
40O
4Na ([M + Na]
+): 475.2810, found 475.2811 (
Figures S73–S75).
Data for
6c: colorless oil, yield: 15%; IR cm
−1: 2359, 1716, 1704, 1206;
1H NMR (600 MHz, CDCl
3):
δ 3.62 (s, 3H, OC
H3), 3.33–3.27 (m, 2H, COC
H2COCH
3), 2.57 (d,
J = 13.8 Hz, 1H), 2.52–2.48 (m, 1H), 2.43 (d,
J = 16.8 Hz, 1H), 2.36–2.15 (m, 7H), 2.09–1.97 (m, 2H), 1.83–1.63 (m, 4H), 1.54–1.50 (m, 1H), 1.40–1.33 (m, 2H), 1.22–1.17 (m, 1H), 0.95–0.92 (m, 1H), 0.91–0.89 (m, 9H, H-16, 17, 19), 0.68 (s, 3H, H-18);
13C NMR (150 MHz, CDCl
3):
δ 201.2 (COCH
2COCH
3), 175.9 (C-20), 166.5 (
COCH
2COCH
3), 129.9 (C-9), 129.2 (C-8), 88.0 (C-13), 52.5 (C-5), 51.4 (OCH
3), 51.3 (COCH
2CO
CH
3), 48.6 (C-10), 42.0 (C-3), 36.2 (C-14), 34.8 (C-1), 33.8 (C-4), 32.1 (C-15), 32.1 (C-12), 31.8 (C-7), 30.2 (C-18), 28.2 (C-25), 21.7 (C-11), 20.3 (C-2), 20.1 (C-19), 18.2 (C-6), 17.5 (C-16), 17.2 (C-17);HRMS
m/z calcd for C
25H
38O
5Na ([M + Na]
+): 441.2611, found 441.2612 (
Figures S76–S78).




 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}