
Atomic-Resolution Experimental Structural Biology and Molecular Dynamics Simulations of Hyaluronan and Its Complexes
Abstract
:1. Physical, Chemical, and Biological Properties of Hyaluronan
2. Atomic-Resolution Experimental Structural Biology of Hyaluronan and Its Complexes
3. Atomic-Resolution Molecular Dynamics Simulations of Hyaluronan and Its Complexes
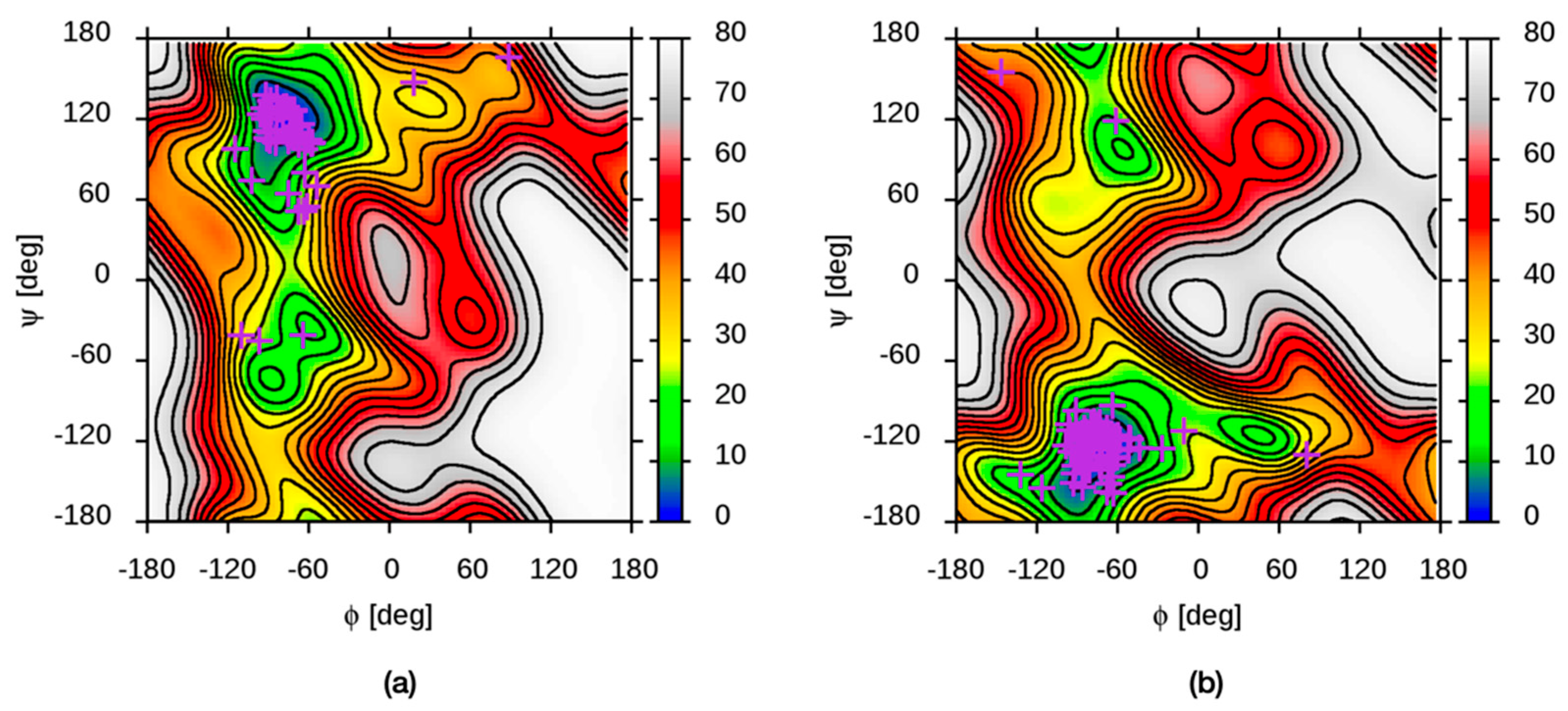
3.1. Molecular Dynamics Simulation Methodology
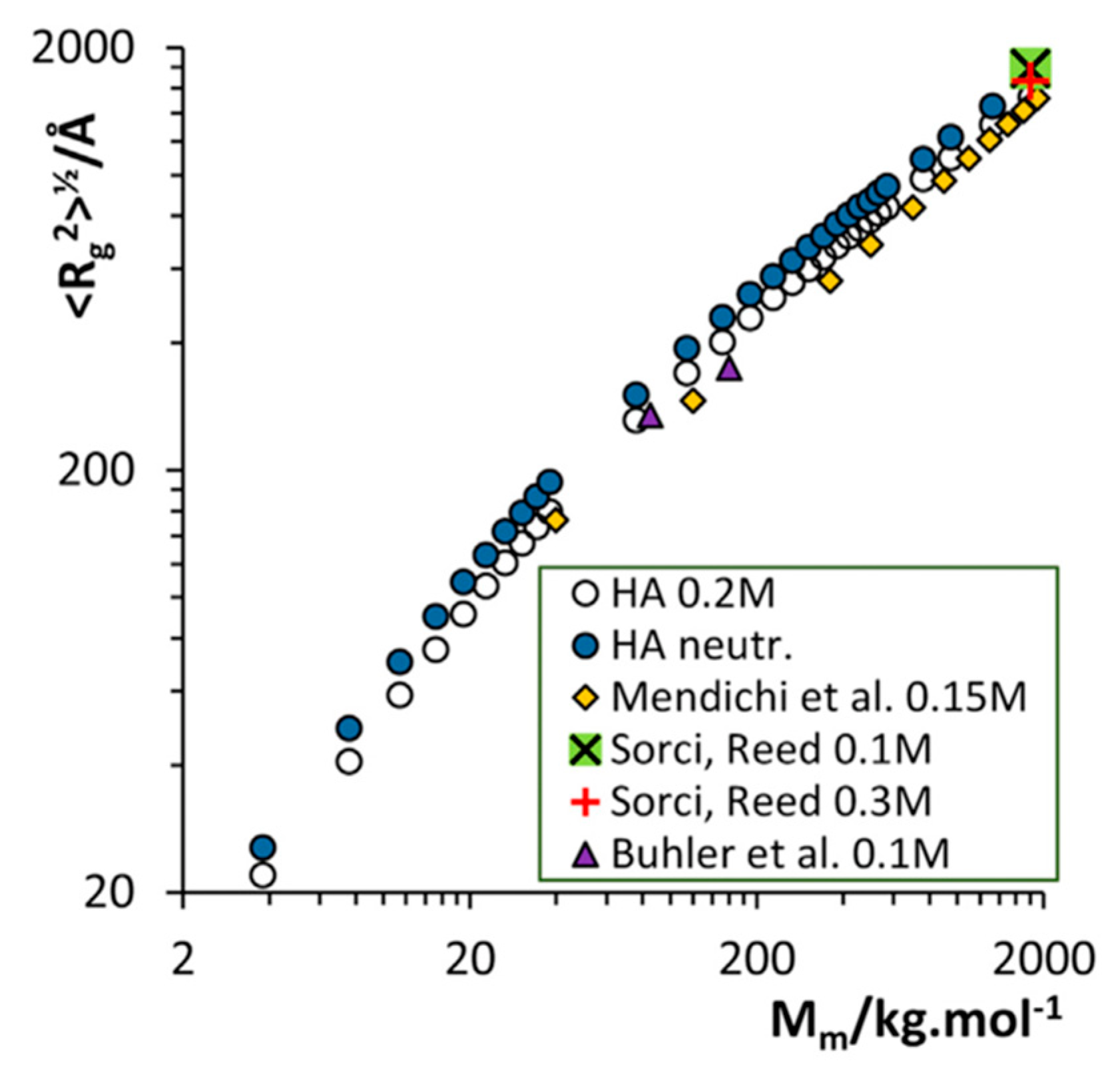
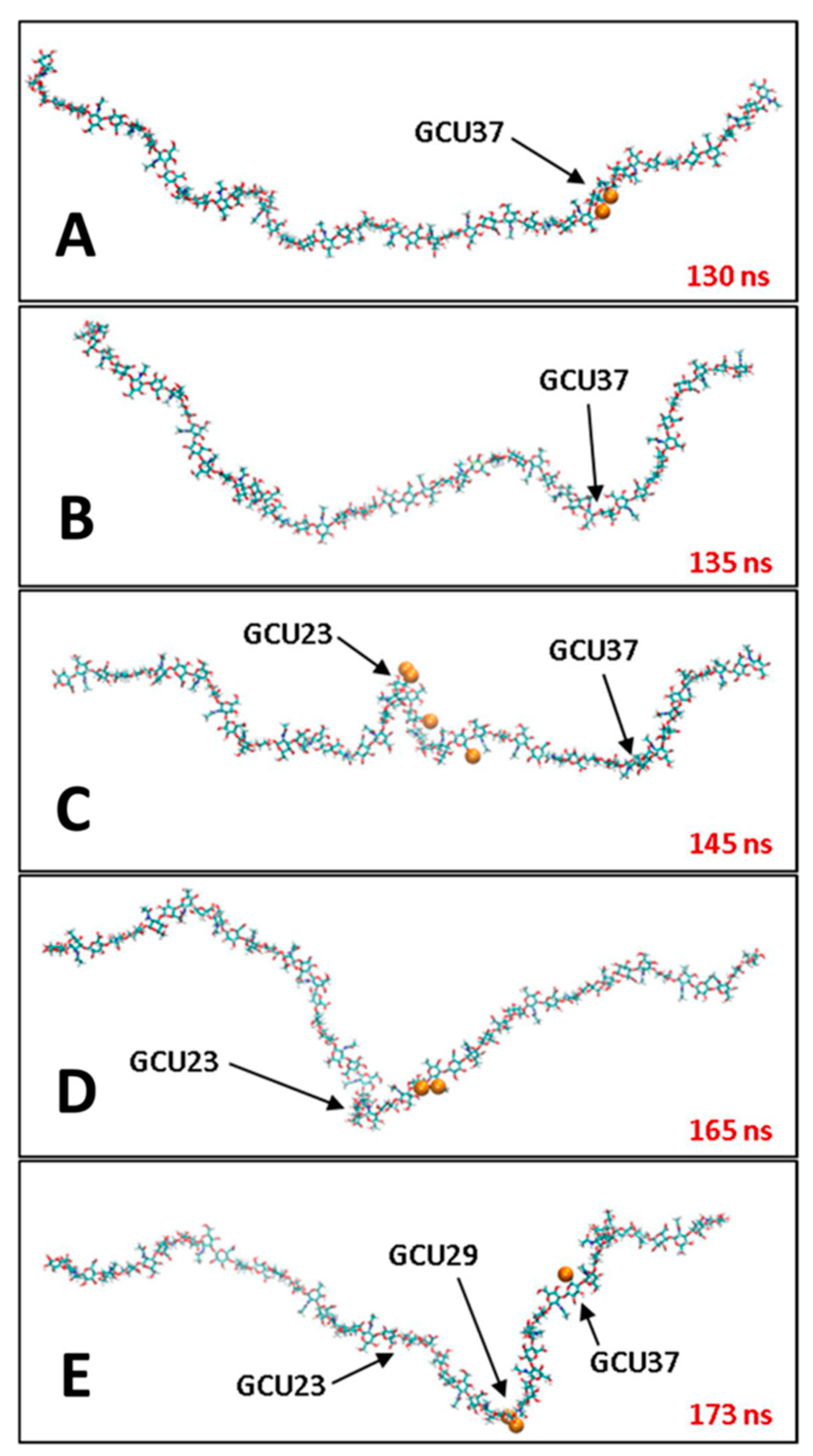
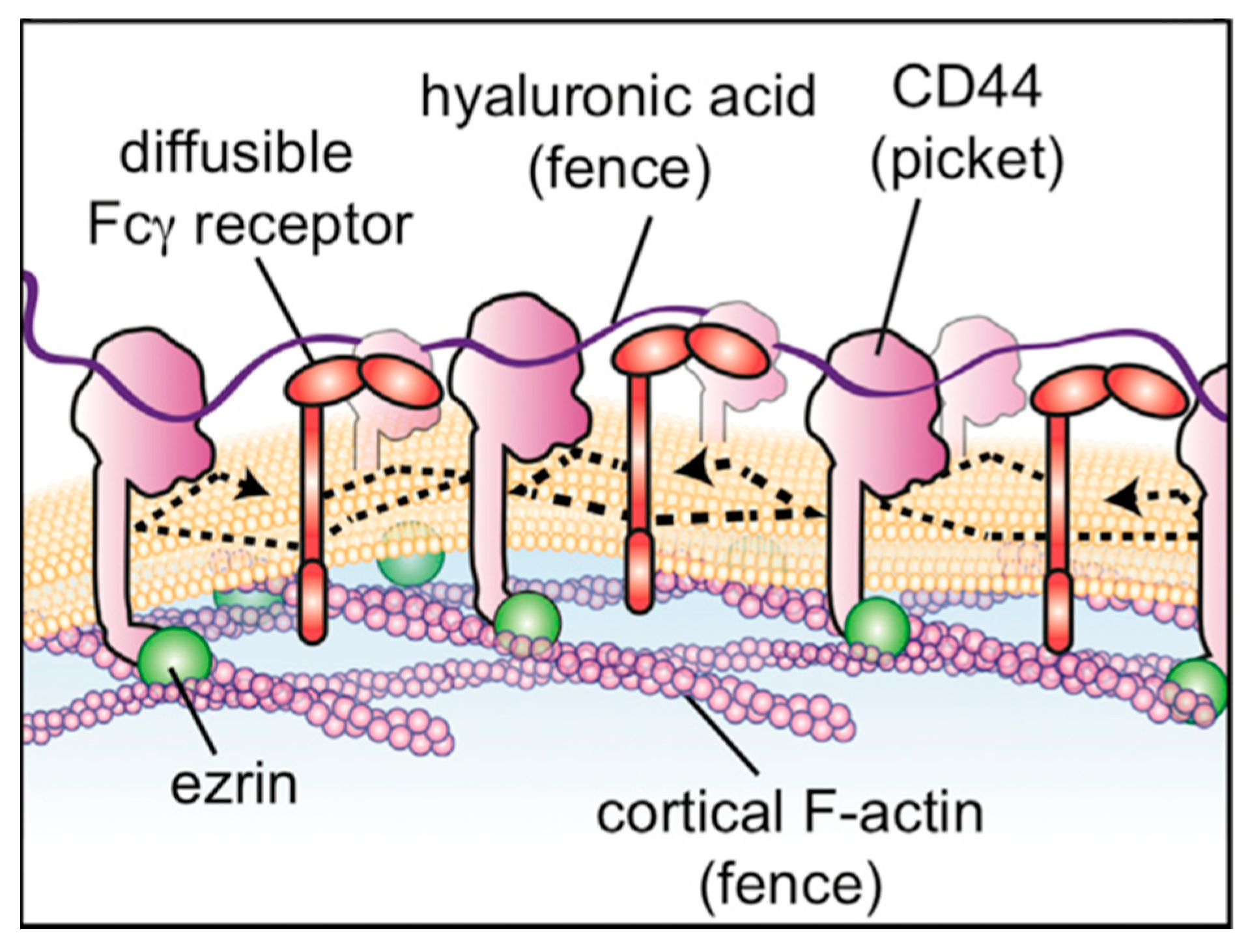
3.2. Molecular Dynamics Simulations of Aqueous Hyaluronan and Its Interactions with Itself, Other Glycosaminoglycans, and Ions
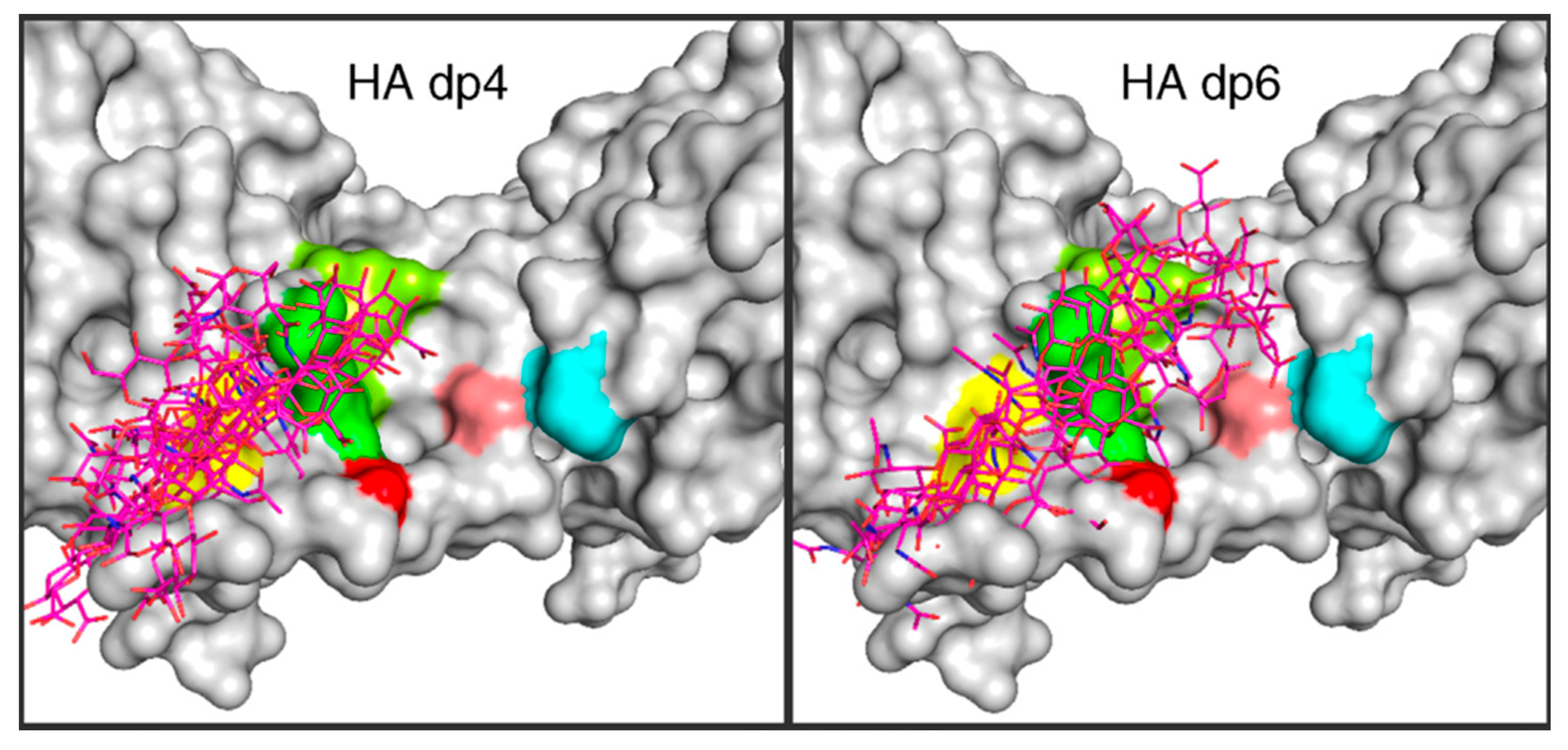
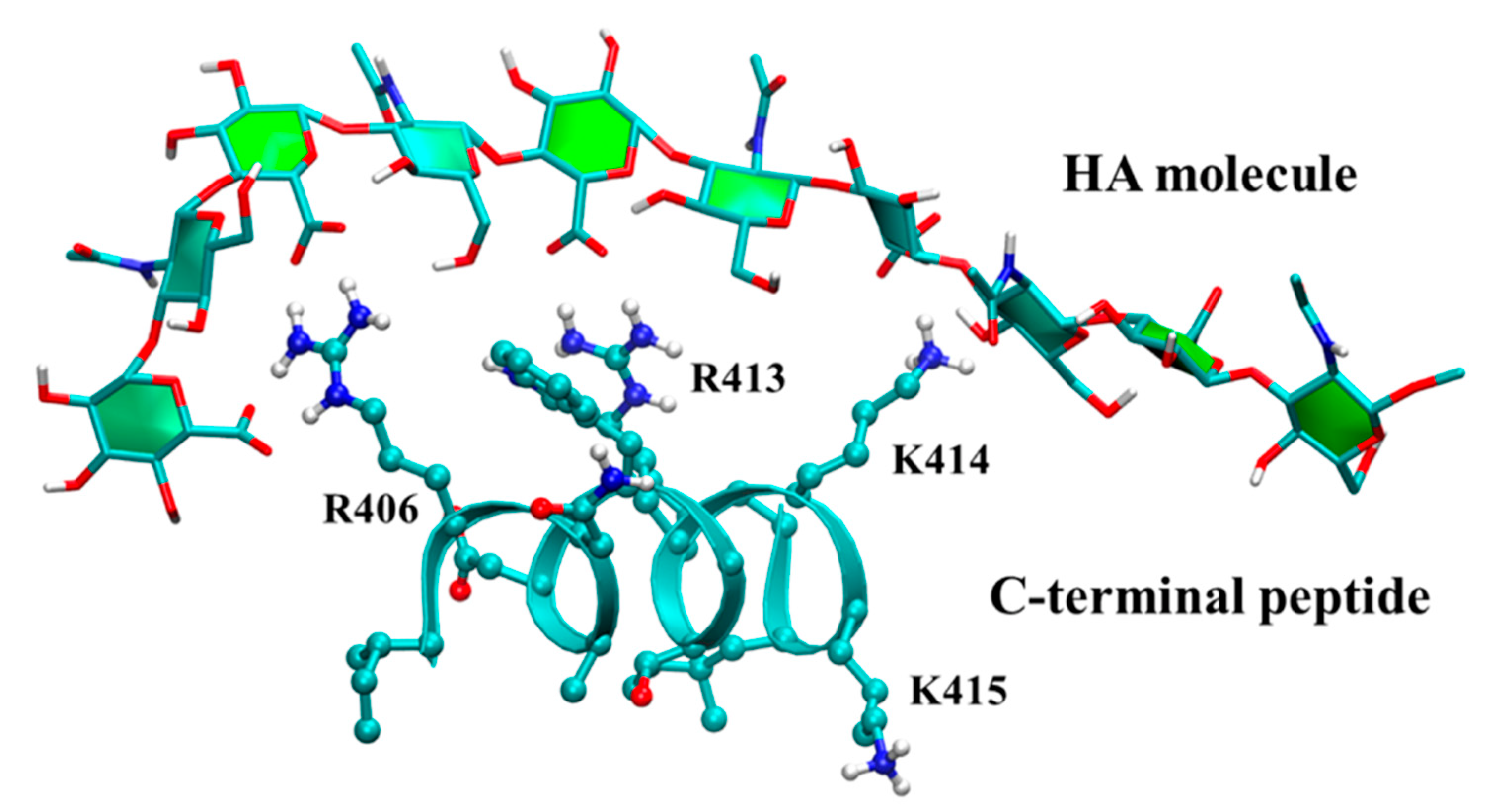
3.3. Molecular Dynamics Simulations of Hyaluronan–Protein Complexes
3.4. Molecular Dynamics Simulations of Hyaluronan–Lipid Complexes
3.5. Molecular Dynamics Simulations of Hyaluronan in the Context of Pharmaceuticals
4. Conclusions and Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simpson, M.; Schaefer, L.; Hascall, V.; Esko, J.D. Hyaluronan. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; pp. 205–216. [Google Scholar]
- Merry, C.L.R.; Lindahl, U.; Couchman, J.; Esko, J.D. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; pp. 217–232. [Google Scholar]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [PubMed]
- Evanko, S.P.; Tammi, M.I.; Tammi, R.H.; Wight, T.N. Hyaluronan-dependent pericellular matrix. Adv. Drug Deliv. Rev. 2007, 59, 1351–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicker, K.T.; Gurski, L.A.; Pradhan-Bhatt, S.; Witt, R.L.; Farach-Carson, M.C.; Jia, X. Hyaluronan: A simple polysaccharide with diverse biological functions. Acta Biomater. 2014, 10, 1558–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Chanmee, T.; Itano, N. Hyaluronan: Metabolism and function. Biomolecules 2020, 10, 1525. [Google Scholar] [CrossRef] [PubMed]
- Cowman, M.K. Hyaluronan and Hyaluronan Fragments. Adv. Carbohydr. Chem. Biochem. 2017, 74, 1–59. [Google Scholar] [CrossRef]
- Skandalis, S.S.; Karalis, T.; Heldin, P. Intracellular hyaluronan: Importance for cellular functions. Semin. Cancer Biol. 2020, 62, 20–30. [Google Scholar] [CrossRef]
- Damodarasamy, M.; Reed, M.J. Aging and hyaluronan. In Hyaluronan: Biological and Medical Implications; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2014; pp. 15–30. [Google Scholar]
- Bohaumilitzky, L.; Huber, A.K.; Stork, E.M.; Wengert, S.; Woelfl, F.; Boehm, H. A trickster in disguise: Hyaluronan’s ambivalent roles in the matrix. Front. Oncol. 2017, 7, 242. [Google Scholar] [CrossRef] [Green Version]
- Knudson, W.; Ishizuka, S.; Terabe, K.; Askew, E.B.; Knudson, C.B. The pericellular hyaluronan of articular chondrocytes. Matrix Biol. 2019, 78–79, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Zakusilo, F.T.; Kerry O’Banion, M.; Gelbard, H.A.; Seluanov, A.; Gorbunova, V. Matters of size: Roles of hyaluronan in CNS aging and disease. Ageing Res. Rev. 2021, 72, 101485. [Google Scholar] [CrossRef] [PubMed]
- Sinova, R.; Pavlik, V.; Ondrej, M.; Velebny, V.; Nesporova, K. Hyaluronan: A key player or just a bystander in skin photoaging? Exp. Dermatol. 2022, 31, 442–458. [Google Scholar] [CrossRef]
- Lompardia, S.; Pibuel, M.; Diaz, M.; Poodts, D.; Hajos, S. Senescence modulation as a key process in the dual role of hyaluronan in cancer: The deforestation allegory. Glycobiology 2022, 32, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan as an immune regulator in human diseases. Physiol. Rev. 2011, 91, 221–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandoch, M.; Bollyky, P.L.; Fischer, J.W. Hyaluronan: A Master Switch Between Vascular Homeostasis and Inflammation. Circ. Res. 2018, 122, 1341–1343. [Google Scholar] [CrossRef] [PubMed]
- Medina, C.O.; Nagy, N.; Bollyky, P.L. Extracellular matrix and the maintenance and loss of peripheral immune tolerance in autoimmune insulitis. Curr. Opin. Immunol. 2018, 55, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.G. Hyaluronan in the lymphatics: The key role of the hyaluronan receptor LYVE-1 in leucocyte trafficking. Matrix Biol. 2019, 78–79, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Nagy, N.; Kuipers, H.F.; Marshall, P.L.; Wang, E.; Kaber, G.; Bollyky, P.L. Hyaluronan in immune dysregulation and autoimmune diseases. Matrix Biol. 2019, 78–79, 292–313. [Google Scholar] [CrossRef]
- Krolikoski, M.; Monslow, J.; Pure, E. The CD44-HA axis and inflammation in atherosclerosis: A temporal perspective. Matrix Biol. 2019, 78–79, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Tavianatou, A.G.; Caon, I.; Franchi, M.; Piperigkou, Z.; Galesso, D.; Karamanos, N.K. Hyaluronan: Molecular size-dependent signaling and biological functions in inflammation and cancer. FEBS J. 2019, 286, 2883–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queisser, K.A.; Mellema, R.A.; Petrey, A.C. Hyaluronan and Its Receptors as Regulatory Molecules of the Endothelial Interface. J. Histochem. Cytochem. 2021, 69, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Jackson, D.G. Hyaluronan and Its Receptors: Key Mediators of Immune Cell Entry and Trafficking in the Lymphatic System. Cells 2021, 10, 2061. [Google Scholar] [CrossRef]
- Tolg, C.; McCarthy, J.B.; Yazdani, A.; Turley, E.A. Hyaluronan and RHAMM in Wound Repair and the “cancerization” of Stromal Tissues. BioMed Res. Int. 2014, 2014, 103923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindaraju, P.; Todd, L.; Shetye, S.; Monslow, J.; Pure, E. CD44-dependent inflammation, fibrogenesis, and collagenolysis regulates extracellular matrix remodeling and tensile strength during cutaneous wound healing. Matrix Biol. 2019, 75–76, 314–330. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Short, W.D.; Keswani, S.G.; Wang, X. Immunologic Roles of Hyaluronan in Dermal Wound Healing. Biomolecules 2021, 11, 1234. [Google Scholar] [CrossRef]
- Tolg, C.; Messam, B.J.; McCarthy, J.B.; Nelson, A.C.; Turley, E.A. Hyaluronan Functions in Wound Repair That Are Captured to Fuel Breast Cancer Progression. Biomolecules 2021, 11, 1551. [Google Scholar] [CrossRef] [PubMed]
- Seluanov, A.; Gladyshev, V.N.; Vijg, J.; Gorbunova, V. Mechanisms of cancer resistance in long-lived mammals. Nat. Rev. Cancer 2018, 18, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tolg, C.; Turley, E. Dissecting the dual nature of hyaluronan in the tumor microenvironment. Front. Immunol. 2019, 10, 947. [Google Scholar] [CrossRef] [Green Version]
- Passi, A.; Vigetti, D.; Buraschi, S.; Iozzo, R.V. Dissecting the role of hyaluronan synthases in the tumor microenvironment. FEBS J. 2019, 286, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Caon, I.; Bartolini, B.; Parnigoni, A.; Carava, E.; Moretto, P.; Viola, M.; Karousou, E.; Vigetti, D.; Passi, A. Revisiting the hallmarks of cancer: The role of hyaluronan. Semin. Cancer Biol. 2020, 62, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Roedig, H.; Damiescu, R.; Zeng-Brouwers, J.; Kutija, I.; Trebicka, J.; Wygrecka, M.; Schaefer, L. Danger matrix molecules orchestrate CD14/CD44 signaling in cancer development. Semin. Cancer Biol. 2020, 62, 31–47. [Google Scholar] [CrossRef]
- Toole, B.P. The CD147-HYALURONAN Axis in Cancer. Anat. Rec. 2020, 303, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Karalis, T.; Skandalis, S.S. Hyaluronan network: A driving force in cancer progression. Am. J. Physiol. Cell Physiol. 2022, 323, C145–C158. [Google Scholar] [CrossRef] [PubMed]
- Cowman, M.K.; Lee, H.G.; Schwertfeger, K.L.; McCarthy, J.B.; Turley, E.A. The Content and Size of Hyaluronan in Biological Fluids and Tissues. Front. Immunol. 2015, 6, 261. [Google Scholar] [CrossRef] [Green Version]
- Tiessen, A.; Perez-Rodriguez, P.; Delaye-Arredondo, L.J. Mathematical modeling and comparison of protein size distribution in different plant, animal, fungal and microbial species reveals a negative correlation between protein size and protein number, thus providing insight into the evolution of proteomes. BMC Res. Notes 2012, 5, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmbeck, S.M.; Petillo, P.A.; Lerner, L.E. The solution conformation of hyaluronan: A combined NMR and molecular dynamics study. Biochemistry 1994, 33, 14246–14255. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, V.; Morando, M.A.; Silipo, A.; Nurisso, A.; Perez, S.; Imberty, A.; Canada, F.J.; Parrilli, M.; Jimenez-Barbero, J.; De Castro, C. Insights on the conformational properties of hyaluronic acid by using NMR residual dipolar couplings and MD simulations. Glycobiology 2010, 20, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Sattelle, B.M.; Almond, A. Is N-acetyl-D-glucosamine a rigid 4C1 chair? Glycobiology 2011, 21, 1651–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guvench, O.; Martin, D.; Greene, M. Pyranose Ring Puckering Thermodynamics for Glycan Monosaccharides Associated with Vertebrate Proteins. Int. J. Mol. Sci. 2022, 23, 473. [Google Scholar] [CrossRef] [PubMed]
- IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Symbols for specifying the conformation of polysaccharide chains. Recommendations 1981. Eur. J. Biochem. 1983, 131, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Day, A.J. The structure and regulation of hyaluronan-binding proteins. Biochem. Soc. Trans. 1999, 27, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.A.; Vega, A.; Riedl, M.; Collins, R.F.; Ostrowski, P.P.; Woods, E.C.; Bertozzi, C.R.; Tammi, M.I.; Lidke, D.S.; Johnson, P.; et al. Transmembrane Pickets Connect Cyto- and Pericellular Skeletons Forming Barriers to Receptor Engagement. Cell 2018, 172, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Toole, B.P.; Hascall, V.C. Hyaluronan and the Aggregating Proteoglycans. In The Extracellular Matrix: An Overview; Mecham, R.P., Ed.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2011; pp. 147–195. [Google Scholar]
- Xu, Y.; Zheng, Y.; Fan, J.S.; Yang, D. A new strategy for structure determination of large proteins in solution without deuteration. Nat. Methods 2006, 3, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Kalodimos, C.G. NMR Studies of Large Proteins. J. Mol. Biol. 2017, 429, 2667–2676. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Guss, J.M.; Hukins, D.W.L.; Smith, P.J.C.; Winter, W.T.; Arnott, S.; Moorhouse, R.; Rees, D.A. Hyaluronic acid: Molecular conformations and interactions in two sodium salts. J. Mol. Biol. 1975, 95, 359–364. [Google Scholar] [CrossRef]
- Ponnuraj, K.; Jedrzejas, M.J. Mechanism of hyaluronan binding and degradation: Structure of Streptococcus pneumoniae hyaluronate lyase in complex with hyaluronic acid disaccharide at 1.7 A resolution. J. Mol. Biol. 2000, 299, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Boju, L.; Tkalec, L.; Su, H.; Yang, H.O.; Gunay, N.S.; Linhardt, R.J.; Kim, Y.S.; Matte, A.; Cygler, M. Active site of chondroitin AC lyase revealed by the structure of enzyme-oligosaccharide complexes and mutagenesis. Biochemistry 2001, 40, 2359–2372. [Google Scholar] [CrossRef]
- Jedrzejas, M.J.; Mello, L.V.; de Groot, B.L.; Li, S. Mechanism of hyaluronan degradation by Streptococcus pneumoniae hyaluronate lyase. Structures of complexes with the substrate. J. Biol. Chem. 2002, 277, 28287–28297. [Google Scholar] [CrossRef] [Green Version]
- Mello, L.V.; De Groot, B.L.; Li, S.; Jedrzejas, M.J. Structure and flexibility of Streptococcus agalactiae hyaluronate lyase complex with its substrate. Insights into the mechanism of processive degradation of hyaluronan. J. Biol. Chem. 2002, 277, 36678–36688. [Google Scholar] [CrossRef] [Green Version]
- Nukui, M.; Taylor, K.B.; McPherson, D.T.; Shigenaga, M.K.; Jedrzejas, M.J. The function of hydrophobic residues in the catalytic cleft of Streptococcus pneumoniae hyaluronate lyase. Kinetic characterization of mutant enzyme forms. J. Biol. Chem. 2003, 278, 3079–3088. [Google Scholar] [CrossRef]
- Almond, A.; DeAngelis, P.L.; Blundell, C.D. Hyaluronan: The local solution conformation determined by NMR and computer modeling is close to a contracted left-handed 4-fold helix. J. Mol. Biol. 2006, 358, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Banerji, S.; Wright, A.J.; Noble, M.; Mahoney, D.J.; Campbell, I.D.; Day, A.J.; Jackson, D.G. Structures of the Cd44-hyaluronan complex provide insight into a fundamental carbohydrate-protein interaction. Nat. Struct. Mol. Biol. 2007, 14, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.K.; Finzel, B.C. Fragment-based identification of an inducible binding site on cell surface receptor CD44 for the design of protein-carbohydrate interaction inhibitors. J. Med. Chem. 2014, 57, 2714–2725. [Google Scholar] [CrossRef] [PubMed]
- Oiki, S.; Sato, M.; Mikami, B.; Murata, K.; Hashimoto, W. Substrate recognition by bacterial solute-binding protein is responsible for import of extracellular hyaluronan and chondroitin sulfate from the animal host. Biosci. Biotechnol. Biochem. 2019, 83, 1946–1954. [Google Scholar] [CrossRef]
- Deivanayagam, C.; Schormann, N. Crystal Structure of Truncated Bacteriophage Hyaluronan Lyase HylP in Complex with Unsaturated Hyaluronan Tetra-Saccharides. 2020. Available online: https://www.rcsb.org/structure/6WWX (accessed on 25 August 2022).
- Deivanayagam, C.; Schormann, N. Crystal Structure of Truncated Streptococcal Bacteriophage Hyaluronidase Complexed with Unsaturated Hyaluronan Hexa-Saccharides. 2020. Available online: https://www.rcsb.org/structure/6WXA (accessed on 25 August 2022).
- Deivanayagam, C.; Schormann, N. Crystal Structure of Full-Length Streptococcal Bacteriophage Hyaluronidase in Complex with Unsaturated Hyaluronan Octa-Saccharides. 2020. Available online: https://www.rcsb.org/structure/6X3M (accessed on 25 August 2022).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wang, J.; Hou, T. Application of molecular dynamics simulations in molecular property prediction II: Diffusion coefficient. J. Comput. Chem. 2011, 32, 3505–3519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Tingjun, H. Application of Molecular Dynamics Simulations in Molecular Property Prediction I: Density and Heat of Vaporization. J. Chem. Theory Comput. 2011, 7, 2151–2165. [Google Scholar] [CrossRef] [Green Version]
- Comer, J.; Gumbart, J.C.; Hénin, J.; Lelievre, T.; Pohorille, A.; Chipot, C. The adaptive biasing force method: Everything you always wanted to know but were afraid to ask. J. Phys. Chem. B 2015, 119, 1129–1151. [Google Scholar] [CrossRef] [Green Version]
- Valsson, O.; Tiwary, P.; Parrinello, M. Enhancing Important Fluctuations: Rare Events and Metadynamics from a Conceptual Viewpoint. Annu. Rev. Phys. Chem. 2016, 67, 159–184. [Google Scholar] [CrossRef]
- Miao, Y.; McCammon, J.A. Unconstrained Enhanced Sampling for Free Energy Calculations of Biomolecules: A Review. Mol. Simul. 2016, 42, 1046–1055. [Google Scholar] [CrossRef]
- Yang, Y.I.; Shao, Q.; Zhang, J.; Yang, L.; Gao, Y.Q. Enhanced sampling in molecular dynamics. J. Chem. Phys. 2019, 151, 070902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Shao, X.; Cai, W.; Chipot, C. Taming Rugged Free Energy Landscapes Using an Average Force. Acc. Chem. Res. 2019, 52, 3254–3264. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Chen, H.; Blazhynska, M.; Goulard Coderc de Lacam, E.; Szczepaniak, F.; Pavlova, A.; Shao, X.; Gumbart, J.C.; Dehez, F.; Roux, B.; et al. Accurate determination of protein:ligand standard binding free energies from molecular dynamics simulations. Nat. Protoc. 2022, 17, 1114–1141. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Pall, S.; Zhmurov, A.; Bauer, P.; Abraham, M.; Lundborg, M.; Gray, A.; Hess, B.; Lindahl, E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153, 134110. [Google Scholar] [CrossRef]
- Jones, D.; Allen, J.E.; Yang, Y.; Drew Bennett, W.F.; Gokhale, M.; Moshiri, N.; Rosing, T.S. Accelerators for Classical Molecular Dynamics Simulations of Biomolecules. J. Chem. Theory Comput. 2022, 18, 4047–4069. [Google Scholar] [CrossRef]
- Sattelle, B.M.; Almond, A. Less is more when simulating unsulfated glycosaminoglycan 3D-structure: Comparison of GLYCAM06/TIP3P, PM3-CARB1/TIP3P, and SCC-DFTB-D/TIP3P predictions with experiment. J. Comput. Chem. 2010, 31, 2932–2947. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; Greene, S.N.; Kamath, G.; Brady, J.W.; Venable, R.M.; Pastor, R.W.; Mackerell, A.D., Jr. Additive empirical force field for hexopyranose monosaccharides. J. Comput. Chem. 2008, 29, 2543–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guvench, O.; Hatcher, E.R.; Venable, R.M.; Pastor, R.W.; MacKerell, A.D., Jr. CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J. Chem. Theory Comput. 2009, 5, 2353–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, F.W., II; Mackerell, A.D., Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Tessier, M.B.; Pederson, K.; Wang, X.; Venot, A.P.; Boons, G.J.; Prestegard, J.H.; Woods, R.J. Extension and validation of the GLYCAM force field parameters for modeling glycosaminoglycans. Can. J. Chem. 2016, 94, 927–935. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Rusu, V.H.; Verli, H.; Lins, R.D. GROMOS 53A6GLYC, an Improved GROMOS Force Field for Hexopyranose-Based Carbohydrates. J. Chem. Theory Comput. 2012, 8, 4681–4690. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Verli, H.; Lins, R.D. Extension and validation of the GROMOS 53A6(GLYC) parameter set for glycoproteins. J. Comput. Chem. 2014, 35, 2087–2095. [Google Scholar] [CrossRef]
- Hansen, H.S.; Hünenberger, P.H. A reoptimized GROMOS force field for hexopyranose-based carbohydrates accounting for the relative free energies of ring conformers, anomers, epimers, hydroxymethyl rotamers, and glycosidic linkage conformers. J. Comput. Chem. 2011, 32, 998–1032. [Google Scholar] [CrossRef]
- Plazinski, W.; Lonardi, A.; Hunenberger, P.H. Revision of the GROMOS 56A6(CARBO) force field: Improving the description of ring-conformational equilibria in hexopyranose-based carbohydrates chains. J. Comput. Chem. 2016, 37, 354–365. [Google Scholar] [CrossRef]
- Panczyk, K.; Gaweda, K.; Drach, M.; Plazinski, W. Extension of the GROMOS 56a6CARBO/CARBO_R Force Field for Charged, Protonated, and Esterified Uronates. J. Phys. Chem. B 2018, 122, 3696–3710. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Liu, B. Chapter 3—Control techniques of molecular dynamics simulation. In Molecular Dynamics Simulation; Zhou, K., Liu, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 67–96. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Roux, B. Simulation of Osmotic Pressure in Concentrated Aqueous Salt Solutions. J. Phys. Chem. Lett. 2010, 1, 183–189. [Google Scholar] [CrossRef]
- Guvench, O.; Whitmore, E.K. Sulfation and Calcium Favor Compact Conformations of Chondroitin in Aqueous Solutions. ACS Omega 2021, 6, 13204–13217. [Google Scholar] [CrossRef]
- Jing, Z.; Liu, C.; Cheng, S.Y.; Qi, R.; Walker, B.D.; Piquemal, J.P.; Ren, P. Polarizable Force Fields for Biomolecular Simulations: Recent Advances and Applications. Annu. Rev. Biophys. 2019, 48, 371–394. [Google Scholar] [CrossRef]
- Lemkul, J.A. Pairwise-additive and polarizable atomistic force fields for molecular dynamics simulations of proteins. Prog. Mol. Biol. Transl. Sci. 2020, 170, 1–71. [Google Scholar] [CrossRef]
- Dauber-Osguthorpe, P.; Hagler, A.T. Biomolecular force fields: Where have we been, where are we now, where do we need to go and how do we get there? J. Comput. Aided Mol. Des. 2019, 33, 133–203. [Google Scholar] [CrossRef]
- Hagler, A.T. Force field development phase II: Relaxation of physics-based criteria… or inclusion of more rigorous physics into the representation of molecular energetics. J. Comput. Aided Mol. Des. 2019, 33, 205–264. [Google Scholar] [CrossRef]
- Antila, H.S.; Salonen, E. Polarizable force fields. Methods Mol. Biol. 2013, 924, 215–241. [Google Scholar] [CrossRef]
- Pandey, P.; Aytenfisu, A.H.; MacKerell, A.D., Jr.; Mallajosyula, S.S. Drude Polarizable Force Field Parametrization of Carboxylate and N-Acetyl Amine Carbohydrate Derivatives. J. Chem. Theory Comput. 2019, 15, 4982–5000. [Google Scholar] [CrossRef]
- Sterling, J.D.; Jiang, W.; Botello-Smith, W.M.; Luo, Y.L. Ion Pairing and Dielectric Decrement in Glycosaminoglycan Brushes. J. Phys. Chem. B 2021, 125, 2771–2780. [Google Scholar] [CrossRef] [PubMed]
- Almond, A. Multiscale modeling of glycosaminoglycan structure and dynamics: Current methods and challenges. Curr. Opin. Struct. Biol. 2018, 50, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathe, M.; Rutledge, G.C.; Grodzinsky, A.J.; Tidor, B. A coarse-grained molecular model for glycosaminoglycans: Application to chondroitin, chondroitin sulfate, and hyaluronic acid. Biophys. J. 2005, 88, 3870–3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samsonov, S.A.; Bichmann, L.; Pisabarro, M.T. Coarse-grained model of glycosaminoglycans. J. Chem. Inf. Model. 2015, 55, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Sattelle, B.M.; Shakeri, J.; Cliff, M.J.; Almond, A. Proteoglycans and their heterogeneous glycosaminoglycans at the atomic scale. Biomacromolecules 2015, 16, 951–961. [Google Scholar] [CrossRef]
- Kumar, R.; Lee, Y.K.; Jho, Y.S. Martini Coarse-Grained Model of Hyaluronic Acid for the Structural Change of Its Gel in the Presence of Monovalent and Divalent Salts. Int. J. Mol. Sci. 2020, 21, 4602. [Google Scholar] [CrossRef] [PubMed]
- Bathe, M.; Rutledge, G.C.; Grodzinsky, A.J.; Tidor, B. Osmotic pressure of aqueous chondroitin sulfate solution: A molecular modeling investigation. Biophys. J. 2005, 89, 2357–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yesylevskyy, S.O.; Schafer, L.V.; Sengupta, D.; Marrink, S.J. Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput. Biol. 2010, 6, e1000810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, J.E.; Cummings, C.; Brass, A.; Chen, Y. Secondary and tertiary structures of hyaluronan in aqueous solution, investigated by rotary shadowing-electron microscopy and computer simulation. Hyaluronan is a very efficient network-forming polymer. Biochem. J. 1991, 274 Pt 3, 699–705. [Google Scholar] [CrossRef]
- Scott, J.E.; Chen, Y.; Brass, A. Secondary and tertiary structures involving chondroitin and chondroitin sulphates in solution, investigated by rotary shadowing/electron microscopy and computer simulation. Eur. J. Biochem. 1992, 209, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Blundell, C.D.; DeAngelis, P.L.; Day, A.J.; Almond, A. Use of 15N-NMR to resolve molecular details in isotopically-enriched carbohydrates: Sequence-specific observations in hyaluronan oligomers up to decasaccharides. Glycobiology 2004, 14, 999–1009. [Google Scholar] [CrossRef]
- Almond, A.; DeAngelis, P.L.; Blundell, C.D. Dynamics of hyaluronan oligosaccharides revealed by 15N relaxation. J. Am. Chem. Soc. 2005, 127, 1086–1087. [Google Scholar] [CrossRef] [PubMed]
- Donati, A.; Magnani, A.; Bonechi, C.; Barbucci, R.; Rossi, C. Solution structure of hyaluronic acid oligomers by experimental and theoretical NMR, and molecular dynamics simulation. Biopolymers 2001, 59, 434–445. [Google Scholar] [CrossRef]
- Almond, A.; Sheehan, J.K.; Brass, A. Molecular dynamics simulations of the two disaccharides of hyaluronan in aqueous solution. Glycobiology 1997, 7, 597–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almond, A.; Brass, A.; Sheehan, J.K. Dynamic exchange between stabilized conformations predicted for hyaluronan tetrasaccharides: Comparison of molecular dynamics simulations with available NMR data. Glycobiology 1998, 8, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almond, A.; Sheehan, J.K. Predicting the molecular shape of polysaccharides from dynamic interactions with water. Glycobiology 2003, 13, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cilurzo, F.; Vistoli, G.; Gennari, C.G.; Selmin, F.; Gardoni, F.; Franze, S.; Campisi, M.; Minghetti, P. The role of the conformational profile of polysaccharides on skin penetration: The case of hyaluronan and its sulfates. Chem. Biodivers. 2014, 11, 551–561. [Google Scholar] [CrossRef]
- Almond, A.; Brass, A.; Sheehan, J.K. Deducing polymeric structure from aqueous molecular dynamics simulations of oligosaccharides: Predictions from simulations of hyaluronan tetrasaccharides compared with hydrodynamic and X-ray fibre diffraction data. J. Mol. Biol. 1998, 284, 1425–1437. [Google Scholar] [CrossRef]
- Ingr, M.; Kutalkova, E.; Hrncirik, J. Hyaluronan random coils in electrolyte solutions-a molecular dynamics study. Carbohydr. Polym. 2017, 170, 289–295. [Google Scholar] [CrossRef]
- Jordan, R.C.; Brant, D.A.; Cesàro, A. A Monte Carlo study of the amylosic chain conformation. Biopolymers 1978, 17, 2617–2632. [Google Scholar] [CrossRef]
- Furlan, S.; La Penna, G.; Perico, A.; Cesaro, A. Hyaluronan chain conformation and dynamics. Carbohydr. Res. 2005, 340, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, E.K.; Martin, D.; Guvench, O. Constructing 3-Dimensional Atomic-Resolution Models of Nonsulfated Glycosaminoglycans with Arbitrary Lengths Using Conformations from Molecular Dynamics. Int. J. Mol. Sci. 2020, 21, 7699. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, E.K.; Vesenka, G.; Sihler, H.; Guvench, O. Efficient Construction of Atomic-Resolution Models of Non-Sulfated Chondroitin Glycosaminoglycan Using Molecular Dynamics Data. Biomolecules 2020, 10, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsyk, V.; Plazinski, W. Conformational Properties of Glycosaminoglycan Disaccharides: A Molecular Dynamics Study. J. Phys. Chem. B 2021, 125, 10900–10916. [Google Scholar] [CrossRef] [PubMed]
- Giubertoni, G.; Pérez de Alba Ortíz, A.; Bano, F.; Zhang, X.; Linhardt, R.J.; Green, D.E.; DeAngelis, P.L.; Koenderink, G.H.; Richter, R.P.; Ensing, B.; et al. Strong Reduction of the Chain Rigidity of Hyaluronan by Selective Binding of Ca2+ Ions. Macromolecules 2021, 54, 1137–1146. [Google Scholar] [CrossRef]
- Taweechat, P.; Pandey, R.B.; Sompornpisut, P. Conformation, flexibility and hydration of hyaluronic acid by molecular dynamics simulations. Carbohydr. Res. 2020, 493, 108026. [Google Scholar] [CrossRef]
- Kutalkova, E.; Hrncirik, J.; Witasek, R.; Ingr, M. Effect of solvent and ions on the structure and dynamics of a hyaluronan molecule. Carbohydr. Polym. 2020, 234, 115919. [Google Scholar] [CrossRef]
- Samantray, S.; Olubiyi, O.O.; Strodel, B. The Influences of Sulphation, Salt Type, and Salt Concentration on the Structural Heterogeneity of Glycosaminoglycans. Int. J. Mol. Sci. 2021, 22, 11529. [Google Scholar] [CrossRef] [PubMed]
- Andrysiak, T.; Beldowski, P.; Siodmiak, J.; Weber, P.; Ledzinski, D. Hyaluronan-Chondroitin Sulfate Anomalous Crosslinking Due to Temperature Changes. Polymers 2018, 10, 560. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Dubey, D.K. Effect of aggrecan degradation on the nanomechanics of hyaluronan in extra-fibrillar matrix of annulus fibrosus: A molecular dynamics investigation. J. Mech. Behav. Biomed. Mater. 2020, 107, 103752. [Google Scholar] [CrossRef] [PubMed]
- Kolarikova, A.; Kutalkova, E.; Bus, V.; Witasek, R.; Hrncirik, J.; Ingr, M. Salt-dependent intermolecular interactions of hyaluronan molecules mediate the formation of temporary duplex structures. Carbohydr. Polym. 2022, 286, 119288. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Adnan, A. Effect of Shock-Induced Cavitation Bubble Collapse on the damage in the Simulated Perineuronal Net of the Brain. Sci. Rep. 2017, 7, 5323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmud, K.; Hasan, F.; Khan, M.I.; Adnan, A. Shock-Induced Damage Mechanism of Perineuronal Nets. Biomolecules 2021, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Rigden, D.J.; Littlejohn, J.E.; Joshi, H.V.; de Groot, B.L.; Jedrzejas, M.J. Alternate structural conformations of Streptococcus pneumoniae hyaluronan lyase: Insights into enzyme flexibility and underlying molecular mechanism of action. J. Mol. Biol. 2006, 358, 1165–1178. [Google Scholar] [CrossRef] [Green Version]
- Joshi, H.V.; Jedrzejas, M.J.; de Groot, B.L. Domain motions of hyaluronan lyase underlying processive hyaluronan translocation. Proteins 2009, 76, 30–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Zhang, H.; Xu, D. Initial events in the degradation of hyaluronan catalyzed by hyaluronate lyase from Streptococcus [corrected] pneumoniae: QM/MM simulation. J. Phys. Chem. B 2012, 116, 11166–11172. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Xu, D. Catalytic mechanism of hyaluronate lyase from Streptococcus pneumonia [corrected]: Quantum mechanical/molecular mechanical and density functional theory studies. J. Phys. Chem. B 2013, 117, 10161–10172. [Google Scholar] [CrossRef]
- Jamison, F.W., II; Foster, T.J.; Barker, J.A.; Hills, R.D., Jr.; Guvench, O. Mechanism of binding site conformational switching in the CD44-hyaluronan protein-carbohydrate binding interaction. J. Mol. Biol. 2011, 406, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Plazinski, W.; Knys-Dzieciuch, A. Interactions between CD44 protein and hyaluronan: Insights from the computational study. Mol. BioSyst. 2012, 8, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Bandyopadhyay, S. Conformational flexibility of a protein-carbohydrate complex and the structure and ordering of surrounding water. Phys. Chem. Chem. Phys. 2012, 14, 6628–6638. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Bandyopadhyay, S. Restricted dynamics of water around a protein-carbohydrate complex: Computer simulation studies. J. Chem. Phys. 2012, 137, 055102. [Google Scholar] [CrossRef] [PubMed]
- Plazinski, W.; Knys-Dzieciuch, A. The ‘order-to-disorder’ conformational transition in CD44 protein: An umbrella sampling analysis. J. Mol. Graph. Model. 2013, 45, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Favreau, A.J.; Faller, C.E.; Guvench, O. CD44 receptor unfolding enhances binding by freeing basic amino acids to contact carbohydrate ligand. Biophys. J. 2013, 105, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Suzuki, M.; Ogino, S.; Umemoto, R.; Nishida, N.; Shimada, I. Mechanical force effect on the two-state equilibrium of the hyaluronan-binding domain of CD44 in cell rolling. Proc. Natl. Acad. Sci. USA 2015, 112, 6991–6996. [Google Scholar] [CrossRef] [Green Version]
- Vuorio, J.; Vattulainen, I.; Martinez-Seara, H. Atomistic fingerprint of hyaluronan-CD44 binding. PLoS Comput. Biol. 2017, 13, e1005663. [Google Scholar] [CrossRef] [Green Version]
- Lintuluoto, M.; Horioka, Y.; Hongo, S.; Lintuluoto, J.M.; Fukunishi, Y. Molecular Dynamics Simulation Study on Allosteric Regulation of CD44-Hyaluronan Binding as a Force Sensing Mechanism. ACS Omega 2021, 6, 8045–8055. [Google Scholar] [CrossRef]
- Patel, S.; Shaikh, F.; Devaraji, V.; Radadiya, A.; Shah, K.; Shah, A.; Rawal, R. Insights into the structural perturbations of spliced variants of CD44: A modeling and simulation approach. J. Biomol. Struct. Dyn. 2017, 35, 354–367. [Google Scholar] [CrossRef]
- Faller, C.E.; Guvench, O. Terminal sialic acids on CD44 N-glycans can block hyaluronan binding by forming competing intramolecular contacts with arginine sidechains. Proteins 2014, 82, 3079–3089. [Google Scholar] [CrossRef] [Green Version]
- Guvench, O. Revealing the Mechanisms of Protein Disorder and N-Glycosylation in CD44-Hyaluronan Binding Using Molecular Simulation. Front. Immunol. 2015, 6, 305. [Google Scholar] [CrossRef]
- Pichert, A.; Samsonov, S.A.; Theisgen, S.; Thomas, L.; Baumann, L.; Schiller, J.; Beck-Sickinger, A.G.; Huster, D.; Pisabarro, M.T. Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology 2012, 22, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Hintze, V.; Samsonov, S.A.; Anselmi, M.; Moeller, S.; Becher, J.; Schnabelrauch, M.; Scharnweber, D.; Pisabarro, M.T. Sulfated glycosaminoglycans exploit the conformational plasticity of bone morphogenetic protein-2 (BMP-2) and alter the interaction profile with its receptor. Biomacromolecules 2014, 15, 3083–3092. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.J.; Casalini, T.; Hulsart-Billstrom, G.; Wang, S.; Oommen, O.P.; Salvalaglio, M.; Larsson, S.; Hilborn, J.; Varghese, O.P. Synthetic design of growth factor sequestering extracellular matrix mimetic hydrogel for promoting in vivo bone formation. Biomaterials 2018, 161, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panitz, N.; Theisgen, S.; Samsonov, S.A.; Gehrcke, J.P.; Baumann, L.; Bellmann-Sickert, K.; Kohling, S.; Pisabarro, M.T.; Rademann, J.; Huster, D.; et al. The structural investigation of glycosaminoglycan binding to CXCL12 displays distinct interaction sites. Glycobiology 2016, 26, 1209–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunze, G.; Gehrcke, J.P.; Pisabarro, M.T.; Huster, D. NMR characterization of the binding properties and conformation of glycosaminoglycans interacting with interleukin-10. Glycobiology 2014, 24, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Gehrcke, J.P.; Pisabarro, M.T. Identification and characterization of a glycosaminoglycan binding site on interleukin-10 via molecular simulation methods. J. Mol. Graph. Model. 2015, 62, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Thones, S.; Rother, S.; Wippold, T.; Blaszkiewicz, J.; Balamurugan, K.; Moeller, S.; Ruiz-Gomez, G.; Schnabelrauch, M.; Scharnweber, D.; Saalbach, A.; et al. Hyaluronan/collagen hydrogels containing sulfated hyaluronan improve wound healing by sustained release of heparin-binding EGF-like growth factor. Acta Biomater. 2019, 86, 135–147. [Google Scholar] [CrossRef]
- Penk, A.; Baumann, L.; Huster, D.; Samsonov, S.A. NMR and molecular modeling reveal specificity of the interactions between CXCL14 and glycosaminoglycans. Glycobiology 2019, 29, 715–725. [Google Scholar] [CrossRef]
- Koehler, L.; Ruiz-Gomez, G.; Balamurugan, K.; Rother, S.; Freyse, J.; Moller, S.; Schnabelrauch, M.; Kohling, S.; Djordjevic, S.; Scharnweber, D.; et al. Dual Action of Sulfated Hyaluronan on Angiogenic Processes in Relation to Vascular Endothelial Growth Factor-A. Sci. Rep. 2019, 9, 18143. [Google Scholar] [CrossRef] [Green Version]
- Koehler, L.; Samsonov, S.; Rother, S.; Vogel, S.; Kohling, S.; Moeller, S.; Schnabelrauch, M.; Rademann, J.; Hempel, U.; Pisabarro, M.T.; et al. Sulfated Hyaluronan Derivatives Modulate TGF-beta1:Receptor Complex Formation: Possible Consequences for TGF-beta1 Signaling. Sci. Rep. 2017, 7, 1210. [Google Scholar] [CrossRef]
- Balamurugan, K.; Koehler, L.; Durig, J.N.; Hempel, U.; Rademann, J.; Hintze, V.; Pisabarro, M.T. Structural insights into the modulation of PDGF/PDGFR-beta complexation by hyaluronan derivatives. Biol. Chem. 2021, 402, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Kerrigan, J.E. Molecular dynamics simulations in drug design. Methods Mol. Biol. 2013, 993, 95–113. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef] [Green Version]
- Kogut, M.M.; Marcisz, M.; Samsonov, S.A. Modeling glycosaminoglycan-protein complexes. Curr. Opin. Struct. Biol. 2022, 73, 102332. [Google Scholar] [CrossRef] [PubMed]
- Samsonov, S.A.; Gehrcke, J.P.; Pisabarro, M.T. Flexibility and explicit solvent in molecular-dynamics-based docking of protein-glycosaminoglycan systems. J. Chem. Inf. Model. 2014, 54, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Mobius, K.; Nordsieck, K.; Pichert, A.; Samsonov, S.A.; Thomas, L.; Schiller, J.; Kalkhof, S.; Teresa Pisabarro, M.; Beck-Sickinger, A.G.; Huster, D. Investigation of lysine side chain interactions of interleukin-8 with heparin and other glycosaminoglycans studied by a methylation-NMR approach. Glycobiology 2013, 23, 1260–1269. [Google Scholar] [CrossRef] [Green Version]
- Tatsinkam, A.J.; Mulloy, B.; Rider, C.C. Mapping the heparin-binding site of the BMP antagonist gremlin by site-directed mutagenesis based on predictive modelling. Biochem. J. 2015, 470, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Kunze, G.; Kohling, S.; Vogel, A.; Rademann, J.; Huster, D. Identification of the Glycosaminoglycan Binding Site of Interleukin-10 by NMR Spectroscopy. J. Biol. Chem. 2016, 291, 3100–3113. [Google Scholar] [CrossRef]
- Sawant, K.V.; Poluri, K.M.; Dutta, A.K.; Sepuru, K.M.; Troshkina, A.; Garofalo, R.P.; Rajarathnam, K. Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci. Rep. 2016, 6, 33123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, P.R.B.; Sawant, K.V.; Iwahara, J.; Garofalo, R.P.; Desai, U.R.; Rajarathnam, K. Lysines and Arginines play non-redundant roles in mediating chemokine-glycosaminoglycan interactions. Sci. Rep. 2018, 8, 12289. [Google Scholar] [CrossRef] [Green Version]
- Fogolari, F.; Zuccato, P.; Esposito, G.; Viglino, P. Biomolecular electrostatics with the linearized Poisson-Boltzmann equation. Biophys. J. 1999, 76, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lange, A.W.; Herbert, J.M. Improving Generalized Born Models by Exploiting Connections to Polarizable Continuum Models. II. Corrections for Salt Effects. J. Chem. Theory Comput. 2012, 8, 4381–4392. [Google Scholar] [CrossRef]
- Beldowski, P.; Przybylek, M.; Raczynski, P.; Dedinaite, A.; Gorny, K.; Wieland, F.; Dendzik, Z.; Sionkowska, A.; Claesson, P.M. Albumin-Hyaluronan Interactions: Influence of Ionic Composition Probed by Molecular Dynamics. Int. J. Mol. Sci. 2021, 22, 12360. [Google Scholar] [CrossRef] [PubMed]
- Sionkowski, P.; Beldowski, P.; Kruszewska, N.; Weber, P.; Marciniak, B.; Domino, K. Effect of Ion and Binding Site on the Conformation of Chosen Glycosaminoglycans at the Albumin Surface. Entropy 2022, 24, 811. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cheng, F.; Yu, H.; Wang, J.; Guo, Z.; Stephanopoulos, G. Key Role of the Carboxyl Terminus of Hyaluronan Synthase in Processive Synthesis and Size Control of Hyaluronic Acid Polymers. Biomacromolecules 2017, 18, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Maloney, F.P.; Kuklewicz, J.; Corey, R.A.; Bi, Y.; Ho, R.; Mateusiak, L.; Pardon, E.; Steyaert, J.; Stansfeld, P.J.; Zimmer, J. Structure, substrate recognition and initiation of hyaluronan synthase. Nature 2022, 604, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Kognole, A.A.; Payne, C.M. Inhibition of mammalian glycoprotein YKL-40: Identification of the physiological ligand. J. Biol. Chem. 2017, 292, 2624–2636. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Gomez, G.; Vogel, S.; Moller, S.; Pisabarro, M.T.; Hempel, U. Glycosaminoglycans influence enzyme activity of MMP2 and MMP2/TIMP3 complex formation—Insights at cellular and molecular level. Sci. Rep. 2019, 9, 4905. [Google Scholar] [CrossRef]
- Jin, L.; Syrovets, T.; Scheller, J.S.; Zhang, X.; Simmet, T. Comparative Study on Hyaluronic Acid Binding to Murine SAA1.1 and SAA2.2. ACS Omega 2019, 4, 13388–13399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.; Eckersley, E.L.; Berger, B.; Klauda, J.B. Probing the pH Effects on Sugar Binding to a Polysaccharide Lyase. J. Phys. Chem. B 2019, 123, 7123–7136. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, K.; Han, W. Prediction of hyaluronic acid target on sucrase-isomaltase (SI) with reverse docking and molecular dynamics simulations for inhibitors binding to SI. PLoS ONE 2021, 16, e0255351. [Google Scholar] [CrossRef]
- Zhang, H.; Cai, W.; Shao, X. Regulation of aquaporin-3 water permeability by hyaluronan. Phys. Chem. Chem. Phys. 2021, 23, 25706–25711. [Google Scholar] [CrossRef] [PubMed]
- Beldowski, P.; Przybylek, M.; Sionkowska, A.; Cysewski, P.; Gadomska, M.; Musial, K.; Gadomski, A. Effect of Chitosan Deacetylation on Its Affinity to Type III Collagen: A Molecular Dynamics Study. Materials 2022, 15, 463. [Google Scholar] [CrossRef] [PubMed]
- Bojarski, K.K.; Sage, J.; Lalmanach, G.; Lecaille, F.; Samsonov, S.A. In silico and in vitro mapping of specificity patterns of glycosaminoglycans towards cysteine cathepsins B, L, K, S and V. J. Mol. Graph. Model. 2022, 113, 108153. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.J.; Elder, R.M.; Neumann, A.J.; Jayaraman, A.; Bryant, S.J. Interaction of hyaluronan binding peptides with glycosaminoglycans in poly(ethylene glycol) hydrogels. Biomacromolecules 2014, 15, 1132–1141. [Google Scholar] [CrossRef]
- Satish, L.; Santra, S.; Tsurkan, M.V.; Werner, C.; Jana, M.; Sahoo, H. Conformational changes of GDNF-derived peptide induced by heparin, heparan sulfate, and sulfated hyaluronic acid—Analysis by circular dichroism spectroscopy and molecular dynamics simulation. Int. J. Biol. Macromol. 2021, 182, 2144–2150. [Google Scholar] [CrossRef]
- Siodmiak, J.; Beldowski, P.; Auge, W.K.; Ledzinski, D.; Smigiel, S.; Gadomski, A. Molecular Dynamic Analysis of Hyaluronic Acid and Phospholipid Interaction in Tribological Surgical Adjuvant Design for Osteoarthritis. Molecules 2017, 22, 1436. [Google Scholar] [CrossRef] [PubMed]
- Beldowski, P.; Weber, P.; Andrysiak, T.; Auge Ii, W.K.; Ledzinski, D.; De Leon, T.; Gadomski, A. Anomalous Behavior of Hyaluronan Crosslinking Due to the Presence of Excess Phospholipids in the Articular Cartilage System of Osteoarthritis. Int. J. Mol. Sci. 2017, 18, 2779. [Google Scholar] [CrossRef] [PubMed]
- Beldowski, P.; Andrysiak, T.; Mrela, A.; Pawlak, Z.; Auge, W.K., II; Gadomski, A. The Anomalies of Hyaluronan Structures in Presence of Surface Active Phospholipids-Molecular Mass Dependence. Polymers 2018, 10, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beldowski, P.; Weber, P.; Dedinaite, A.; Claesson, P.M.; Gadomski, A. Physical crosslinking of hyaluronic acid in the presence of phospholipids in an aqueous nano-environment. Soft Matter 2018, 14, 8997–9004. [Google Scholar] [CrossRef] [PubMed]
- Beldowski, P.; Kruszewska, N.; Yuvan, S.; Dendzik, Z.; Goudoulas, T.; Gadomski, A. Capstan-like mechanism in hyaluronan-phospholipid systems. Chem. Phys. Lipids 2018, 216, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Beldowski, P.; Mazurkiewicz, A.; Topolinski, T.; Malek, T. Hydrogen and Water Bonding between Glycosaminoglycans and Phospholipids in the Synovial Fluid: Molecular Dynamics Study. Materials 2019, 12, 2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beldowski, P.; Yuvan, S.; Dedinaite, A.; Claesson, P.M.; Poschel, T. Interactions of a short hyaluronan chain with a phospholipid membrane. Colloids Surf. B Biointerfaces 2019, 184, 110539. [Google Scholar] [CrossRef] [PubMed]
- Zander, T.; Garamus, V.M.; Dedinaite, A.; Claesson, P.M.; Beldowski, P.; Gorny, K.; Dendzik, Z.; Wieland, D.C.F.; Willumeit-Romer, R. Influence of the Molecular Weight and the Presence of Calcium Ions on the Molecular Interaction of Hyaluronan and DPPC. Molecules 2020, 25, 3907. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Ziolek, R.M.; Gazzarrini, E.; Owen, D.M.; Lorenz, C.D. On the interaction of hyaluronic acid with synovial fluid lipid membranes. Phys. Chem. Chem. Phys. 2019, 21, 9845–9857. [Google Scholar] [CrossRef]
- Payne, W.M.; Svechkarev, D.; Kyrychenko, A.; Mohs, A.M. The role of hydrophobic modification on hyaluronic acid dynamics and self-assembly. Carbohydr. Polym. 2018, 182, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Svechkarev, D.; Kyrychenko, A.; Payne, W.M.; Mohs, A.M. Probing the self-assembly dynamics and internal structure of amphiphilic hyaluronic acid conjugates by fluorescence spectroscopy and molecular dynamics simulations. Soft Matter 2018, 14, 4762–4771. [Google Scholar] [CrossRef] [PubMed]
- Franze, S.; Rama, F.; Rocco, P.; Debernardi, M.; Bincoletto, V.; Arpicco, S.; Cilurzo, F. Rationalizing the Design of Hyaluronic Acid-Decorated Liposomes for Targeting Epidermal Layers: A Combination of Molecular Dynamics and Experimental Evidence. Mol. Pharm. 2021, 18, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Kutalkova, E.; Hrncirik, J.; Witasek, R.; Ingr, M.; Huerta-Angeles, G.; Hermannova, M.; Velebny, V. The rate and evenness of the substitutions on hyaluronan grafted by dodecanoic acid influenced by the mixed-solvent composition. Int. J. Biol. Macromol. 2021, 189, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Kianipour, S.; Ansari, M.; Farhadian, N.; Moradi, S.; Shahlaei, M. A molecular dynamics study on using of naturally occurring polymers for structural stabilization of erythropoietin at high temperature. J. Biomol. Struct. Dyn. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Moradi, S.; Mirzaei, S.; Khosravi, R.; Farhadian, N.; Hosseininezhadian Koushki, E.; Shahlaei, M. Computational investigation on the effects of pharmaceutical polymers on the structure and dynamics of interleukin2 in heat stress. J. Biomol. Struct. Dyn. 2021, 39, 4536–4546. [Google Scholar] [CrossRef] [PubMed]
- Lee, H. Effect of polyelectrolyte size on multilayer conformation and dynamics at different temperatures and salt concentrations. J. Mol. Graph. Model. 2016, 70, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Hao Feng, Y.; Ling Liu, J.; Zhu, D.D.; Hao, Y.Y.; Dong Guo, X. Multiscale simulations of drug distributions in polymer dissolvable microneedles. Colloids Surf. B Biointerfaces 2020, 189, 110844. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.H.; Zhang, X.P.; Li, W.X.; Guo, X.D. Stability and Diffusion Properties of Insulin in Dissolvable Microneedles: A Multiscale Simulation Study. Langmuir 2021, 37, 9244–9252. [Google Scholar] [CrossRef] [PubMed]
- GhavamiNejad, A.; Lu, B.; Samarikhalaj, M.; Liu, J.F.; Mirzaie, S.; Pereira, S.; Zhou, L.; Giacca, A.; Wu, X.Y. Transdermal delivery of a somatostatin receptor type 2 antagonist using microneedle patch technology for hypoglycemia prevention. Drug Deliv. Transl. Res. 2022, 12, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Farag, M.M.; Abd El Malak, N.S.; Yehia, S.A.; Ahmed, M.A. Hyaluronic Acid Conjugated Metformin-Phospholipid Sonocomplex: A Biphasic Complexation Approach to Correct Hypoxic Tumour Microenvironment. Int. J. Nanomed. 2021, 16, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Bhardwaj, T.; Kumar, A.; Gehi, B.R.; Kapuganti, S.K.; Garg, N.; Nath, G.; Giri, R. Reprofiling of approved drugs against SARS-CoV-2 main protease: An in-silico study. J. Biomol. Struct. Dyn. 2022, 40, 3170–3184. [Google Scholar] [CrossRef]
- Nag, N.; Chetri, P.B.; Uversky, V.N.; Giri, R.; Tripathi, T. Chapter 31—Experimental methods to study intrinsically disordered proteins. In Advances in Protein Molecular and Structural Biology Methods; Tripathi, T., Dubey, V.K., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 505–533. [Google Scholar]
- Shrestha, U.R.; Smith, J.C.; Petridis, L. Full structural ensembles of intrinsically disordered proteins from unbiased molecular dynamics simulations. Commun. Biol. 2021, 4, 243. [Google Scholar] [CrossRef]
- Chu, X.; Nagpal, S.; Muñoz, V. Molecular Simulations of Intrinsically Disordered Proteins and Their Binding Mechanisms. In Protein Folding: Methods and Protocols; Muñoz, V., Ed.; Springer US: New York, NY, USA, 2022; pp. 343–362. [Google Scholar]
- Gong, X.; Zhang, Y.; Chen, J. Advanced Sampling Methods for Multiscale Simulation of Disordered Proteins and Dynamic Interactions. Biomolecules 2021, 11, 1416. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description of Available Coordinates 2 | Method | PDB ID [Reference] |
|---|---|---|
| 8-mer from sodium fiber | Fiber diffraction | 3HYA [49] |
| 3-mer | Solution NMR | 1HUA [37] |
| Two 2-mers + Streptococcus pneumoniae hyaluronate lyase | X-ray diffraction | 1C82 [50] |
| 2-mer + chondroitin AC lyase | X-ray diffraction | 1HM3 [51] |
| 6-mer + Streptococcus pneumoniae hyaluronate lyase | X-ray diffraction | 1LOH [52] |
| 4-mer + Streptococcus pneumoniae hyaluronate lyase | X-ray diffraction | 1LXK [52] |
| 6-mer + Streptococcus agalactiae hyaluronate lyase | X-ray diffraction | 1LXM [53] |
| 6-mer + Streptococcus pneumoniae hyaluronate lyase W291A/W292A mutant | X-ray diffraction | 1N7Q [54] |
| 6-mer + Streptococcus pneumoniae hyaluronate lyase W291A/W292A/F343V mutant | X-ray diffraction | 1N7R [54] |
| 8-mer | Solution NMR | 2BVK [55] |
| 7-mer + murine CD44 hyaluronan binding domain | X-ray diffraction | 2JCQ [56] |
| 7-mer + murine CD44 hyaluronan binding domain | X-ray diffraction | 2JCR [56] |
| 4-mer + murine CD44 hyaluronan binding domain | X-ray diffraction | 4MRD [57] |
| 2-mer + Streptobacillus moniliformis solute-binding protein | X-ray diffraction | 6INZ [58] |
| Six 4-mers + bacteriophage hyaluronan lyase | X-ray diffraction | 6WWX [59] |
| Two 4-mers + one 5-mer + two 6-mers + bacteriophage hyaluronan lyase | X-ray diffraction | 6WXA [60] |
| Three 4-mers + three 8-mers + bacteriophage hyaluronan lyase | X-ray diffraction | 6X3M [61] |
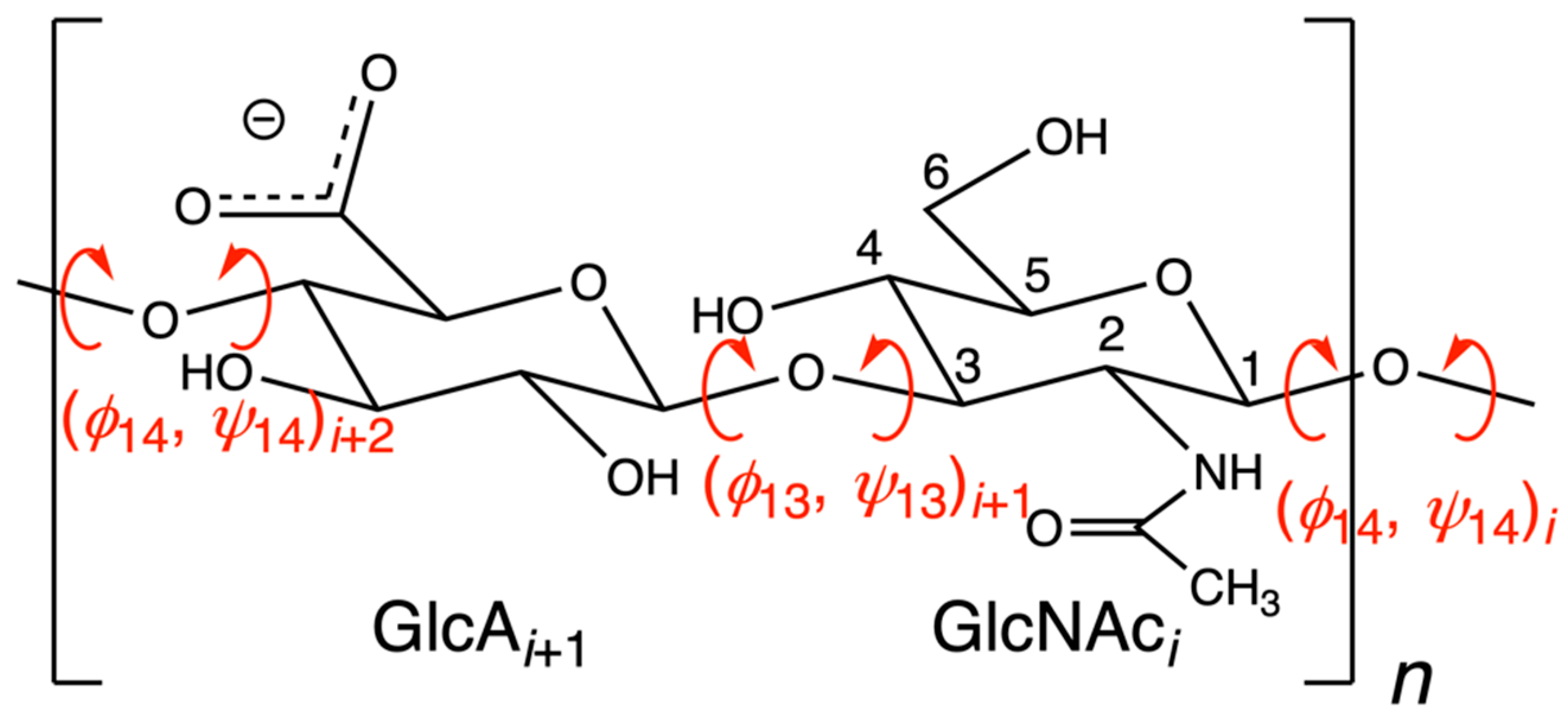
| Index | i | i + 1 | i + 2 | i + 3 | i + 4 | i + 5 | i + 6 | i + 7 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDB ID | Chain | ϕ14 | ψ14 | ϕ13 | ψ13 | ϕ14 | ψ14 | ϕ13 | ψ13 | ϕ14 | ψ14 | ϕ13 | ψ13 | ϕ14 | ψ14 | ϕ13 | ψ13 |
| 3HYA | A | −76.8 | 129.1 | −52.2 | −126.4 | −80.8 | 127.6 | −50.6 | −118.6 | −76.8 | 129.1 | −52.2 | −126.4 | −80.8 | 127.6 | ||
| 1HUA | model 2 | −73.8 | 126.8 | −79.8 | −106.5 | ||||||||||||
| 1C82 | B | −63.0 | −116.5 | ||||||||||||||
| C | −94.8 | −127.6 | |||||||||||||||
| 1HM3 | D | −72.9 | −126.1 | ||||||||||||||
| 1LOH | B | −81.2 | −141.2 | −58.4 | 101.8 | −60.4 | −131.6 | −67.7 | 51.2 | −63.4 | −158.5 | ||||||
| 1LXK | B | −83.1 | −139.3 | −60.1 | 104.4 | −58.5 | −118.9 | ||||||||||
| 1LXM | B | −98.7 | −123.2 | −57.5 | 102.3 | −51.6 | −117.5 | −54.3 | 69.7 | −61.4 | 119.2 | ||||||
| 1N7Q | B | −94.4 | −122.9 | −63.2 | 80.0 | −60.0 | −115.6 | −63.9 | 51.3 | −67.0 | −151.5 | ||||||
| 1N7R | B | −89.2 | −128.0 | −63.2 | 97.5 | −62.5 | −132.1 | −61.3 | 54.5 | −68.1 | −157.7 | ||||||
| 2BVK | A | −71.2 | 126.0 | −68.1 | −110.3 | −71.1 | 126.6 | −68.1 | −110.3 | −71.1 | 126.6 | −68.0 | −110.3 | −71.1 | 126.6 | ||
| 2JCQ | B | −86.2 | −154.1 | −73.7 | 108.2 | −69.0 | −114.6 | −88.6 | 133.7 | −78.1 | −119.4 | −68.9 | 114.5 | ||||
| 2JCR | B | −91.9 | −138.1 | −71.2 | 107.9 | −71.3 | −117.1 | −79.0 | 129.0 | −82.5 | −123.6 | −75.4 | 103.6 | ||||
| 4MRD | B | −93.1 | −151.0 | −79.4 | 111.9 | −67.5 | −132.8 | ||||||||||
| 6INZ | B | −92.5 | −143.8 | ||||||||||||||
| 6WWX | D | −79.2 | −119.9 | −96.2 | 123.5 | −79.8 | −131.3 | ||||||||||
| E | −73.4 | −106.0 | −84.8 | 102.4 | −95.7 | −111.7 | |||||||||||
| F | −77.9 | −105.5 | −91.4 | 105.7 | −85.1 | −110.2 | |||||||||||
| G | −85.2 | −116.1 | −88.7 | 123.5 | −86.2 | −126.4 | |||||||||||
| H | −79.7 | −124.1 | −93.6 | 128.0 | −85.7 | −131.7 | |||||||||||
| I | −76.5 | −108.2 | −84.6 | 105.5 | −73.2 | −112.4 | |||||||||||
| 6WXA | F | −83.8 | −113.2 | −91.7 | 111.0 | −91.2 | −97.0 | ||||||||||
| H | −77.6 | −106.5 | −89.2 | 103.9 | −66.3 | −116.6 | |||||||||||
| E | −71.9 | −130.6 | −90.1 | 128.0 | −90.3 | −124.2 | −84.1 | 135.4 | |||||||||
| D | −83.1 | −119.3 | −93.2 | 120.1 | −80.2 | −130.8 | −92.2 | 137.8 | −66.7 | −125.7 | |||||||
| G | −82.5 | −123.9 | −88.1 | 125.8 | −80.1 | −123.4 | −84.0 | 124.2 | −64.5 | −125.4 | |||||||
| 6X3M | J | −78.6 | −123.1 | −88.5 | 109.7 | −81.1 | −130.5 | ||||||||||
| N | −27.1 | −125.3 | −75.5 | 64.3 | −116.6 | −154.8 | |||||||||||
| O | −64.0 | −137.7 | −102.8 | 74.1 | −10.8 | −112.1 | |||||||||||
| K | −43.8 | −124.8 | −67.6 | 101.0 | 80.7 | −130.0 | −97.1 | −45.6 | −96.6 | −107.3 | 88.7 | 166.0 | −64.7 | −146.5 | |||
| L | −63.2 | −132.7 | −64.5 | −41.4 | −132.4 | −144.8 | −110.4 | −41.6 | −72.3 | −141.1 | −67.8 | 109.7 | −73.5 | −137.0 | |||
| M | −146.9 | 155.6 | 17.9 | 147.3 | −87.4 | −128.9 | −65.6 | 116.2 | −64.1 | −93.3 | −115.1 | 97.6 | −73.0 | −135.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guvench, O. Atomic-Resolution Experimental Structural Biology and Molecular Dynamics Simulations of Hyaluronan and Its Complexes. Molecules 2022, 27, 7276. https://doi.org/10.3390/molecules27217276
Guvench O. Atomic-Resolution Experimental Structural Biology and Molecular Dynamics Simulations of Hyaluronan and Its Complexes. Molecules. 2022; 27(21):7276. https://doi.org/10.3390/molecules27217276
Chicago/Turabian StyleGuvench, Olgun. 2022. "Atomic-Resolution Experimental Structural Biology and Molecular Dynamics Simulations of Hyaluronan and Its Complexes" Molecules 27, no. 21: 7276. https://doi.org/10.3390/molecules27217276
APA StyleGuvench, O. (2022). Atomic-Resolution Experimental Structural Biology and Molecular Dynamics Simulations of Hyaluronan and Its Complexes. Molecules, 27(21), 7276. https://doi.org/10.3390/molecules27217276