

Comparison of Intermolecular Interactions of Irreversible and Reversible Inhibitors with Bruton’s Tyrosine Kinase via Molecular Dynamics Simulations

Abstract
:1. Introduction
2. Results
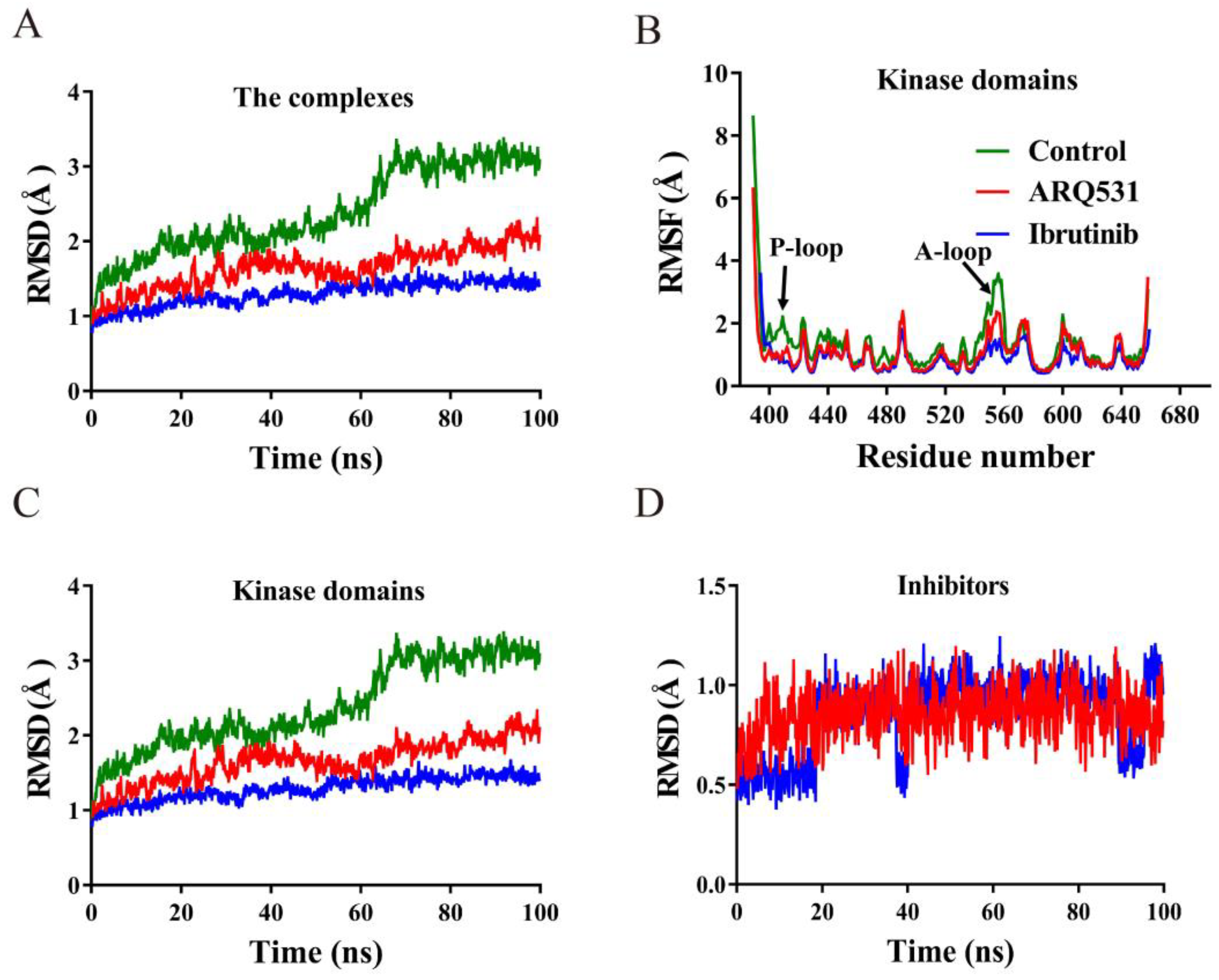
2.1. Conformational Stability of Simulated Systems
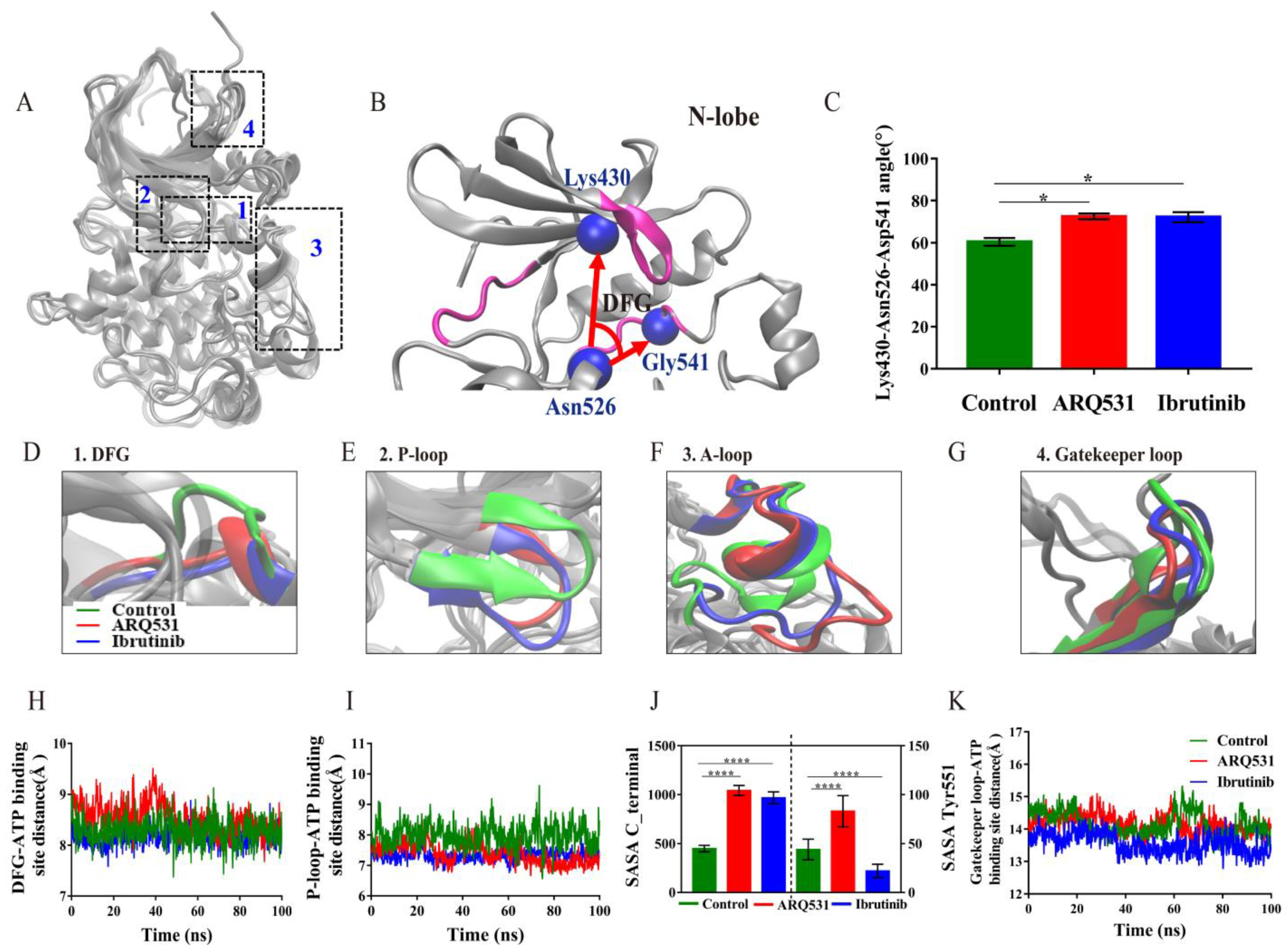
2.2. Local Conformational Changes in BTK upon Binding of Different Small Molecule Inhibitors
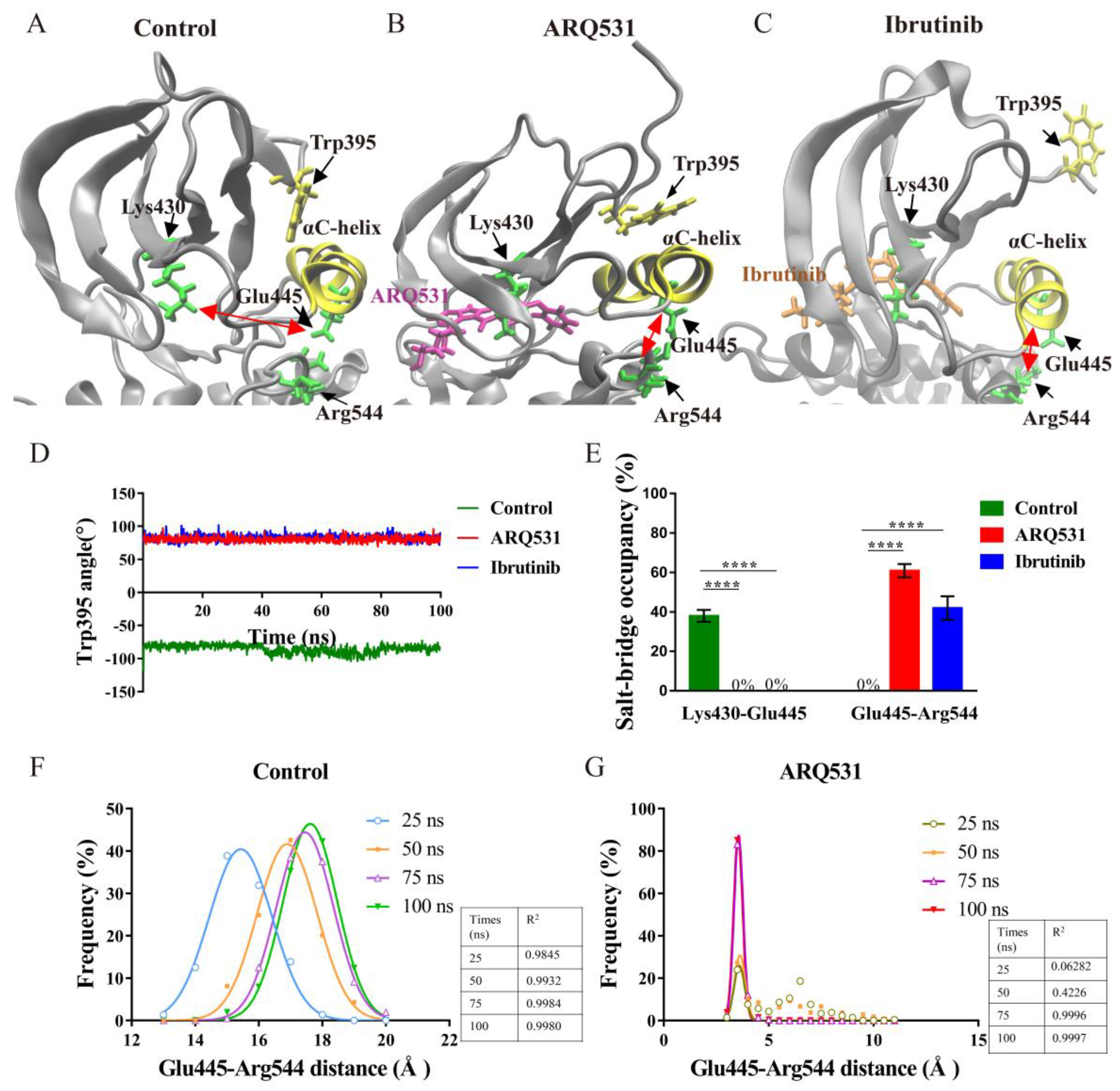
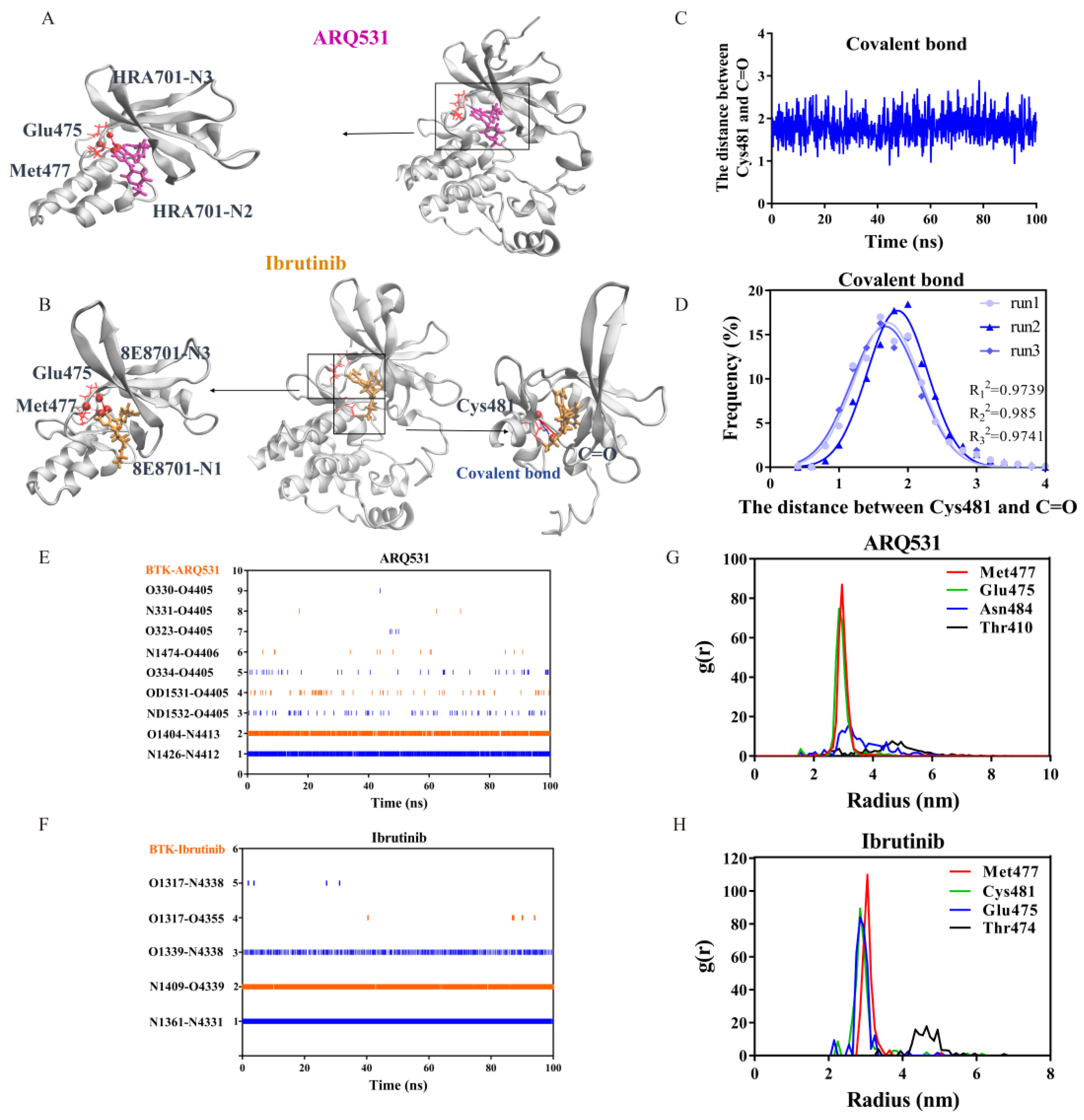
2.3. Key Intermolecular Interactions in BTK Kinase Domain and Inhibitor Complexes
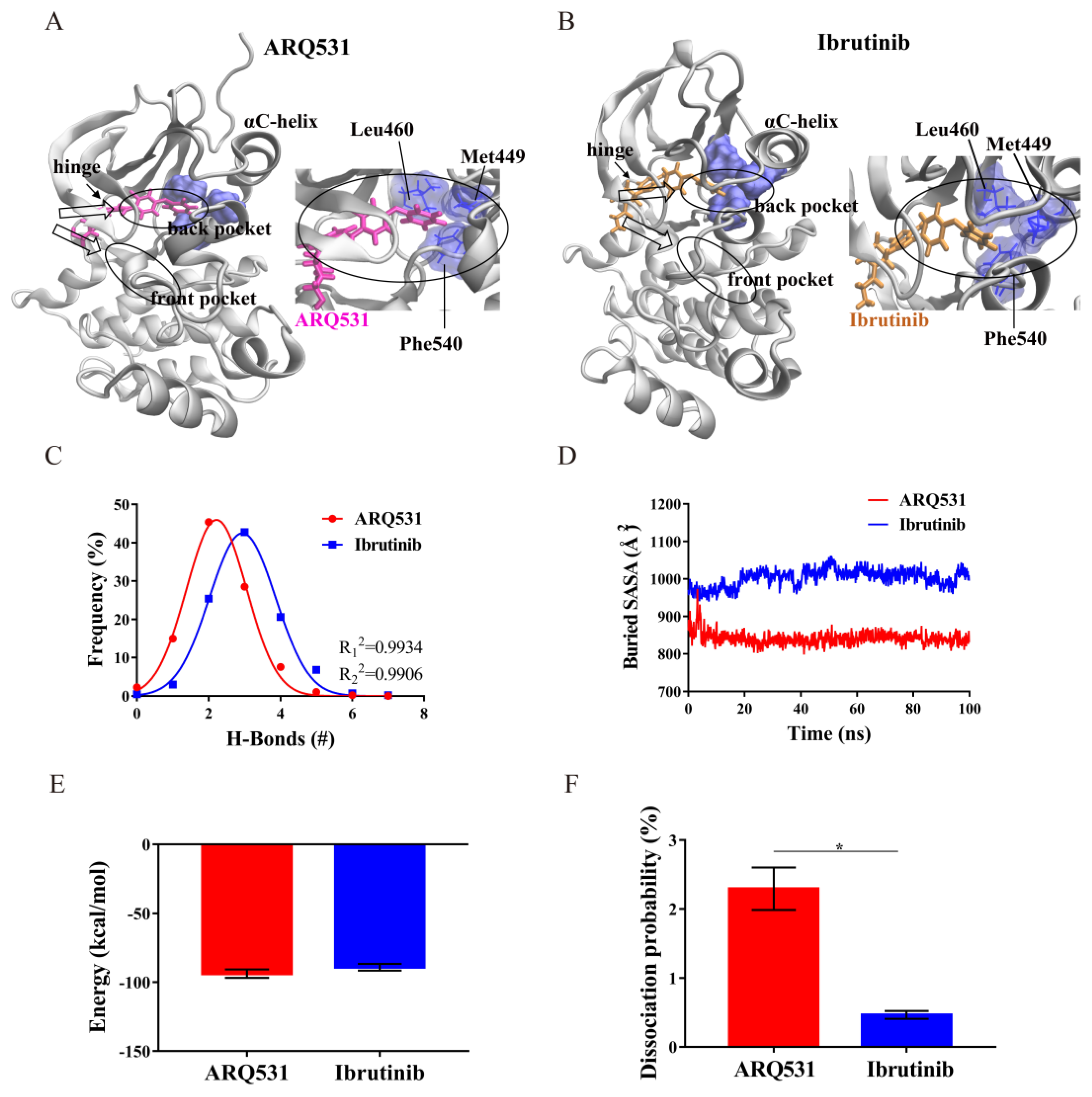
2.4. Binding Modes and Interaction Energies of Small Molecule Inhibitors
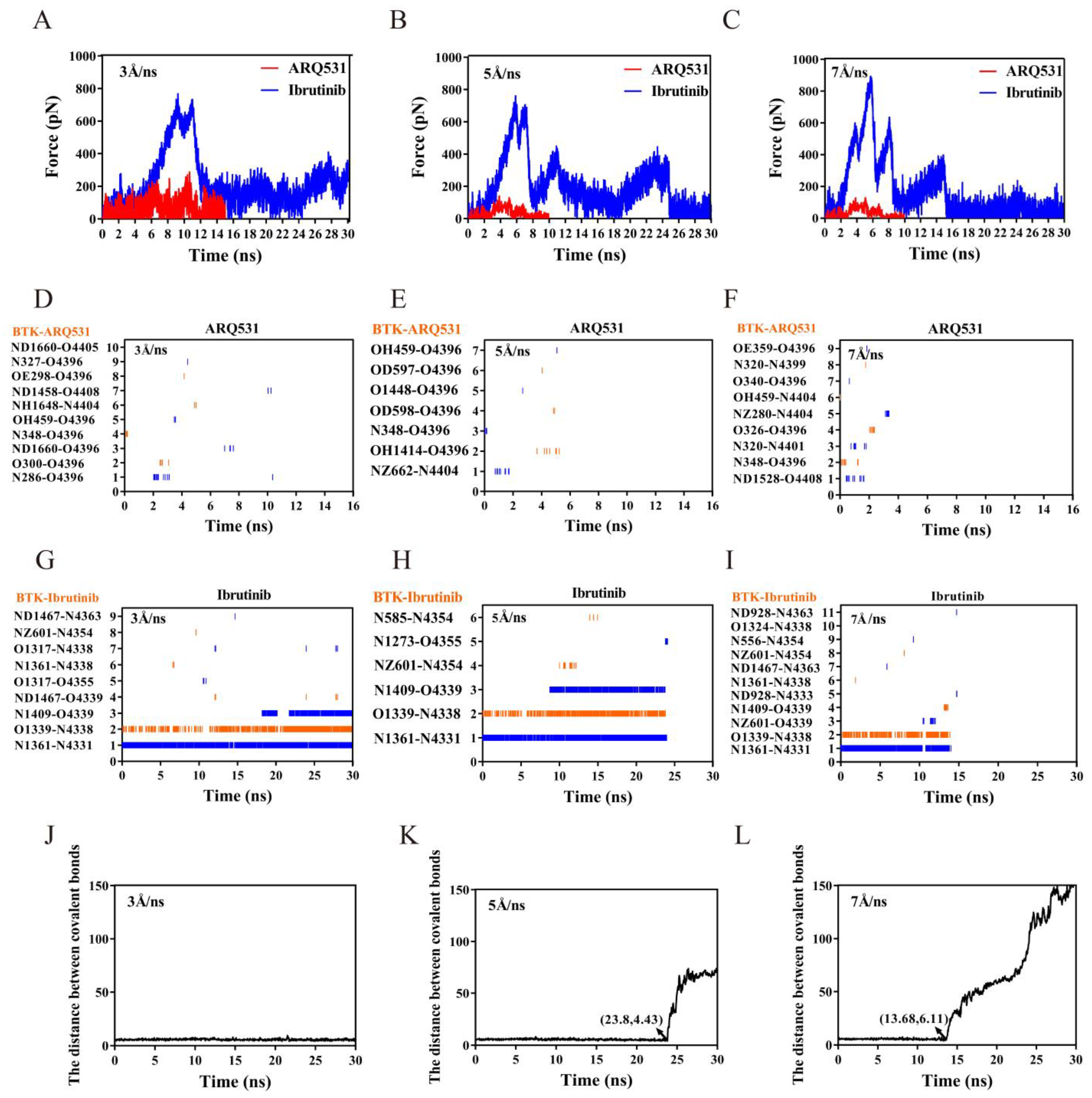
2.5. Dissociation Kinetics of Inhibitors
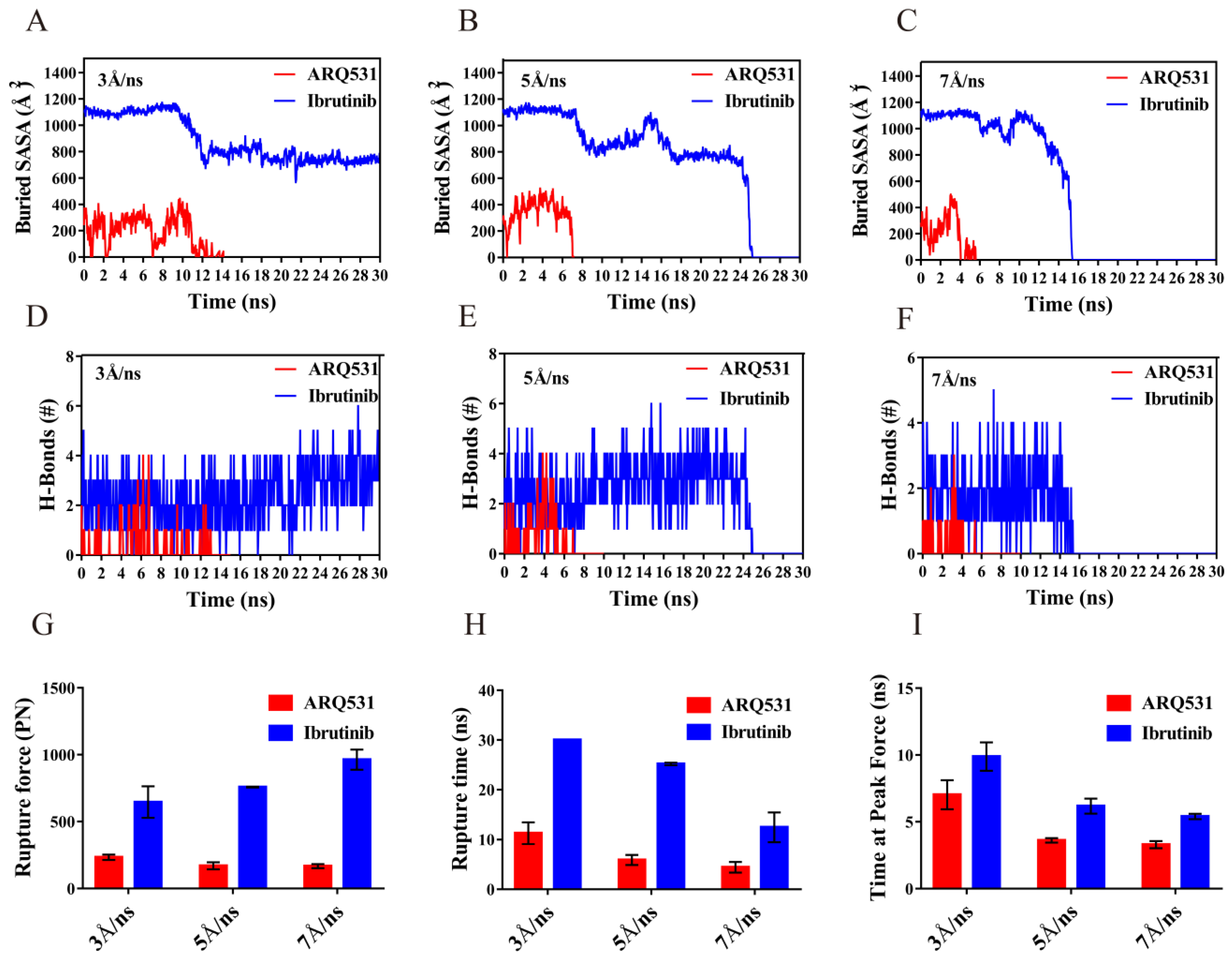
2.6. Quantification of Dissociation of Different Inhibitors from BTK Kinase Domain
3. Discussion
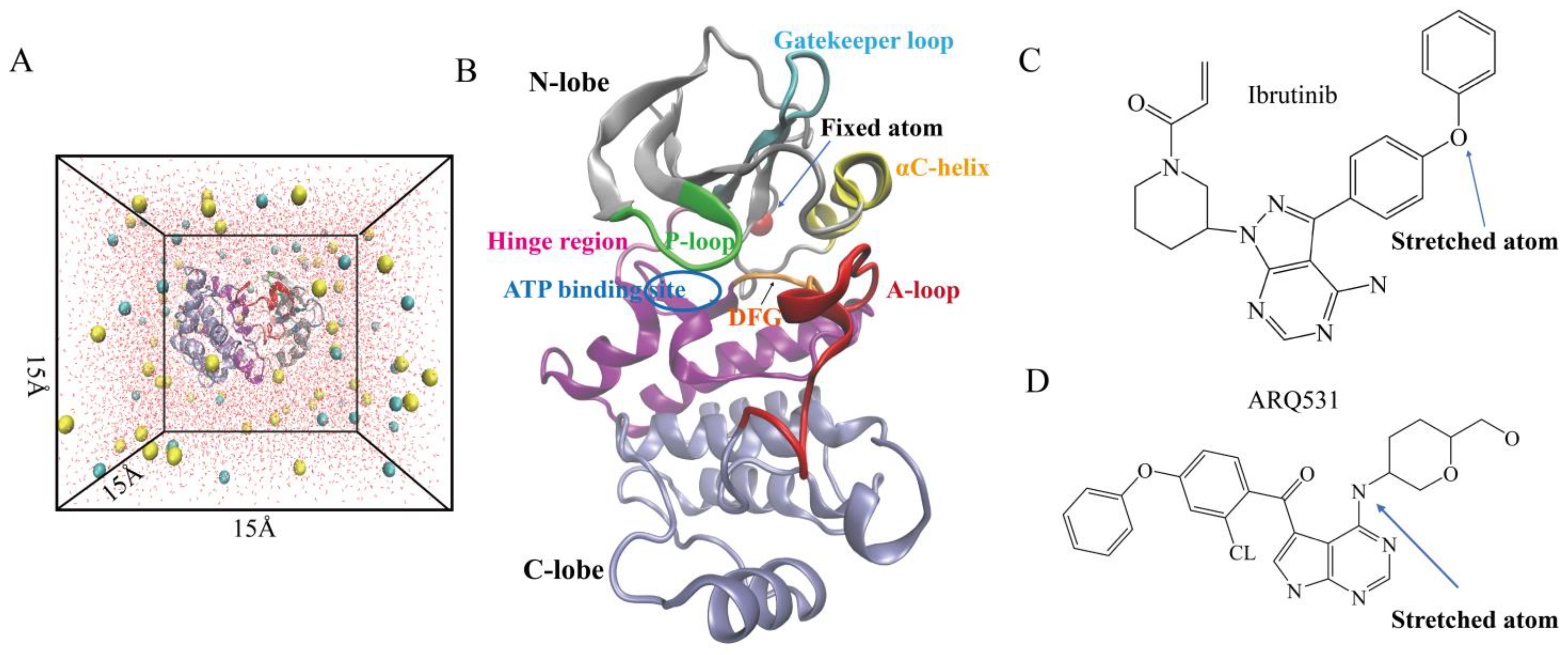
4. Materials and Methods
4.1. System Setup
4.2. Molecular Dynamics Simulations
4.3. Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mohamed, A.J.; Yu, L.; Backesjo, C.M.; Vargas, L.; Faryal, R.; Aints, A.; Christensson, B.; Berglof, A.; Vihinen, M.; Nore, B.F.; et al. Bruton’s tyrosine kinase (Btk): Function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009, 228, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, S.; Saffran, D.C.; Rawlings, D.J.; Parolini, O.; Allen, R.C.; Klisak, I.; Sparkes, R.S.; Kubagawa, H.; Mohandas, T.; Quan, S.; et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. 1993. J. Immunol. 2012, 188, 2936–2947. [Google Scholar] [PubMed]
- Rawlings, D.J.; Saffran, D.C.; Tsukada, S.; Largaespada, D.A.; Grimaldi, J.C.; Cohen, L.; Mohr, R.N.; Bazan, J.F.; Howard, M.; Copeland, N.G.; et al. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science 1993, 261, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Vogan, E.M.; Nocka, L.M.; Rosen, C.E.; Zorn, J.A.; Harrison, S.C.; Kuriyan, J. Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. Elife 2015, 4, e06074. [Google Scholar] [CrossRef]
- Qiu, S.M.; Liu, Y.F.; Li, Q.H. A mechanism for localized dynamics-driven activation in Bruton’s tyrosine kinase. Roy. Soc. Open Sci. 2021, 8, 210066. [Google Scholar] [CrossRef]
- Hopkins, B.T.; Bame, E.; Bajrami, B.; Black, C.; Bohnert, T.; Boiselle, C.; Burdette, D.; Burns, J.C.; Delva, L.; Donaldson, D.; et al. Discovery and Preclinical Characterization of BIIB091, a Reversible, Selective BTK Inhibitor for the Treatment of Multiple Sclerosis. J. Med. Chem. 2022, 65, 1206–1224. [Google Scholar] [CrossRef]
- Dinh, M.; Grunberger, D.; Ho, H.; Tsing, S.Y.; Shaw, D.; Lee, S.; Barnett, J.; Hill, R.J.; Swinney, D.C.; Bradshaw, J.M. Activation mechanism and steady state kinetics of Bruton’s tyrosine kinase. J. Biol. Chem. 2007, 282, 8768–8776. [Google Scholar] [CrossRef] [Green Version]
- Kuglstatter, A.; Wong, A.; Tsing, S.; Lee, S.W.; Lou, Y.; Villasenor, A.G.; Bradshaw, J.M.; Shaw, D.; Barnett, J.W.; Browner, M.F. Insights into the conformational flexibility of Bruton’s tyrosine kinase from multiple ligand complex structures. Protein Sci. 2011, 20, 428–436. [Google Scholar] [CrossRef] [Green Version]
- Sultan, M.M.; Denny, R.A.; Unwalla, R.; Lovering, F.; Pande, V.S. Millisecond dynamics of BTK reveal kinome-wide conformational plasticity within the apo kinase domain. Sci. Rep. 2017, 7, 15604. [Google Scholar] [CrossRef] [Green Version]
- Block, H.; Zarbock, A. The role of the tec kinase Bruton’s tyrosine kinase (BTK) in leukocyte recruitment. Int. Rev. Immunol. 2012, 31, 104–118. [Google Scholar] [CrossRef]
- Singh, S.P.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef]
- Liu, X.J.; Liu, X.; Pang, X.J.; Yuan, X.Y.; Yu, G.X.; Li, Y.R.; Guan, Y.F.; Zhang, Y.B.; Song, J.; Zhang, Q.R.; et al. Progress in the development of small molecular inhibitors of the Bruton’s tyrosine kinase (BTK) as a promising cancer therapy. Biorg. Med. Chem. 2021, 47, 116358. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Ibrutinib inhibition of Bruton’s tyrosine kinase (BTK) in the treatment of B cell neoplasms. Pharmacol. Res. 2016, 113, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Narita, Y.; Nagane, M.; Mishima, K.; Terui, Y.; Arakawa, Y.; Yonezawa, H.; Asai, K.; Fukuhara, N.; Sugiyama, K.; Shinojima, N.; et al. Phase I/II study of tirabrutinib, a second-generation Bruton’s tyrosine kinase inhibitor, in relapsed/refractory primary central nervous system lymphoma. Neuro Oncol. 2021, 23, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Whang, J.A.; Chang, B.Y. Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov. Today 2014, 19, 1200–1204. [Google Scholar] [CrossRef]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat. Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef]
- Akinleye, A.; Chen, Y.M.; Mukhi, N.; Song, Y.P.; Liu, D.L. Ibrutinib and novel BTK inhibitors in clinical development. J. Hematol. Oncol. 2013, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTK(C481S)-mediated resistance to Ibrutinib in chronic lymphocytic leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.J.; Zhang, M.Z.; Liu, D.L. Acalabrutinib (ACP-196): A selective second-generation BTK inhibitor. J. Hematol. Oncol. 2016, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- Reiff, S.D.; Mantel, R.; Smith, L.L.; Greene, J.T.; Muhowski, E.M.; Fabian, C.A.; Goettl, V.M.; Tran, M.; Harrington, B.K.; Rogers, K.A.; et al. The BTK inhibitor ARQ 531 targets Ibrutinib-resistant CLL and Richter transformation. Cancer Discov. 2018, 8, 1300–1315. [Google Scholar] [CrossRef]
- Johnson, A.R.; Kohli, P.B.; Katewa, A.; Gogol, E.; Belmont, L.D.; Choy, R.; Penuel, E.; Burton, L.; Eigenbrot, C.; Yu, C.; et al. Battling BTK mutants with noncovalent inhibitors that overcome Cys481 and Thr474 mutations. ACS Chem. Biol. 2016, 11, 2897–2907. [Google Scholar] [CrossRef]
- Reiff, S.D.; Muhowski, E.M.; Guinn, D.; Lehman, A.; Fabian, C.A.; Cheney, C.; Mantel, R.; Smith, L.; Johnson, A.J.; Young, W.B.; et al. Noncovalent inhibition of C481S Bruton’s tyrosine kinase by GDC-0853: A new treatment strategy for ibrutinib-resistant CLL. Blood 2018, 132, 1039–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirley, M. Bruton Tyrosine Kinase Inhibitors in B-Cell Malignancies: Their Use and Differential Features. Target. Oncol. 2021, 17, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Jebaraj, B.M.C.; Muller, A.; Dheenadayalan, R.P.; Endres, S.; Roessner, P.M.; Seyfried, F.; Walliser, C.; Wist, M.; Qi, J.; Tausch, E.; et al. Evaluation of vecabrutinib as a model for non-covalent BTK/ITK inhibition for treatment of chronic lymphocytic leukemia. Blood 2022, 139, 859–875. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, D.J.; Liu, Y.T.; Arduini, R.M.; Hession, C.A.; Miatkowski, K.; Wildes, C.P.; Cullen, P.F.; Hong, V.; Hopkins, B.T.; Mertsching, E.; et al. Structures of human Bruton’s tyrosine kinase in active and inactive conformations suggest a mechanism of activation for TEC family kinases. Protein Sci. 2010, 19, 429–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, R.E.; Wales, T.E.; Fulton, D.B.; Engen, J.R.; Andreotti, A.H. Achieving a graded immune response: BTK adopts a range of active/inactive conformations dictated by multiple interdomain contacts. Structure 2017, 25, 1481–1494.e4. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Liu, G.; Zhou, H.; Fang, X.; Fang, Y.; Wu, J. Computer prediction of paratope on antithrombotic antibody 10B12 and epitope on platelet glycoprotein VI via molecular dynamics simulation. Biomed. Eng. Online 2016, 15, 152. [Google Scholar] [CrossRef] [Green Version]
- Bodor, C.; Kotmayer, L.; Laszlo, T.; Takacs, F.; Barna, G.; Kiss, R.; Sebestyen, E.; Nagy, T.; Hegyi, L.L.; Mikala, G.; et al. Screening and monitoring of the BTK(C481S) mutation in a real-world cohort of patients with relapsed/refractory chronic lymphocytic leukaemia during ibrutinib therapy. Br. J. Haematol. 2021, 194, 355–364. [Google Scholar] [CrossRef]
- Lamichhane, T.R.; Ghimire, M.P. Evaluation of SARS-CoV-2 main protease and inhibitor interactions using dihedral angle distributions and radial distribution function. Heliyon 2021, 7, e08220. [Google Scholar] [CrossRef]
- Joseph, R.E.; Amatya, N.; Fulton, D.B.; Engen, J.R.; Wales, T.E.; Andreotti, A. Differential impact of BTK active site inhibitors on the conformational state of full-length BTK. Elife 2020, 9, e60470. [Google Scholar] [CrossRef]
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Tian, D.; Ren, X.; Ding, S.; Jia, M.; Xin, M.; Thareja, S. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur. J. Med. Chem. 2018, 151, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Yu, Z.; Jia, Y.; Fang, H.; Shao, S.; Huang, L.; Feng, Y. Efficacy and safety of ibrutinib in diffuse large B-cell lymphoma: A single-arm meta-analysis. Crit. Rev. Oncol. Hematol. 2020, 152, 103010. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Ibrutinib: A review in chronic lymphocytic leukaemia. Drugs 2017, 77, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.J.; Kulkarni, V.M. Exploration of structural requirements for the inhibition of VEGFR-2 tyrosine kinase: Binding site analysis of type II, ‘DFG-out’ inhibitors. J. Biomol Struct Dyn. 2022, 40, 5712–5727. [Google Scholar] [CrossRef]
- Estupinan, H.Y.; Wang, Q.; Berglof, A.; Schaafsma, G.C.P.; Shi, Y.; Zhou, L.; Mohammad, D.K.; Yu, L.; Vihinen, M.; Zain, R.; et al. BTK gatekeeper residue variation combined with cysteine 481 substitution causes super-resistance to irreversible inhibitors acalabrutinib, ibrutinib and zanubrutinib. Leukemia 2021, 35, 1317–1329. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein data bank (PDB): The single global macromolecular structure archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.J.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem. 2017, 38, 1114–1124. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Fang, X.; Fang, Y.; Liu, L.; Liu, G.; Wu, J. Mapping paratope on antithrombotic antibody 6B4 to epitope on platelet glycoprotein Ibalpha via molecular dynamic simulations. PLoS ONE 2012, 7, e42263. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Klauda, J.B. Update of the CHARMM36 united atom chain model for hydrocarbons and phospholipids. J. Phys. Chem. B 2020, 124, 6797–6812. [Google Scholar] [CrossRef] [PubMed]
- Isralewitz, B.; Gao, M.; Schulten, K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001, 11, 224–230. [Google Scholar] [CrossRef]
- Florin, E.L.; Moy, V.T.; Gaub, H.E. Adhesion forces between individual ligand-receptor pairs. Science 1994, 264, 415–417. [Google Scholar] [CrossRef] [Green Version]
- Chng, C.P.; Kitao, A. Mechanical unfolding of bacterial flagellar filament protein by molecular dynamics simulation. J. Mol. Graph. Model. 2010, 28, 548–554. [Google Scholar] [CrossRef]
- Sargsyan, K.; Grauffel, C.; Lim, C. How molecular size impacts RMSD applications in molecular dynamics simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]
- Margreitter, C.; Oostenbrink, C. MDplot: Visualise molecular dynamics. R J. 2017, 9, 164–186. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hou, T. Develop and test a solvent accessible surface area-based model in conformational entropy calculations. J. Chem. Inf. Model. 2012, 52, 1199–1212. [Google Scholar] [CrossRef] [Green Version]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef]
- Herman, S.E.M.; Montraveta, A.; Niemann, C.U.; Mora-Jensen, H.; Gulrajani, M.; Krantz, F.; Mantel, R.; Smith, L.L.; McClanahan, F.; Harrington, B.K.; et al. The Bruton Tyrosine Kinase (BTK) Inhibitor Acalabrutinib Demonstrates Potent On-Target Effects and Efficacy in Two Mouse Models of Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 2831–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Sun, Z.J.; Liu, Y.; Guo, M.M.; Zhang, Y.L.; Zhou, D.P.; Zhang, B.; Su, D.; Zhang, S.; Han, J.; et al. BGB-3111 is a novel and highly selective Bruton’s tyrosine kinase (BTK) inhibitor. Cancer Res. 2015, 75, 2597. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Song, Y.P.; Li, Z.J.; Yang, S.M.; Liu, L.H.; Hu, Y.; Zhang, W.; Zhou, J.F.; Gao, S.J.; Ding, K.Y.; et al. Safety, Tolerability and Efficacy of Orelabrutinib, Once a Day, to Treat Chinese Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia/Small Cell Leukemia. Blood 2019, 134, 4319. [Google Scholar] [CrossRef]
- Francesco, M.R.; Wong, M.; LaStant, J.; Finkle, D.; Loewenstein, N.; Macsata, R.; Lindstrom, M.M.; Shu, J.; Ton, T.; Zhu, J.; et al. PRN2246, a potent and selective blood brain barrier penetrating BTK inhibitor, exhibits efficacy in central nervous system immunity. Mult. Scler. J. 2017, 23, 989. [Google Scholar]
- Evans, E.K.; Tester, R.; Aslanian, S.; Karp, R.; Sheets, M.; Labenski, M.T.; Witowski, S.R.; Lounsbury, H.; Chaturvedi, P.; Mazdiyasni, H.; et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J. Pharmacol. Exp. Ther. 2013, 346, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Kokabee, L.; Wang, X.; Sevinsky, C.J.; Wang, W.L.; Cheu, L.; Chittur, S.V.; Karimipoor, M.; Tenniswood, M.; Conklin, D.S. Bruton’s tyrosine kinase is a potential therapeutic target in prostate cancer. Cancer Biol. Ther. 2015, 16, 1604–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.Y.; Park, K.T.; Jang, S.Y.; Lee, K.H.; Byun, J.Y.; Suh, K.H.; Lee, Y.M.; Kim, Y.H.; Hwang, K.W. HM71224, a selective Bruton’s tyrosine kinase inhibitor, attenuates the development of murine lupus. Arthritis Res. Ther. 2017, 19, 211. [Google Scholar] [CrossRef]
- Watterson, S.H.; Liu, Q.; Beaudoin, B.M.; Batt, D.G.; Li, L.; Pattoli, M.A.; Skala, S.; Cheng, L.; Obermeier, M.T.; Moore, R.; et al. Discovery of Branebrutinib (BMS-986195): A Strategy for Identifying a Highly Potent and Selective Covalent Inhibitor Providing Rapid In Vivo Inactivation of Bruton’s Tyrosine Kinase (BTK). J. Med. Chem. 2019, 62, 3228–3250. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, S.; Ghosh, S.; Sudbeck, E.A.; Zheng, Y.; Downs, S.; Hupke, M.; Uckun, F.M. Rational design and synthesis of a novel anti-leukemic agent targeting Bruton’s tyrosine kinase (BTK), LFM-A13 [alpha-cyano-beta-hydroxy-beta-methyl-N-(2, 5-dibromophenyl)propenamide]. J. Biol. Chem. 1999, 274, 9587–9599. [Google Scholar] [CrossRef] [Green Version]
- Labenski, M.; Chaturvedi, P.; Evans, E.; Mazdiyazni, H.; Sheets, M.; Aslanian, S.; Nacht, M.; Petter, R.; Singh, J.; Westlin, W. In vitro reactivity assessment of covalent drugs targeting Bruton’s tyrosine kinase. Drug Metab. Rev. 2011, 43, 140. [Google Scholar]
- Goess, C.; Harris, C.M.; Murdock, S.; McCarthy, R.W.; Sampson, E.; Twomey, R.; Mathieu, S.; Mario, R.; Perham, M.; Goedken, E.R.; et al. ABBV-105, a selective and irreversible inhibitor of Bruton’s tyrosine kinase, is efficacious in multiple preclinical models of inflammation. Mod. Rheumatol. 2019, 29, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.D.; Qiu, H.; Askew, B.C.; Bender, A.T.; Brugger, N.; Camps, M.; Dhanabal, M.; Dutt, V.; Eichhorn, T.; Gardberg, A.S.; et al. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J. Med. Chem. 2019, 62, 7643–7655. [Google Scholar] [CrossRef]
- Pulz, R.; Angst, D.; Dawson, J.; Gessier, F.; Gutmann, S.; Hersperger, R.; Hinniger, A.; Janser, P.; Koch, G.; Revesz, L.; et al. Design of Potent and Selective Covalent Inhibitors of Bruton’s Tyrosine Kinase Targeting an Inactive Conformation. ACS Med. Chem. Lett. 2019, 10, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Metrick, C.M.; Gu, C.; Hoemberger, M.; Bajrami, B.; Bame, E.; Huang, J.; Mingueneau, M.; Murugan, P.; Santoro, J.C.; et al. Optimization of a novel piperazinone series as potent selective peripheral covalent BTK inhibitors. Bioorg. Med. Chem. Lett. 2022, 60, 128549. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Li, Q.; Zhao, M.; Fan, G.; Zhao, J.; Zhang, D.; Yang, S.; Zhang, S.; Gao, D.; Mao, L.; et al. Discovery of 1-Amino-1H-imidazole-5-carboxamide Derivatives as Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitors. J. Med. Chem. 2021, 64, 16242–16270. [Google Scholar] [CrossRef]
- Eathiraj, S.; Savage, R.; Yu, Y.; Schwartz, B.; Woyach, J.; Johnson, A.; Reiff, S.; Abbadessa, G. Targeting Ibrutinib-Resistant BTK-C481S Mutation with ARQ 531, a Reversible Non-Covalent Inhibitor of BTK. Clin. Lymphoma Myeloma Leukemia 2016, 16, S47–S48. [Google Scholar] [CrossRef]
- Erickson, R.I.; Schutt, L.K.; Tarrant, J.M.; McDowell, M.; Liu, L.; Johnson, A.R.; Lewin-Koh, S.C.; Hedehus, M.; Ross, J.; Carano, R.A.; et al. Bruton’s Tyrosine Kinase Small Molecule Inhibitors Induce a Distinct Pancreatic Toxicity in Rats. J. Pharmacol. Exp. Ther. 2017, 360, 226–238. [Google Scholar] [CrossRef] [Green Version]
- Gomez, E.B.; Isabel, L.; Rosendahal, M.S.; Rothenberg, S.M.; Andrews, S.W.; Brandhuber, B.J. Loxo-305, a Highly Selective and Non-Covalent Next Generation BTK Inhibitor, Inhibits Diverse BTK C481 Substitution Mutations. Blood 2019, 134, 4644. [Google Scholar] [CrossRef]
- Burger, J.A. Bruton’s tyrosine kinase (BTK) inhibitors in clinical trials. Curr. Hematol. Malig. Rep. 2014, 9, 44–49. [Google Scholar] [CrossRef]
- Smith, P.F.; Krishnarajah, J.; Nunn, P.A.; Hill, R.J.; Karr, D.; Tam, D.; Masjedizadeh, M.; Funk, J.O.; Gourlay, S.G. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br. J. Clin. Pharmacol. 2017, 83, 2367–2376. [Google Scholar] [CrossRef]
- Ma, B.; Bohnert, T.; Otipoby, K.L.; Tien, E.; Arefayene, M.; Bai, J.; Bajrami, B.; Bame, E.; Chan, T.R.; Humora, M.; et al. Discovery of BIIB068: A Selective, Potent, Reversible Bruton’s Tyrosine Kinase Inhibitor as an Orally Efficacious Agent for Autoimmune Diseases. J. Med. Chem. 2020, 63, 12526–12541. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Kim, Y.; Postelnek, J.; Vu, M.D.; Hu, D.Q.; Liao, C.; Bradshaw, M.; Hsu, J.; Zhang, J.; Pashine, A.; et al. RN486, a selective Bruton’s tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J. Pharmacol. Exp. Ther. 2012, 341, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Di Paolo, J.; Barbosa, J.; Rong, H.; Reif, K.; Wong, H. Antiarthritis effect of a novel Bruton’s tyrosine kinase (BTK) inhibitor in rat collagen-induced arthritis and mechanism-based pharmacokinetic/pharmacodynamic modeling: Relationships between inhibition of BTK phosphorylation and efficacy. J. Pharmacol. Exp. Ther. 2011, 338, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.; Scheerens, H.; Li, S.J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.; Grothaus, P.G.; et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Watterson, S.H.; De Lucca, G.V.; Shi, Q.; Langevine, C.M.; Liu, Q.; Batt, D.G.; Beaudoin Bertrand, M.; Gong, H.; Dai, J.; Yip, S.; et al. Discovery of 6-Fluoro-5-(R)-(3-(S)-(8-fluoro-1-methyl-2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl )-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-1H-carbazole-8- carboxamide (BMS-986142): A Reversible Inhibitor of Bruton’s Tyrosine Kinase (BTK) Conformationally Constrained by Two Locked Atropisomers. J. Med. Chem. 2016, 59, 9173–9200. [Google Scholar]
- De Lucca, G.V.; Shi, Q.; Liu, Q.; Batt, D.G.; Beaudoin Bertrand, M.; Rampulla, R.; Mathur, A.; Discenza, L.; D’Arienzo, C.; Dai, J.; et al. Small Molecule Reversible Inhibitors of Bruton’s Tyrosine Kinase (BTK): Structure-Activity Relationships Leading to the Identification of 7-(2-Hydroxypropan-2-yl)-4-[2-methyl-3-(4-oxo-3,4-dihydroquinazolin-3-yl)phenyl]- 9H-carbazole-1-carboxamide (BMS-935177). J. Med. Chem. 2016, 59, 7915–7935. [Google Scholar]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef]
- Gui, F.; Jiang, J.; He, Z.; Li, L.; Li, Y.; Deng, Z.; Lu, Y.; Wu, X.; Chen, G.; Su, J.; et al. A non-covalent inhibitor XMU-MP-3 overrides ibrutinib-resistant Btk(C481S) mutation in B-cell malignancies. Br. J. Pharmacol. 2019, 176, 4491–4509. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex of BTK with ARQ531 | Complex of BTK with Ibrutinib | ||||||
|---|---|---|---|---|---|---|---|
| No | BTK | ARQ531 | RII | No | BTK | Ibrutinib | RII |
| 1 | Met477 | HRA701 | 0.87 | 1 | Met477 | 8E8701 | 0.92 |
| 2 | Glu475 | HRA701 | 0.80 | 2 | Cys481 | 8E8701 | 0.81 |
| 3 | Asn484 | HRA701 | 0.16 | 3 | Glu475 | 8E8701 | 0.72 |
| 4 | Thr410 | HRA701 | 0.05 | 4 | Thr474 | 8E8701 | 0.01 |
| 5 | Cys481 | HRA701 | 0.02 | ||||
| 6 | Leu408 | HRA701 | 0.004 | ||||
| 7 | Gln412 | HRA701 | 0.001 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Qiu, S.; Sun, D.; Guo, P.; Li, Q. Comparison of Intermolecular Interactions of Irreversible and Reversible Inhibitors with Bruton’s Tyrosine Kinase via Molecular Dynamics Simulations. Molecules 2022, 27, 7451. https://doi.org/10.3390/molecules27217451
Yu X, Qiu S, Sun D, Guo P, Li Q. Comparison of Intermolecular Interactions of Irreversible and Reversible Inhibitors with Bruton’s Tyrosine Kinase via Molecular Dynamics Simulations. Molecules. 2022; 27(21):7451. https://doi.org/10.3390/molecules27217451
Chicago/Turabian StyleYu, Xiangfan, Simei Qiu, Dongshan Sun, Pei Guo, and Quhuan Li. 2022. "Comparison of Intermolecular Interactions of Irreversible and Reversible Inhibitors with Bruton’s Tyrosine Kinase via Molecular Dynamics Simulations" Molecules 27, no. 21: 7451. https://doi.org/10.3390/molecules27217451
APA StyleYu, X., Qiu, S., Sun, D., Guo, P., & Li, Q. (2022). Comparison of Intermolecular Interactions of Irreversible and Reversible Inhibitors with Bruton’s Tyrosine Kinase via Molecular Dynamics Simulations. Molecules, 27(21), 7451. https://doi.org/10.3390/molecules27217451