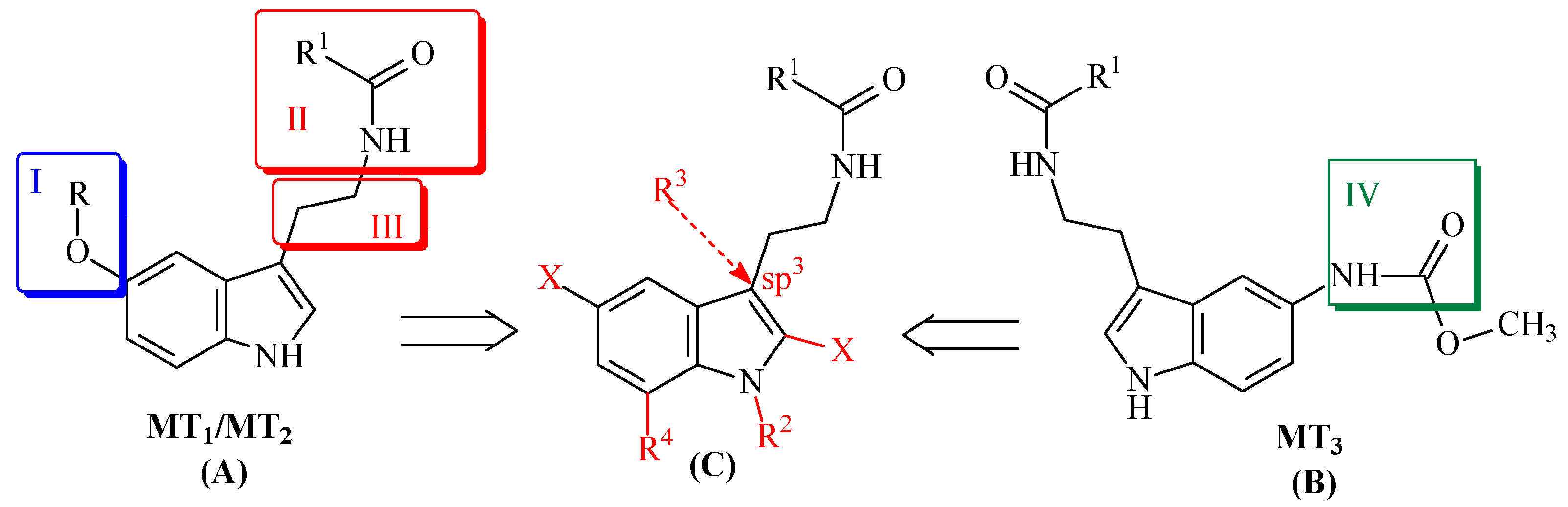
3.1. Chemistry
All solvents were used as received without further purification. The reactions were monitored by thin layer chromatography (TLC) carried out on Merck TLC silica gel plates (60 F254), using UV light for visualization and basic aqueous potassium permanganate or iodine fumes as developing agent. Flash column chromatography purifications were carried out using silica gel 60 (particle size 0.040–0.060 mm).
1H and
13C NMR spectra were recorded at 298 K on Bruker Avance 300 spectrometer with operating frequency of 400.13 and 100.6 MHz, respectively, and calibrated using residual CHCl
3 (δH = 7.26 ppm) and CDCl
3 (δC = 77.16 ppm) or DMSO-d
5 (δH = 2.50 ppm) and DMSO-d
6 (δC = 39.52 ppm) as internal references. NMR data were presented as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, m = multiplet, br. = broad), coupling constant (J) in Hertz (Hz), integration. High-resolution mass spectra (HRMS) were measured on a Thermo Scientific LTQ Orbitrap instrument using nanoelectrospray ionization (nano-ESI). Isatin
1a, 5-methoxyisatin
1b, 5-bromoisatin
1c and 6,7-dimethylisatin
1d were purchased from Merck. The following isatin derivatives were obtained by
N-alkylation using previously described procedure [
28]:
N-methylisatin
1e,
N-benzylisatin
1f,
N-methyl-5-methoxyisatin
1g,
N-benzyl-5-methoxyisatin
1h,
N-methyl-5-bromoisatin
1i,
N-methyl-6,7-dimethylisatin
1j. The following compounds were obtained as previously described: cyano(2-oxoindolin-3-ylidene)acetic acid
2a [
25], cyano(2-oxo-5-methoxy-indolin-3-ylidene)acetic acid
2b [
24], cyano(2-oxo-5-bromoindolin-3-ylidene)acetic acid
2c [
23], 2-(2-oxo-2,3-dihydro-
1H-indol-3-ylidene)acetonitrile
3a, [
25] 2-(1-methyl-2-oxo-2,3-dihydro-
1H-indol-3-ylidene)acetonitrile
3e [
24], 2-(5-methoxy-1-methyl-2-oxo-2,3-dihydro-
1H-indol-3-ylidene)acetonitrile
3g [
29], 2’-oxo-1’,2’-dihydrospiro[cyclopropane-1,3′-indol]-3-carbonitrile (
6a) [
19], 5′-methoxy-2’-oxo-1’,2’-dihydrospiro[cyclopropane-1,3′-indol]-3-carbonitrile (
6b) [
19], 2-chloromelatonin
7d [
26].
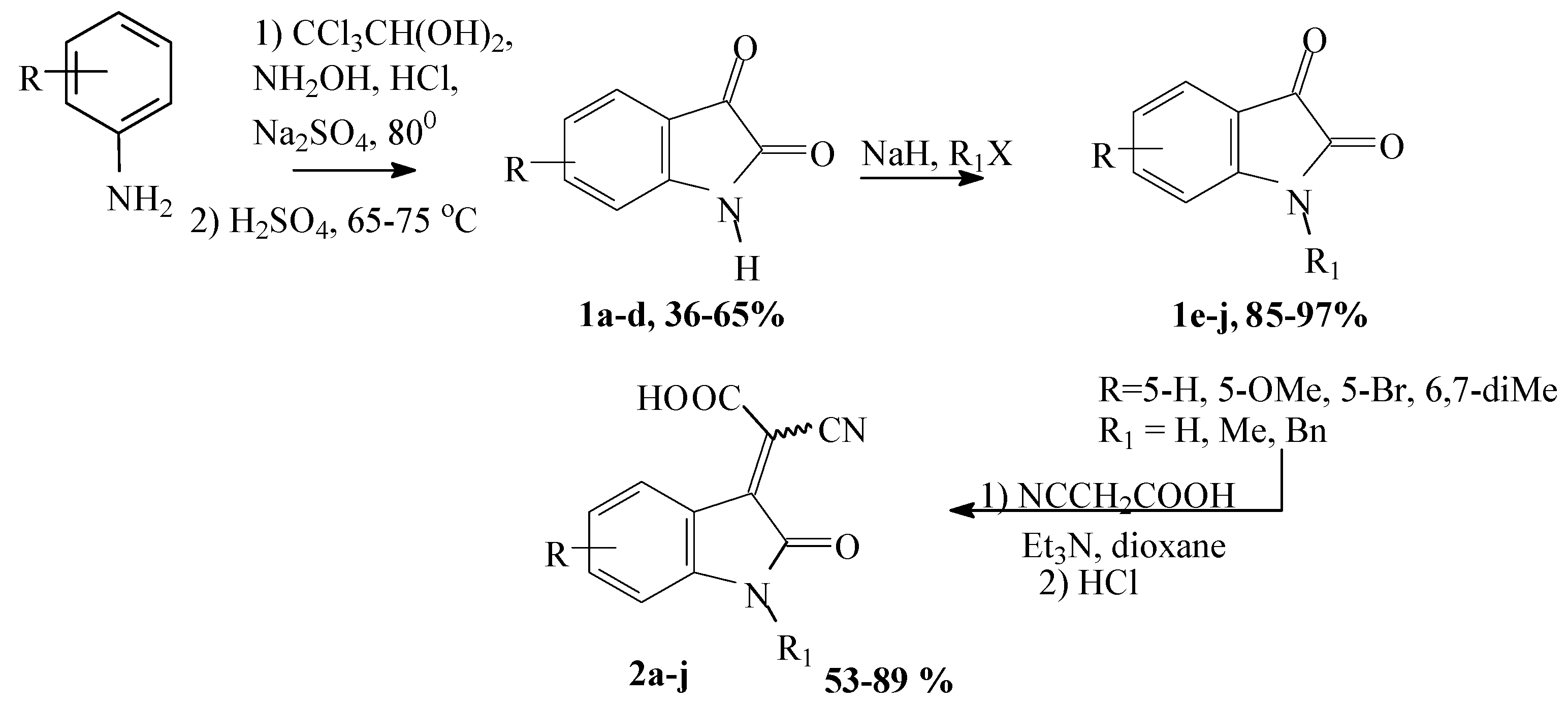
3.1.1. General Procedure for Synthesis of cyano(2-oxoindolin-3-ylidene)acetic Acids (2)
Isatin 1 (1 eq.) was dissolved in warm abs. dioxane (ca. 3 mL dioxane per 1 g of isatin) and mixture of cyanoacetic acid (1eq) and triethylamine (1.2 eq) in dioxane (ca. 1 mL dioxane per 1 g of acid) was added. Reaction mixture was vigorously stirred for 4–5 h, and then, reaction was terminated by addition of 30 mL conc. HCl. The reaction mixture was stored at room temperature for 1–5 days until precipitate was obtained. The precipitate was filtered and washed with cold water. All obtained compounds were sufficiently pure (according to 1H NMR) and could be used in further synthesis without additional purification. The following compounds were obtained according to this procedure:
From 8.00 g (0.046 mol) of 6,7-dimethylisatin (1d), 3.90 g (0.046 mol) of cyanoacetic acid, 6.70 mL (0.055 mol) of triethylamine in 28 mL of 1,4-dioxane brown powder (5.95 g, yield 54%) was obtained; m.p. = 222–230 °C. NMR 1H (DMSO-d6): δ 1.52 (s, 3H), 1.67 (s, 3H), 6.26 (d, J = 7.6 Hz, 1H), 7.27 (d, J = 7.5, 1H), 10.8 (s, 1H). NMR 13C (DMSO-d6): δ 13.46, 17.91, 25.44, 50.79, 119.83, 122.07, 125.44, 128.42, 130.01, 147.88, 151.41, 166.22, 173.57. Elemental analysis found (%): C 64.42, H 4.18, N 11.51, calculated for C13H10N2O3 (%): C 64.46, H 4.16, N 11.56.
From 6.87 g (0.043 mol) of N-methylisatin (1e), 3.62 g (0.043 mol) of cyanoacetic acid, 5.68 mL (0.052 mol) of triethylamine in 35 mL of 1,4-dioxane dark-cherry solid was obtained (7.40 g, 75% yield); m.p. = 177–178 °C. NMR 1H (DMSO-d6): δ 3.13 (s, 3H), 6.85 (d, J = 7.8, 1H), 7.05 (t, J = 7.6, 1H), 7.41 (t, J = 7.6, 1H), 7.95 (d, J = 7.8, 1H). NMR 13C (DMSO-d6): δ 26.32, 108.45, 109.59, 114.63 (CN), 122.97, 123.39, 124.71, 134.63, 138.79, 145.67, 161.79, 164.05. Elemental analysis found (%): C 63.21, H 3.68, N 12.11, calculated for C12H8N2O3 (%): C 63.16, H 3.53, N 12.28.
From 7.56 g (0.032 mol) of N-benzylisatin (1f), 2.9 g (0.032 mol) of cyanoacetic acid, 7.70 mL (0.056 mol) of triethylamine and 50 mL in 1,4-dioxane brown powder was obtained (7.98 g, 82% yield); m.p. = 169–173 °C. NMR 1H (CDCl3): δ 4.55 ** (s, 2H), 4.58 * (s, 2H), 6.45 ** (d, J = 8.1, 1H), 6.42 * (d, J = 8.1, 1H), 6.64 * (t, J = 7.6, 1H), 6.75 ** (t, J = 7.8, 1H), 6.95 (m, 5H), 7.02 ** (t, J = 7.8, 1H), 7.72 ** (d, J = 7.6, 1H), 7.91 * (d, J = 7.8, 1H). **—major isomer, *—minor isomer. Ratio of isomers was 4:1. NMR 13C (CDCl3): δ 43.69, 108.49, 110.28, 114.61, 114.78, 123.46, 124.67, 134.46, 127.31, 128.93, 129.4, 138.54, 134.87, 144.8, 161.85, 164.35. Elemental analysis found (%): C 70.07, H 3.92, N 9.22, calculated for C18H12N2O3 (%): C 71.05, H 3.97, N 9.21.
From 3.32 g (0.0173 mol) of N-methyl-5-methoxyisatin (1g), 1.51 g (0.0178 mol) of cyanoacetic acid, 3.0 mL (0.0238 mol) of triethylamine in 18 mL of 1,4-dioxane dark-violet solid (3.08 g, yield 69%) was obtained; m.p. = 182–183 °C. NMR 1H (CDCl3): δ major isomer: 3.16 (s, 3H), 3.71 (s, 3H), 6.79 (d, J = 8.8, 1H), 6.99 (dd, J = 8.6, J = 2.3, 1H), 7.82 (d, J = 2.8, 1H), δ minor isomer: 3.05 (s, 3H), 3.64 (s, 3H), 6.61 (d, J = 8.3, 1H), 6.89 (dd, J = 8.6, J = 2.5, 1H), 7.78 (d, J = 2.8, 1H), Ratio of isomers was 10:1. NMR 13C (CDCl3): δ major isomer 26.48, 55.40, 110.67, 112.09, 114.21, 116.21, 119.13, 120.65, 137.95, 141.96, 150.60, 156.74, 159.94. Elemental analysis found (%): C 60.57, H 3.86, N 10.22, calculated for C13H10N2O4 (%): C 60.47, H 3.90, N 10.85.
From 7.42 g (0.028 mol) of N-benzyl-5-methoxyisatin (1h), 2.47 g (0.029 mol) of cyanoacetic acid, 4.20 mL (0.03 mol) of triethylamine in 45 mL of 1,4-dioxane violet powder was obtained (6.42 g, yield 68%); m.p. = 152–153 °C. NMR 1H (DMSO-d6): δ 3.69 ** (s, 3H), 3.65 * (s, 3H), 4.79 ** (s, 2H), 4.81 * (s, 2H), 6.63 ** (d, J = 8.6, 1H), 6.6 * (d, J = 8.59, 1H), 6.85 ** (dd, J = 8.6, J = 2.5, 1H), 6.84 * (dd, J = 8.6, J = 2.5, 1H); 7.21 (m, 5H); 7.54 ** (d, J = 2.5, 1H); 7.81 * (d, J = 2.5, 1H). **—major isomer, *—minor isomer. Ratio of isomers was 4:1. NMR 13C (DMSO-d6): δ 43.71, 55.83, 108.75, 111.10, 114.61, 115.33, 119.72, 120.63, 127.35, 128.45, 129.97, 135.02, 138.53, 139.15, 156.06, 161.73, 164.08. Elemental analysis found (%): C 68.33, H 4.18, N 8.32, calculated for C18H12N2O3 (%): C 68.26, H 4.22, N 8.38.
From 5.13 g (0.021 mol) of N-methyl-5-bromoisatin (1i), 1.82 g (0.021 mol) of cyanoacetic acid, 2.80 mL (0.027 mol) of triethylamine in 17 mL of 1,4-dioxane violet powder was obtained (5.90 g, yield 89%); m.p. = 137–138 °C. NMR 1H (CDCl3): δ 3.14 (s, 3H); 6.95 (d, J = 8.4, 1H), 7.61 (dd, J = 8.4, J = 1.9, 1H), 8.28 (d, J = 1.9, 1H). NMR 13C (CDCl3): δ 21.48, 105.49, 109.20, 115.26, 122.47, 127.07, 132.57, 139.57, 140.36, 156.42, 158.11, 166.82. Elemental analysis found (%): C 47.00, H 3.83, N 9.17, calculated for C12H7BrN2O3 (%): C 46.93, H 2.30, N 9.12.
From 5.00 g (0.027 mol) of N-methyl-6,7-dimethylisatin (1j), 2.30 g (0.027 mol) of cyanoacetic acid, 3.50 mL (0.028 mol) of triethylamine in 17 mL of 1,4-dioxane light brown solid (3.76 g, yield 59%) was obtained; m.p. = 177–180 °C. NMR 1H (CDCl3): δ 3.21 (s, 3H), 2.34 (s, 3H), 2.55 (s, 3H), 3.59 (s, 3H), 7.12 (s, 1H), 8.17 (s, 1H). NMR 13C (CDCl3): δ 14.01, 21.51, 30.57, 95.23, 116.48, 118.22, 119.38, 119.89, 122.37, 125.01, 143.08, 143.79, 145.08, 168.75. Elemental analysis found (%): C 65.53, H 4.80, N 10.85, calculated for C14H12N2O3 (%): C 65.62, H 4.72, N 10.93.
3.1.2. General Procedure for Synthesis (2-oxoindolin-3-ylidene)acetonitriles (3)
Solution of compound 2 in pyridine was heated using water bath for 2 h. Then, reaction mixture was cooled, and acetic acid was added. Precipitate was collected, washed with cold water and dried. According to this procedure, the following compounds were obtained:
1.00 g (4.1 mmol) of cyano(5-methoxy-2-oxo-indolin-3-ylidene)acetic acid (2b) in 2 mL of pyridine was heated for 2 h before the 10 mL of acetic acid was added. Dark red precipitate (0.52 g, yield 64%) was obtained; m.p. = 290–292 °C. NMR 1H (DMSO-d6): δ 3.73 (s, 3H), 6.47 (s, 1H), 6.79 (d, J = 8.6, 1H), 7.03 (dd, J = 8.6, J = 2.5, 1H), 7.35 (d, J = 2.3, 1H), 10.65 (s, 1H). IR, cm−1: 1600 (Ar), 1620 (C=CH-CN), 1730 (NHC(O)), 2220 (CN), 3150–3300 (NH). Elemental analysis found (%): C 65.95, H 4.03, N 13.92; calculated for C11H8N2O2 (%): C 66.00, H 4.03, N 13.99.
5.00 g (0.017 mol) of cyano(5-bromo-2-oxoindolin-3-ylidene)acetic acid 2c in 20 mL of pyridine was heated for 2 h, and then 50 mL of acetic acid was added. Dark red precipitate was obtained, 2.84 g, yield 66%; m.p. = 238–239 °C. NMR 1H (CDCl3): δ 6.49 ** (s, 1H), 6.59 * (s, 1H), 6.79 ** (d, J = 8.2, 1H), 6.85 * (d, J = 8.2, 1H), 7.50 ** (d, J = 8.2, 1H), 7.57 * (d, J = 8.2, 1H), 7.84 * (s, 1H), 7.93 ** (s, 1H), 10.84 ** (s, 1H), 10.99 * (s, 1H). **—major isomer, *—minor isomer. Ratio of isomers was 10:1. NMR 13C (CDCl3): δ 99.91, 113.36, 114.04 (CN), 116.92, 121.73, 126.48, 136.45, 143.02, 144.28, 165.92 (C=O).
5.85 g (0.029 mol) of cyano(6,7-dimethyl-2-oxo-indolin-3-ylidene)acetic acid 2d and 12 mL of pyridine were heated for 2 h, and then 70 mL of acetic acid was added. Resulting 3.11 g of dark red precipitate had been obtained, yield 63%; m.p. = 207 °C. NMR 1H (CDCl3): δ 1.71 (s, 3H), 1.85 (s, 3H), 5.89 (s, 1H), 6.40 (d, J = 7.6, 1H), 7.23 (d, J = 7.6, 1H), 10.1 (s, 1H). NMR 13C (CDCl3): δ 12.85, 20.23, 94.18, 116.34 (CN), 117.13, 119.04, 121.57, 123.55, 143.35, 144.93, 167.96 (C2 = O). Elemental analysis found (%): C 72.65, H 5.04, N 14.10, calculated for C12H10N2O (%): C 72.71, H 5.08, N 14.13.
7.80 g (0.026 mol) of cyano(N-benzyl-2-oxoindolin-3-ylidene)acetic acid 2f in 20 mL of pyridine was heated for 2 h, and then 90 mL of acetic acid was added. Dark red precipitate was obtained (4.50 g, yield 68%); m.p. = 155 °C. NMR 1H (CDCl3): δ 4.93 (s, 2H), 6.39 (s, 1H), 6.75 (d, J = 7.8, 1H), 7.1 (t, J = 7.6, 1H), 7.33 (m, 6H), 8.09 (d, J = 7.6, 1H). NMR 13C (CDCl3): δ 43.55 (CH2), 98.85 (=C(CN)), 110.34 (C7), 116.73 (CN), 119.24 (C5), 123.25 (C4), 124.47 (C3a), 127.47, 127.92, 128.96, 133.88 (CBn), 135.97 (C6), 143.08 (C3), 145.3 (C3b), 165.17 (C2=O). IR, cm−1: 2220 (-CN), 1715 (CO), 1610 (C=C). Elemental analysis found (%): C 78.50, H 4.64, N 10.79, calculated for C17H12N2O (%): C 78.44, H 4.65, N 10.76.
2.90 g (0.0086 mol) of cyano(N-benzyl-2-oxo-5-methoxyindolin-3-ylidene)acetic acid 2h in 6 mL of pyridine was heated for 2 h, and then 27 mL of acetic acid was added. Dark red precipitate was obtained, 1.71 g, yield 91%; m.p. = 126–127 °C. NMR 1H (DMSO-d6): δ 3.72 (s, 3H), 4.88 (s, 2H), 6.67 (s, 1H), 6.9 (d, J = 8.8, 1H), 7.01 (dd, J = 2.5, J = 8.6, 1H), 7.31 (m, 5H), 7.44 (d, J = 2.5, 1H). NMR 13C (DMSO-d6): δ 43.93 (CH2), 55.85 (MeO), 97.82 (=C(CN)), 110.57 (C4), 116.03 (C7), 119.36 (CN), 119.88 (C6), 124.88 (C3a), 127.24, 127.91, 128.89, 134.99 (CBn), 138.59 (C3), 143.78 (C3b), 154.33 (C5), 165.22 (C2=O). IR, cm−1: 2230 (-CN), 1710 (CONH), 1620 (C=C). Elemental analysis found (%): C 74.44, H 4.81, N 9.66, calculated for C18H14N2O2 (%): C 74.47, H 4.86, N 9.65.
1.04 g (0.0049 mol) of cyano(N-methyl-2-oxo-5-bromoindolin-3-ylidene)acetic acid 2i and 4 mL of pyridine were heated for 2 h, then 20 mL of acetic acid was added. Resulting red precipitate was obtained, single isomer, yield 0.43 g, 54%; m.p. = 195–196 °C. NMR 1H (CDCl3): δ 3.23 (s, 3H), 6.38 (s, 1H), 6.74 (d, J = 8.3, 1H), 7.57 (dd, J = 1.5, J = 8.3, 1H), 8.16 (d, J = 1.5, 1H). NMR 13C (CDCl3): δ 26.42, 99.10, 110.32, 115.56, 120.67, 125.03, 127.66, 136.27, 142.33, 144.54, 164.70. Elemental analysis found (%): C 50.34, H 2.71, N 10.66, calculated for C11H7BrN2O (%): C 50.22, H 2.68, N 10.65.
1.00 g (0.015 mol) of cyano(1,6,7-trimethyl-2-oxoindolin-3-ylidene)acetic acid 2j and 4 mL of pyridine were heated, and then 20 mL of acetic acid was added. Resulting 0.45 g of dark violet precipitate had been obtained, mixture of isomers Z:E = 1:10, yield 52%; m.p. = 187 °C. Major isomer: NMR 1H (CDCl3): 2.34 (s, 3H), 2.43 (s, 3H), 3.50 (s, 3H), 6.17 (s, 1H), 6.90 (d, J = 7.7, 1H), 7.80 (d, J = 7.7, 1H). NMR 13C (CDCl3): 13.96, 21.46, 30.52, 95.18, 116.43 (CN), 118.17, 119.84, 122.32, 124.96, 143.03, 143.74, 145.02, 166.70 (C2 = O). Minor isomer: NMR 1H (CDCl3): 2.34 (s, 3H), 2.43 (s, 3H), 3.47 (s, 3H), 5.94 (s, 1H), 6.85 (d, J = 7.6, 1H), 7.16 (d, J = 7.7, 1H). Elemental analysis found (%): C 73.57, H 5.66, N 13.19, calculated for C13H12N2O (%): C 73.56, H 5.70, N 13.20.
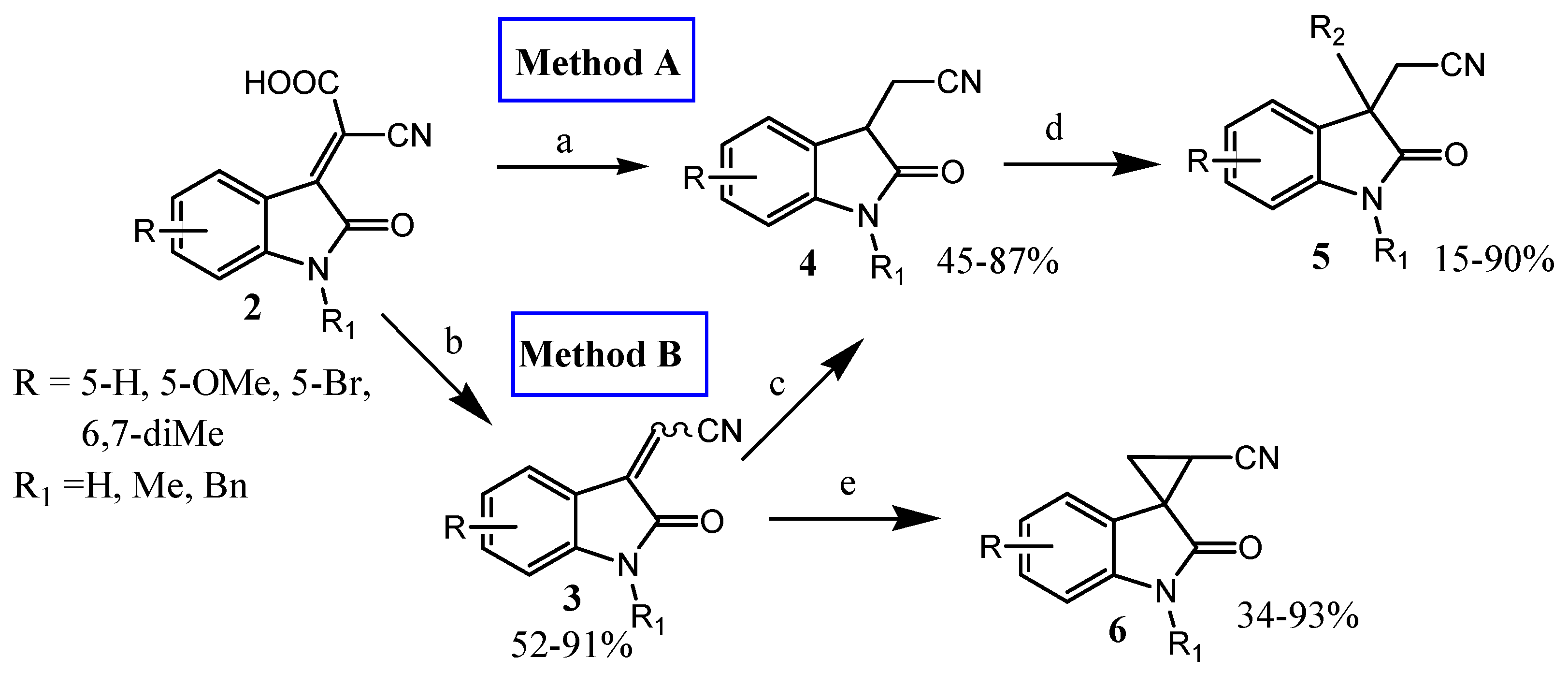
3.1.3. General Procedure for Synthesis of (2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitriles (4)
Method A:
To a solution of 2 in mixture of ethyl acetate and 3N hydrochloric acid, the excess of zinc dust was added, and the reaction mixture was vigorously stirred for 0.5 h. The color of the reaction mixture turned from dark red to pale yellow. Then, organic phase was separated, washed with cold distilled water and dried with Na2SO4. The solution was concentrated in vacuum. The obtained cyano(2-oxo-2,3-dihydro-1H-indol-3-yl)acetic acid was used in the decarboxylation step without additional purification. The residue was dissolved in 2-ethoxyethanol and stirred with reflux for 2–2.5 h. Reaction mixture was concentrated and purified by filtration through silica gel pad with ethyl acetate as eluent. The following compounds were obtained according to this procedure:
2-(2-Oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
4a) [
25]
First, 10.25 g (0.048 mol) of cyano(2-oxo-indolin-3-ylidene)acetic acid
2a, 40 mL of ethyl acetate, 20 mL of 3N HCl, and 4.80 g (0.08 mol) of zinc dust were involved in the reaction. The organic phase was separated and concentrated. The dry residue was dissolved in 30 mL of 2-ethoxyethanol, refluxed for 2 h, concentrated and filtered through silica gel pad using ethyl acetate as eluent. As a result, 5.68 g of beige powder was obtained, yield 69%; m.p. = 155–160 °C (m.p.
lit. 160–161 °C [
25]). NMR
1H (CDCl
3): 2.61 (dd, J = 16.8, J = 8.2, 1H), 2.88 (dd, J = 16.8, J = 8.2, 1H), 3.44 (m, 1H), 6.74 (d, J = 6.5, 1H), 6.84 (t, J = 5.6, 1H), 7.05 (t, J = 6.1, 1H), 7.22 (d, J = 6.3, 1H), 10.03 (s, 1H, NH).
2-(5-Methoxy-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
4b) [
24]
First, 2.54 g (0.01 mol) of cyano(5-methoxy-2-oxo-indolin-3-ylidene)acetic acid
2b, 40 mL of ethyl acetate, 10 mL of 3N HCl and 1.20 g (0.02 mol) of zinc dust were involved in reaction. The organic phase was separated and concentrated. The dry residue was dissolved in 15 mL of 2-ethoxyethanol, refluxed for 2 h concentrated and filtered through silica gel pad using ethyl acetate as eluent. As a result, 1.09 g of beige solid was obtained, yield 57%; m.p. = 179–180 °C (m.p.
lit = 180–181 °C [
24]). NMR
1H (CDCl
3): 2.77 (dd, J = 17.0, J = 9.1, 1H), 3.10 (dd, J = 17.0, J = 4.8, 1H), 3.70 (dd, J = 8.6, J = 4.3, 1H), 3.81 (c, 3H), 6.89 (dd, J = 8.6, J = 1.2, 1H), 7.02 (c, 1H), 7.20 (d, J = 8.6, 1H), 8.45 (s, 1H, NH). IR, cm
−1: 2340 (NH), 2280 (-CN), 1700 (CO). MS-EI, 70 eV, m/z: 202 (25%), 175 (2%), 162 (100%), 176 (100%), 147 (14%), 131 (13%), 119 (18%), 104 (11%), 91 (9%), 77 (14%).
2-(5-Bromo-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
4c) [
23]
First, 5.22 g (0.018 mol) of cyano(5-bromo-2-oxoindolin-3-ylidene)acetic acid
2c, 36 mL of ethyl acetate, 18 mL of 3N HCl and 2.50 g (0.04 mol) of zinc dust were involved in the reaction. The organic phase was separated and concentrated. The dry residue was dissolved in 15 mL of 2-ethoxyethanol, refluxed for 2.5 h concentrated and filtered through silica gel pad using ethyl acetate as eluent. As a result, 2.02 g of beige solid was obtained, yield 45%; m.p. = 209–210 °C (m.p.
lit. 211 °C [
23]). NMR
1H (CDCl
3): 3.11 (dd, 1H, J = 5.8, J = 5.8), 3.26 (dd, 1H, J = 5.9, J = 5.8), 3.88 (d, 1H, J = 5.5), 6.84 (d, 1H, J = 8.6), 7.42 (d, 1H, J = 8.4), 7.60 (s, 1H), 10.74 (s, 1H). NMR
13C (CDCl
3): 17.87, 41.90, 111.94, 113.83, 118.50, 127.67, 130.09, 131.70, 142.63, 176.52. MS-EI (mz, %): 252, 250 (M+, 75%), 212, 210 (100%), 195, 197 (10%), 182, 184 (10%), 143 (5%), 116 (15%), 76 (20%).
2-(1-Methyl-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
4e) [
24]
First, 7.40 g (0.0324 mol) of cyano(
N-methyl-2-oxo-indolin-3-ylidene)acetic acid
2e, 54 mL of ethyl acetate, 27 mL of 3N HCl and 5.40 g (0.076 mol) of zinc dust were involved in reaction. The organic phase was separated and concentrated. The dry residue was dissolved in 30 mL of 2-ethoxyethanol, refluxed for 2 h concentrated and filtered through silica gel pad using ethyl acetate as eluent. As a result, 5.30 g of beige solid was obtained, yield 87%; m.p. = 91 °C (m.p
lit. 89–90 °C [
24], 71–72 °C [
30]). NMR
1H (CDCl
3): 2.64 (dd, J = 16.8, J = 8.6, 1H), 3.00 (dd, J = 16.7, J = 4.8, 1H), 3.14 (s, 3H), 3.58 (dd, J = 8.3, J = 4.8, 1H), 6.83 (d, J = 7.8, 1H), 7.05 (t, J = 7.6, 1H), 7.29 (t, J = 7.6, 1H), 7.42 (t, J = 7.3, 1H). IR, cm
−1: 2230 (-CN), 1725 (NH-CO).
2-(1-Benzyl-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
4f) [
24]
First, 2.35 g (0.0077 mol) of cyano(
N-benzyl-2-oxoindolin-3-ylidene)acetic acid
2f, 15 mL of ethyl acetate, 8 mL of 3N HCl and 1.20 g (0.0177 mol) of zinc dust were involved in reaction. The organic phase was separated and concentrated. The dry residue was dissolved in 20 mL of 2-ethoxyethanol, refluxed for 2 h concentrated and filtered through silica gel pad using ethyl acetate as eluent. As a result, 5.30 g of beige solid was obtained, yield 60%; m.p. = 152–154 °C (m.p.
lit 150–151 °C [
24]). NMR
1H (CDCl
3): 2.76 (d, J = 15.9, 1H), 2.96 (d, J = 15.9, 1H), 3.10 (dd, J = 16.7, J = 4.8, 2H), 3.68 (t, J = 4.6, 1H), 4.88 (d, J = 10.1, 2H), 6.73 (d, J = 7.8, 1H), 7.03 (t, J = 7.3, 1H), 7.22 (m, 5H), 7.44 (d, J = 7.3, 1H). NMR
13C (CDCl
3): 18.97, 41.40, 43.95, 109.64, 117.14, 123.12, 124.21, 127.77, 128.83, 135.21, 126.65, 127.23, 129.25, 143.25, 162.8. IR, cm
−1: 2220 (-CN), 1715 (NH-CO).
2-(1-Methyl-5-methoxy-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
4g) [
29]
First, 2.58 g (0.01 mol) of cyano(
N-methyl-2-oxo-5-methoxy-indolin-3-ylidene)acetic acid
2g, 22 mL of ethyl acetate, 11 mL of 3N HCl and 1.80 g (0.0277 mol) of zinc dust were involved in the reaction. The organic phase was separated and concentrated. The dry residue was dissolved in 20 mL of 2-ethoxyethanol and orange solution refluxed for 1.5 h concentrated and filtered through silica gel pad using ethyl acetate as eluent. As a result, 1.519 g of beige solid was obtained, yield 67%; m.p. = 116–118 °C (m.p.
lit. 115–116 °C [
29,
30]). NMR
1H (CDCl
3): 2.96 (dd, J = 16.9, J = 6.3, 1H), 3.07 (dd, J = 16.9, J = 5.8, 1H), 3.13 (s, 3H), 3.65–6.68 (m, 1H), 3.77 (s, 3H), 6.84 (d, J = 8.6, 1H), 6.89 (dd, J = 2.5, J = 8.6, 1H), 7.06 (d, J = 2.5, 1H). NMR
13C (CDCl
3): 18.85, 26.39, 41.60, 55.60, 108.79, 111.46, 113.60, 116.94, 126.89, 137.47, 156.23, 173.60. IR, cm
−1: 2235 (-CN), 1715 (NH-CO).
2-(1-Benzyl-5-methoxy-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
4h) [
31]
First, 6.50 g (0.019 mol) of cyano(
N-benzyl-2-oxo-5-methoxyindolin-3-ylidene)acetic acid
2h, 65 mL of ethyl acetate, 35 mL of 3N HCl and 4.00 g (0.06 mol) of zinc dust were involved in reaction. The organic phase was separated and concentrated. The dry residue was dissolved in 25 mL of 2-ethoxyethanol, refluxed for 2.5 h concentrated and filtered through silica gel pad using ethyl acetate as eluent. As a result, 3.78 g of beige solid was obtained, yield 69%; m.p. = 143 °C (m.p.
lit 144–145 °C [
31]). NMR
1H (CDCl
3): 2.76 (dd, J = 15.9, 1H), 2.96 (d, J = 15.9, 1H), 3.14 (dd, J = 16.9, J = 4.8, 2H), 3.75 (dd, J = 4.3, J = 8.6, 1H), 3.78 (s, 3H), 4.91 (d, J = 7.3, 2H), 6.67 (d, J = 8.6, 1H), 6.78 (dd, J = 2.5, J = 8.6, 1H), 7.13 (d, J = 1.7, 1H), 7.34 (m, 5H). NMR
13C (CDCl
3): 19.08, 41.72, 44.03, 55.59, 109.99, 111.48, 113.71, 116.88, 124.93, 127.21, 127.75, 128.83, 135.29, 136.69, 156.27, 173.86. IR, cm
−1: 2255 (-CN), 1710 (NH-CO).
2-(5-Bromo-1-methyl-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
4i) [
30]
First, 5.90 g (0.019 mol) of cyano(
N-methyl-2-oxo-5-bromoindolin-3-ylidene)acetic acid
2i, 30 mL of ethyl acetate, 15 mL of 3N HCl and 3.60 g (0.058 mol) of Zn were involved in reaction. The organic phase was separated and concentrated. The dry residue was dissolved in 10 mL of 2-ethoxyethanol and refluxed. As a result, 2.87 g of dark beige solid was obtained, yield 57%; m.p. = 168 °C. Lit. [
31]: brown oil. NMR
1H (CDCl
3): 2.73 (dd, J = 16.8, J = 8.7, 1H), 3.10 (dd, J = 16.9, J = 4.7, 1H), 3.21 (s, 3H), 3.66–3.69 (m, 1H), 6.77 (d, J = 8.3, 1H), 7.49 (d, J = 8.3, 1H), 7.60 (s, 1H). NMR
13C (CDCl
3): 18.68, 26.50, 41.19, 109.99, 115.60, 116.66, 127.25, 127.40, 132.18, 143.11, 173.49. MS-EI (mz, %): 264 (M
+, 70%), 226, 224 (100%), 208, 210 (5%), 186 (10%), 155 (10%), 146 (15%), 117 (50%), 90 (20%), 76 (20%).
First, 3.76 g (0.015 mol) of cyano(1,6,7-trimethyl-2-oxoindolin-3-ylidene)acetic acid 2j, 35 mL of ethyl acetate, 18 mL of 3N HCl and 2.50 g (0.04 mol) of Zn were involved in reaction. The organic phase was separated and concentrated. The dry residue was dissolved in 10 mL of 2-ethoxyethanol and refluxed. Isolation gave 1.80 g of red solid, yield 60%; m.p. = 161 °C. NMR 1H (DMSO-d6): 2.22 (s, 3H), 2.37 (s, 3H), 2.72 (dd, J = 16.9, J = 4.8, 1H), 2.96 (d, J = 15.9, 1H), 3.43 (s, 3H), 3.53 (m, 1H), 6.84 (d, J = 7.1, 1H), 7.11 (d, J = 6.9, 1H). NMR 13C (CDCl3): 13.99, 19.21, 20.83, 30.82, 40.83, 117.43, 119.60, 121.28, 124.32, 124.75, 139.11, 141.98, 176.06.
Method B:
A solution of compound 3 in mixture of ethyl acetate and 3N hydrochloric acid was vigorously stirred with excess of zinc dust for 0.5 h. Then, the organic phase was separated, washed with cold water and dried using Na2SO4. The solvent was removed in vacuum. According to this procedure, the following compounds were obtained:
First, 2.000 g (11.6 mmol) of 2-(1-methyl-2-oxo-2,3-dihydro-1H-indol-3-ylidene)acetonitrile 3e (was dissolved in the mixture of ethyl acetate (15.4 mL) and 3N HCl (7.7 mL) and 1.817 g of zinc dust (0.028 mol) was added. The mixture was vigorously stirred for 0.5 h. Yield of 4e: 1.624 g, 80%. The spectral data of this compound are the same as for compound obtained using method A.
First, 2.500 g, (9.6 mmol) of 2-(1-benzyl-2-oxo-2,3-dihydro-1H-indol-3-ylidene)acetonitrile 3f was dissolved in mixture of ethyl acetate (12.7 mL) and 3N HCl (6.4 mL) and 1.200 g of zinc dust (17.7 mmol) was added. Yield of 4f: 79%, 1.985 g. The spectral data of this compound are the same as for compound obtained using method A.
First, 1.977 g (11.4 mmol) of 2-(1-methyl-5-methoxy-2-oxo-2,3-dihydro-1H-indol-3-ylidene)acetonitrile 3g was dissolved in mixture of ethyl acetate (15 mL) and 3N HCl (7 mL) and 1.800 g of zinc dust (27.7 mmol) was added. Yield of 4g: 72%, 1.430 g. The spectral data of this compound are the same as for the compound obtained using method A.
First, 2.266 g (7.8 mmol) of 2-(1-benzyl-5-methoxy-2-oxo-2,3-dihydro-1H-indol-3-ylidene)acetonitrile 3h was dissolved in mixture of ethyl acetate (10.3 mL) and 3N HCl (5.2 mL) and 1.220 g of zinc dust (18.8 mmol) was added. Yield of 4h: 83%, 1.902 g. The spectral data of this compound are the same as for compound obtained using method A.
First, 0.333 g (1.3 mmol) of 2-(5-bromo-1-methyl-2-oxo-2,3- dihydro-1H-indol-3-ylidene)acetonitrile 3i was dissolved in mixture of ethyl acetate (20 mL) and 3N HCl (10 mL) and 1.400 g of zinc dust (25 mmol) was added. Yield of 4i: 72%, 0.250 g. The spectral data of this compound are the same as for compound obtained using method A.
First, 1.63 g (8 mmol) of 2-(1,6,7-trimethyl-2-oxo-2,3-dihydro-1H-indol-3-ylidene)acetonitrile 3j was dissolved in mixture of ethyl acetate (20 mL) and 3N HCl (10 mL), and 1.20 g of zinc dust (20 mmol) was added. Yield of 4j: 82%, 1.40 g. The spectral data of this compound are the same as for compound obtained using method A.
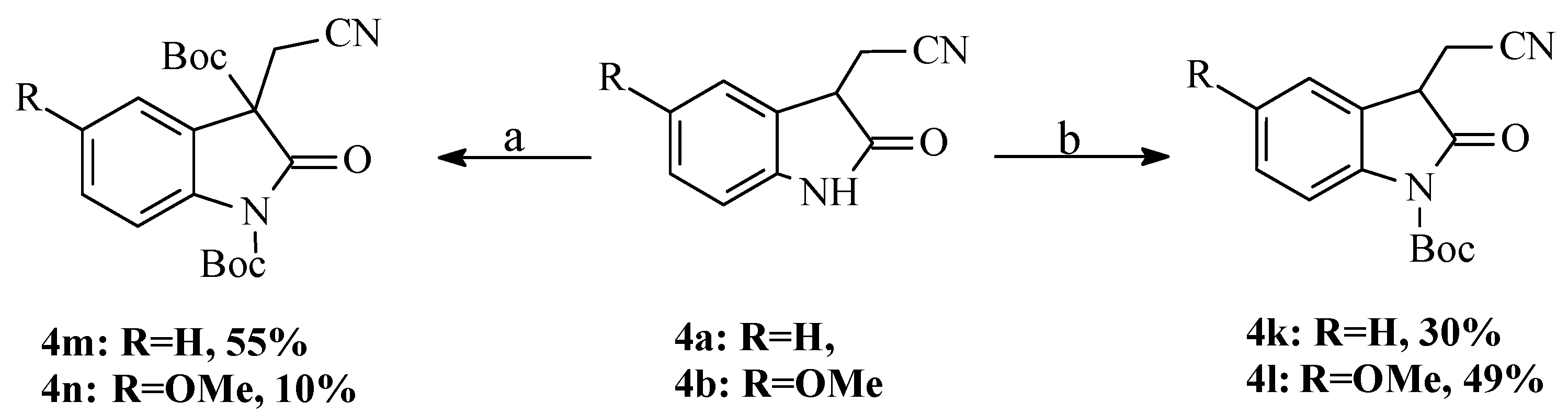
3.1.4. Boc Protection/Alkylation of N-unsubstituted Compounds (4a,b)
2-(2-Oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile (4a) (1.57 g, 1.2 mmol) was dissolved in 20 mL of dry THF and solution was cooled up to −40 °C. After addition of sodium hydride (60% oil suspension) (0.05 g, 1.2 mmol) the reaction mixture was vigorously stirred for 45 min at the same temperature, then 2.06 г (12 mmol) Boc2O was added, and solution was stirred at −40–−20 °C for 45 min and at 25 °C for 30 min. The reaction was terminated with 1 mL of water and organic solvent was evaporated. Dry residue was washed with saturated aqueous solution of K2CO3 and cold water and extracted with CH2Cl2. Compound 4k was obtained as orange oil after filtration through silica gel pad (yield 1.20 g, 38%). NMR 1H (CDCl3): 1.64 (s, 9H), 2.77 (dd, J = 16.9, J = 8.7, 1H), 3.11 (dd, J = 16.9, J = 4.8, 1H), 3.81 (dd, J = 8.7, J = 4.8, 1H), 7.22 (t, J = 7.6, 1H), 7.38 (t, J = 7.6, 1H), 7.51 (d, J = 7.5, 1H), 7.85 (d, J = 8.1, 1H). NMR 13C (CDCl3): 19.13, 27.87, 41.85, 85.85, 115.25, 116.75, 123.78, 124.77, 129.41, 139.91, 148.51, 172.59.
2-(5-Methoxy-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile (4b) (1.00 g, 5 mmol) was dissolved in 20 mL of dry THF and solution was cooled up to −40 °C. After addition of sodium hydride (60% oil suspension) (0.22 g, 5.5 mmol), the reaction mixture was vigorously stirred for 45 min at the same temperature, then 1.09 г (5 mmol) Boc2O was added and solution was stirred at −40–−20 °C for 45 min and at 25 °C for 30 min. The reaction was terminated with addition of 1 mL of water and organic solvent was evaporated. Dry residue was washed with saturated aqueous solution of K2CO3 and cold water and extracted with CH2Cl2. Compound 4l was obtained as orange oil after filtration through silica gel pad (yield 0.74 g, 49%). NMR 1H (CDCl3): 1.46 (s, 9H), 2.76 (dd, J = 16.9, J = 8.2, 1H), 3.03 (dd, J = 16.9, J = 5.2, 1H), 3.73 (s, 3H, OCH3), 3.72–3.75 (m, 1H), 6.81 (dd, J = 9.0, J = 2.6, 1H), 6.99 (d, J = 2.6, 1H), 7.69 (d, J = 9.0, 1H). NMR 13C (CDCl3): 18.82, 27.67, 41.95, 55.28, 84.27, 109.82, 113.86, 115.98, 116.59, 125.47, 133.00, 146.40, 156.75, 172.45. IR, cm−1 (nujol): 2850–3000 (NH), 2260 (CN), 1550, 1650. MS-EI (70 eV), m/z: 302 (M+), 246 (M+-C(CH3)3), 202 (M+-Boc), 175, 158, 146, 57(100%).
2-(2-Oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile (4a) (0.960 g, 0.0052 mol) was dissolved in dry acetonitrile and 0.130 g (0.0113 mol) of DMAP was added. The reaction mixture was vigorously stirred for 5 min and 2.7 g (0.012 mol) of Boc2O was added. The reaction was continued for 80 h at room temperature, then a solvent was evaporated, the residue was washed with aq. NaHCO3 and cold water, dissolved in CH2Cl2 and dried with MgSO4. Orange oil (55%, 1.067 g) was obtained after column chromatography (Rf 0.87, eluent ethyl acetate). NMR 1H (CDCl3): 1.22 (s, 9H), 1.50 (s, 9H), 2.94 (d, J = 16.8, 1H), 3.15 (J = 16.8, 1H), 7.07–7.11 (m, 1H), 7.26–7.30 (m, 2H), 7.78 (d, J = 8.0, 1H). NMR 13C (CDCl3): 13.63, 26.90, 27.40, 56.60 (C3), 84.03, 84.49, 115.10, 115.23, 122.40, 124.30, 124.67, 129.87, 139.79, 147.98, 164.73, 169.74.
2-(5-Methoxy-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile (4b) (0.228 g, 1.13 mmol) was dissolved in dry acetonitrile and 0.028 g (0.23 mmol) of DMAP was added. The reaction mixture was vigorously stirred for 5 min, and 0.800 g (3.6 mmol) of Boc2O was added. The reaction was continued for 40 h at room temperature, then a solvent was evaporated, the residue was washed with aq. NaHCO3 and cold water, dissolved in CH2Cl2 and dried with MgSO4. Orange oil (10%, 0.044 g) was obtained after column chromatography (Rf 0.8, eluent ethyl acetate). NMR 1H (CDCl3): 1.37 (s, 9H), 1.64 (s, 9H), 3.23 (d, J = 16.7, 1H), 3.03 (J = 16.7, 1H), 3.81 (s, 3H), 6.92 (d, 1H), 6.94 (d, 1H), 7.83 (d, J = 9.6, 1H). MS-EI (70 eV), m/z: 402 (2%), 376 (2%,), 364 (2%), 320 (3%), 302 (5%), 264 (10%), 246 (7%), 220 (30%), 202 (7%), 189 (5%), 175 (25%), 158 (8%). Elemental analysis found (%): C 62.68, H 6.62, N 6.91, calculated for C21H26N2O6 (%): C 62.67, H 6.51, N 6.96.
3.1.5. General Procedure of Alkylation of N-Boc/alkyl-(2-oxoindolin-3-yl)acetonitriles
A mixture N-Boc/alkyl-(2-oxoindolin-3yl)acetonitrile, sodium hydride (60% suspension in oil) and dry THF was stirred under argon atmosphere for 30 min. Reaction mixture was cooled with ice bath (0 °C), and excess of alkyl halide was added. After stirring at room temperature for 48 h, the solvent was evaporated and the resulting oil was washed with ice water, dichloromethane (for N-Boc, Bn) or chloroform (for N-Me) and dried with MgSO4. The product was obtained after column chromatography. The following compounds were obtained using this general procedure:
2-(1,3-Dimethyl-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
5a) [
32,
33]
The compound 5a (light-orange oil, Rf = 0.8 in ethyl acetate, yield 56%, 3.16 g) was obtained from 5.30 g (28 mmol) of 2-(1-methyl-2-oxoindolin-3-yl)acetonitrile 4e, 0.75 g (30 mmol) of sodium hydride and 2 mL (30 mmol) of methyl iodide in 30 mL of dry THF. NMR 1H (CDCl3): 1.53 (s, 3H), 2.57 (d, J = 16.7, 1H), 2.86 (d, J = 16.7, 1H), 3.24 (s, 3H), 6.91 (d, J = 7.8, 1H), 7.14 (t, J = 7.8, 1H), 7.34–7.38 (m, 1H), 7.48 (d, J = 7.5, 1H). NMR 13C (CDCl3): 21.22, 24.91, 25.44, 43.93, 107.89, 115.99, 122.02, 122.08, 128.19, 130.14, 141.95, 176.44.
2-(1-Benzyl-3-methyl-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
5b) [
33]
The compound 5b (Rf = 0.7 in hexane:ethyl acetate 8:2 (v/v), yield 57%, 1.100 g) was obtained as yellow oil from 1.780 g (6.7 mmol) of 2-(1-benzyl-2-oxoindolin-3-yl)acetonitrile 4f, 0.177 g (75 mmol) of sodium hydride and 2 mL (30 mmol) of methyl iodide in 35 mL of dry THF. NMR 1H (CDCl3): 1.54 (s, 3H), 2.65 (d, J = 16.6, 1H), 2.86 (d, J = 16.6, 1H), 4.91 (d, J = 15.7, 1H), 4.96 (d, J = 15.8, 1H), 6.80 (d, J = 7.8, 1H), 7.06 (t, J = 7.3, 1H), 7.17–7.31 (m, 6H), 7.45 (d, J = 7.3, 1H). NMR 13C (CDCl3): 22.07, 25.55, 43.30, 44.46, 109.23, 116.27, 122.62, 122.70, 126.69, 127.26, 128.37, 128.57, 130.49, 134.98, 141.36, 177.13.
2-(3-Ethyl-1-methyl-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
5c) [
34]
The compound 5c (Rf = 0.7 in ethyl acetate, yield 15%, 0.317 g) was obtained from 1.780 g (9.6 mmol) of 2-(1-methyl-2-oxoindolin-3-yl)acetonitrile 4e, 0.28 g (12 mmol) of sodium hydride and 1.50 mL (12 mmol) of ethyl bromide in 15 mL of dry THF. NMR 1H (CDCl3): 1.14 (t, J = 7.0, 3H), 1.73 (m, 1H), 1.85 (m, 1H), 2.55 (d, J = 16.5, 1H), 2.78 (d, J = 16.5, 1H), 3.17 (s, 3H), 6.86 (d, J = 7.8, 1H), 7.07 (t, J = 7.8, 1H), 7.20 (t, J = 7.8, 1H), 7.35 (d, J = 7.7, 1H). NMR 13C (CDCl3): 8.38, 25.63, 26.37, 29.41, 49.57, 108.52, 116.57, 122.78, 129.18, 137.47, 143.62, 176.84.
The compound 5d (Rf = 0.75 in ethyl acetate, yield 90%, 4.030 g) was obtained from 3.860 g (18 mmol) of 2-(1-methyl-2-oxo-indolin-3-yl)acetonitrile 4e, 0.54 g (19 mmol) of sodium hydride and 2.10 mL (36 mmol) of chloroacetonitrile in 35 mL of dry THF. NMR 1H (CDCl3): 2.76 (d, J = 16.8, 2H), 2.88 (d, J = 16.8, 2H), 3.10 (s, 3H), 6.88 (d, J = 7.8, 1H), 7.07 (t, J = 7.8, 1H), 7.31 (t, J = 7.7, 1H), 7.43 (d, J = 7.3, 1H). NMR 13C (CDCl3): 23.67, 26.03, 45.16, 108.76, 114.95, 122.82, 122.98, 125.94, 129.84, 142.62, 173.35. Elemental analysis: found for C13H11N3O (%): C 69.26, H 4.99, N 18.62, calculated: 69.32, H 4.92, N 18.65.
2-(1,3-Dimethyl-5-methoxy-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
5e) [
35]
The compound
5e (R
f = 0.7 in chloroform, yield 36%, 0.24 g) was obtained from 0.63 g (2.9 mmol) of 2-(1-methyl-5-methoxy-2-oxoindolin-3-yl)acetonitrile
4g, 0.07 g (3 mmol) of sodium hydride and 1 mL (8 mmol) of methyl iodide in 15 mL of dry THF. M.p. = 84–86 °C (m.p.
lit. = 84–86 °C [
30]), light-brown solid. NMR
1H (CDCl
3): 1.36 (s, 3H), 2.50 (d, J = 16.7, 1H), 2.72 (d, J = 16.7, 1H), 3.07 (s, 3H), 3.66 (s, 3H), 6.70 (d, J = 8.3, 1H). 6.73 (dd, J = 8.6, J = 2.3, 1H), 6.95 (d, J = 2.2, 1H). NMR
13C (CDCl
3): 21.67, 25.55, 25.96, 44.73, 55.21, 108.62, 109.99, 112.71, 116.20, 131.76, 135.61, 155.83, 176.53. IR, cm
−1 (film): 2950 (NH), 2270 (CN), 1720, 1600 (CO). MS-EI (70 eV), m/z: 230 (62%), 190 (100%), 175 (30%), 147 (30%), 111 (20%), 97 (25%), 69 (50%).
2-(1-Benzyl-3-methyl-5-methoxy-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
5f) [
32]
The compound 5f (Rf = 0.7 in ethyl acetate, yield 45%, 0.90 g) was obtained as pale yellow oil from 1.90 g (6.5 mmol) of 2-(1-benzyl-5-methoxy-2-oxoindolin-3-yl)acetonitrile 4h, 0.17 g (7 mmol) of sodium hydride and 2 mL (32 mmol) of methyl iodide in 25 mL of dry THF. NMR 1H (CDCl3): 1.56 (s, 3H), 2.67 (d, J = 16.6, 1H), 2.88 (d, J = 16.6, 1H), 3.74 (s, 3H), 4.88 (d, J = 15.6, 1H), 4.94 (d, J = 15.6, 1H), 6.69 (d, J = 8.6, 1H), 6.74 (dd, J = 8.6, J = 2.4, 1H), 7.09 (d, J = 2.4, 1H), 7.29 (m, 5H). NMR 13C (CDCl3): 22.35, 25.95, 43.71, 55.49, 109.99, 110.25, 113.12, 116.39, 126.92, 127.49, 128.60, 132.00, 134.82, 135.21, 156.16, 177.09.
The compound 5g (Rf = 0.8 in ethyl acetate, yield 62%, 1.16 g) was obtained from 1.67 g (7.7 mmol) of 2-(1-methyl-5-methoxy-2-oxoindolin-3-yl)acetonitrile 4g, 0.20 g (8.3 mmol) of sodium hydride and 1.20 mL (16 mmol) of ethyl bromide in 35 mL of dry THF. NMR 1H (CDCl3): 0.59 (t, J = 7.3, 3H), 1.95–2.00 (m, 2H), 2.59 (d, J = 16.6, 1H), 2.81 (d, J = 16.6, 1H), 3.20 (s, 3H), 3.79 (s, 3H), 6.73–6.87 (m, 2H), 7.01 (s, 1H). NMR 13C (CDCl3): 8.20, 25.39, 26.21, 29.22, 49.80, 55.59, 108.74, 110.39, 113.11, 116.37, 130.06, 136.81, 156.22, 176.25. HRMS-ESI, m/z: 267.1105, calculated for C14H16N2O2Na (M+Na): 267.1104.
The compound 5h (Rf = 0.8 in chloroform:ethyl acetate 1:1 (v/v), yield 20%, 0.220 g) was obtained from 0.940 g (4.3 mmol) of 2-(1-methyl-5-methoxy-2-oxoindolin-3-yl)acetonitrile 4g, 0.115 g (4.7 mmol) of sodium hydride and 1.00 mL (17 mmol) of chloroacetonitrile in 15 mL of dry THF. NMR 1H (CDCl3): 2.81 (d, J = 16.8, 2H), 2.97 (d, J = 16.8, 2H), 3.24 (s, 3H), 3.81 (s, 3H), 6.88 (d, J = 8.6, 1H), 6.95 (dd, J = 8.6, J = 2.4, 1H), 7.16 (d, J = 2.3, 1H). NMR 13C (CDCl3): 24.66, 26.90, 29.68, 46.12, 55.90, 109.98, 110.88, 114.95, 115.06, 127.53, 136.38, 156.80, 173.51. HRMS-ESI, m/z: 278.0900, calculated for C14H13N3O2Na (M+Na): 278.0900.
2-(5-Methoxy-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile (4b) (0.625 g, 3.1 mmol) was dissolved in 20 mL of dry THF and sodium hydride (60% oil suspension) (0.276 g, 6.2 mmol). The reaction mixture was vigorously stirred for 15 min, then 0.58 mL (9.3 mmol) MeI was added and solution was stirred at room temperature for 4 days. The organic solvent was evaporated, and dry residue was washed with cold water, extracted with CH2Cl2 and dried with MgSO4. After column chromatography separation, compounds 5i (Rf = 0.7 in CHCl3:EtOAc = 2:1) and 5e (Rf = 0.85 in CHCl3) were obtained.
Compound 5e was obtained as orange solid (yield 0.124 g, 17%). The spectral data of this compound are the same as for compound obtained from 4g.
Compound (5i) was obtained as bright orange solid (yield 0.163 g, 24%). M.p. 135–140 °C. NMR 1H (CDCl3): 1.53 (s, 3H), 2.65 (d, J = 16.4, 1H), 2.83 (d, J = 16.7, 1H), 3.79 (s, 3H (OMe)), 6.80 (d, J = 6.0, 1H), 6.89 (s, 1H), 7.01 (d, J = 4.7, 1H). NMR 13C (CDCl3): 22.18, 26.18, 45.84, 55.30, 110.34, 111.11, 113.82, 116.57, 132.73, 133.20, 156.26 (C-OMe), 180.06 (C=O). MS-EI (70 eV), m/z: 216 (25%), 191 (100%), 176 (67%), 148 (25%), 118 (20%), 91 (15%), 72 (30%). Elemental analysis found (%): C 66.68, H 6.62, N 12.91, calculated for C12H12N2O2 (%): C 66.65, H 5.59, N 12.96.
2-(5-Bromo-1,3-dimethyl-2-oxo-2,3-dihydro-
1H-indol-3-yl)acetonitrile (
5j) [
36]
The compound
5j (R
f = 0.63 in ethyl acetate, yield 68%, 0.87 g) was obtained from 1.23 g (4.6 mmol) of 2-(5-bromo-1-methyl-2-oxoindolin-3-yl)acetonitrile
4i, 0.123 g (5 mmol) of sodium hydride and 1 mL (16 mmol) of methyl iodide in 25 mL of dry THF. M.p. 102 °C (m.p.
lit. = 101–102 °C [
36]). NMR
1H (CDCl
3): 1.39 (s, 3H), 2.56 (d, J = 16.7, 1H), 2.76 (d, J = 16.7, 1H), 3.11 (s, 3H), 6.72 (d, J = 8.3, 1H), 7.34 (dd, J = 8.2, 1.9, 1H), 7.46 (d, J = 1.9, 1H). NMR
13C (CDCl
3): 21.68, 25.44, 26.12, 44.59, 109.85, 115.09, 115.89, 125.76, 131.50, 132.47, 141.48, 176.27.
2-(1,3,6,7-Tetramethyl-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile (5k)
The compound 5k (Rf = 0.7 in ethyl acetate:petroleum ether 1:2 (v/v)), yield 38%, 1.10 g) was obtained from 2.70 g (12.6 mmol) of 2-(1,6,7-trimethyl-2-oxoindolin-3-yl)acetonitrile 4j, 0.30 g (13 mmol) of sodium hydride and 2 mL (32 mmol) of methyl iodide in 15 mL of dry THF. NMR 1H (CDCl3): 1.46 (s, 3H), 2.31 (s, 3H), 2.48 (s, 3H), 2.55 (d, J = 16.7, 1H), 2.79 (d, J = 16.7, 1H), 3.54 (s, 3H), 6.93 (d, J = 7.6, 1H), 7.18 (d, J = 7.6, 1H). NMR 13C (CDCl3): 14.07, 19.12, 20.78, 26.38, 30.55, 43.99, 116.76, 120.09, 121.23, 129.59, 139.02, 140.73, 142.12, 175.61.
The compound 5l (orange oil, Rf = 0.8 in CH2Cl2, yield 50%, 0.215 g) was obtained from 0.41 g (1.5 mmol) of N-Boc-3-(cyanomethyl)-2-oxoindole 4k, 0.04 g (1.6 mmol) of sodium hydride and 0.5 mL (80 mmol) of methyl iodide in 20 mL of dry THF. NMR 1H (CDCl3): 1.53 (s, 3H), 1.62 (s, 9H), 2.64 (d, J = 16.7, 1H), 2.84 (d, J = 16.7, 1H), 7.18–7.21 (m, 1H), 7.32–7.36 (m, 1H), 7.45 (dd, J = 7.5, J = 1.1, 1H), 7.86 (d, J = 8.2, 1H). NMR 13C (CDCl3): 23.00, 26.47, 27.73, 45.02, 84.62, 115.18, 115.95, 122.64, 124.79, 129.10, 129.55, 138.35, 148.52, 175.84. Elemental analysis found for C16H18N2O3 (%): C 67.24, H 6.40, N 9.59, calculated: 67.12, H 6.34, N 9.78.
The compound 5m (light-orange oil, Rf = 0.8 in petroleum ether, yield 20%, 0.160 g) was obtained from 0.89 g (3.3 mmol) of N-Boc-3-(cyanomethyl)-2-oxindole 4k, 0.078 g (3.3 mmol) of sodium hydride and 2 mL (27 mmol) of ethyl bromide in 20 mL of dry THF. NMR 1H (CDCl3): 0.68 (t, J = 7.3, 3H), 1.47 (s, 9H), 1.97–2.07 (m, 2H), 2.66 (d, J = 16.7, 1H), 2.86 (d, J = 16.7, 1H), 7.37 (t, J = 7.2, 1H), 7.43 (d, J = 7.4, 1H), 7.50 (t, J = 8.1, 1H), 7.89 (d, J = 8.2, 1H). NMR 13C (CDCl3): 8.37, 22.65, 26.09, 29.66, 56.74, 87.74, 115.22, 122.98, 124.49, 125.04, 129.38, 134.03, 139.63, 148.70, 175.65.
The compound 5n (red oil, Rf = 0.89 in chloroform, yield 27%, 0.043 g) was obtained from 0.150 g (5 mmol) of N-Boc-3-(cyanomethyl)-5-methoxy-2-oxindole 4l, 0.022 g (0.92 mmol) of sodium hydride and 0.048 mL (0.75 mmol) of methyl iodide in 7 mL of dry THF. NMR 1H (CDCl3): 1.61 (s, 3H), 1.67 (s, 9H), 3.02 (d, J = 16.4, 1H), 3.26 (d, J = 16.7, 1H), 3.86 (s, 3H (OMe)), 6.93 (d, J = 9.1, 1H), 7.08 (s, 1H), 7.84 (d, J = 8.9, 1H). IR, cm−1 (film): 2250 (CN), 1790, 1770, 1740, 1600 (C=O), 1350–1150 (broad), 1080, 1050, 1020, 890, 860. MS-EI (70 eV), m/z: 316 (36%, M+), 260 (15%), 216 (100%) (M+-Boc), 201 (11%, M+-CH3), 189 (58%), 176 (70%), 158 (22%), 146 (19%), 132 (20%), 117 (27%), 104 (19%), 85 (37%), 56 (86%). Elemental analysis: found for C17H20N2O4(%): C 64.55, H 6.42, N 8.83, calculated: 64.54, H 6.37, N 8.86.
The compound 5o (yellow oil, Rf = 0.7 in chloroform, yield 16%, 0.020 g) was obtained from 0.230 g (0.76 mmol) of N-Boc-3-(cyanomethyl)-5-methoxy-2-oxoindole 4l, 0.034 g (0.83 mmol) of sodium hydride and 0.085 mL (1.14 mmol) of bromoethane in 7 mL of dry THF. NMR 1H (CDCl3): 1.63 (s, 9H), 2.22–2.34 (m, 2H), 3.03 (d, J = 16.9, 1H), 3.27 (d, J = 16.9, 1H), 3.86 (s, 3H), 6.76 (s, 1H), 6.93 (d, J = 9.9, 1H), 7.05 (d, J = 8.5, 1H).
The compound 5p (Rf = 0.7 in chloroform:ethyl acetate 1:1 (v/v), yield 19%, 0.041 g) was obtained from 0.195 g (0.65 mmol) of N-Boc-3-(cyanomethyl)-5-methoxy-2-oxoindole 4l, 0.028 g (0.71 mmol) of sodium hydride and 0.045 mL (0.71 mmol) of chloroacetonitrile in 7 mL of dry THF. NMR 1H (CDCl3): 1.67 (s, 9H), 2.71–2.91 (m, 2H), 2.98–3.12 (m, 2H), 3.82 (s, 3H), 6.69–6.73 (m, 1H), 6.86 (s, 1H), 6.90–6.94 (m, 1H). MS-EI (70 eV), m/z: 341 (2%), 257 (5%,), 241 (7%), 202 (50%), 189 (58%), 162 (100%), 148 (30%), 119 (27%), 77 (60%).
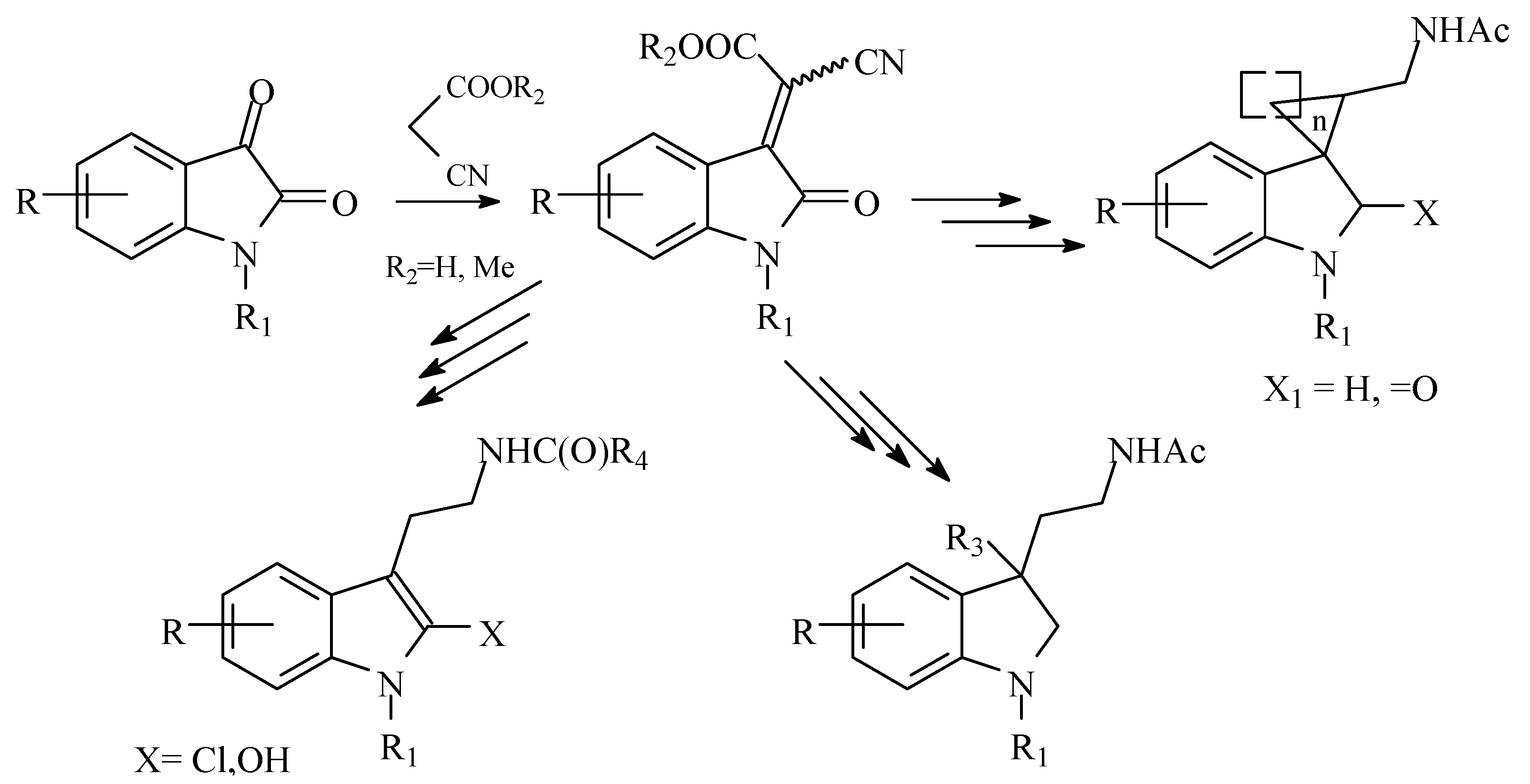
3.1.6. General Procedure for Synthesis of spiroindolin-3-ylacetonitriles (6)
N-nitrosomethylurea was added portionwise to the solution of potassium hydroxide in Et
2O/water mixture at 0–5 °C to obtain diazomethane. The ether layer was separated and dried with KOH for 20 min. The diazomethane solution was dried by filtering through anhydrous Na
2SO
4 pad, then added to the solid compound
3, and reaction mixture was vigorously stirred for 12 h until color disappeared. The reaction mixture was concentrated, and white solid residue was dissolved in toluene and heated under reflux for 8 h. The toluene was evaporated, dry residue was washed with ether, and filtered and dried in air. The following compounds were obtained using this general procedure, described by us for
6a,
6b [
19]:
5′-Bromo-2′-oxo-1′,2′-dihydrospiro[cyclopropane-1,3′-indole]-3-carbonitrile (
6c) [
37]
The diazomethane solution was obtained from 10.50 g (100 mmol) of N-nitrosomethylurea, 21.20 g (380 mmol) of KOH in 57 mL of water and 137 mL of diethyl ether. After filtration through anhydrous sodium sulfate pad, the diazomethane solution was added to 2.84 g (10.8 mmol) of 2-(5-bromo-2-oxo-2,3-dihydro-1H-indol-3-ylidene)acetonitrile (3c). The reaction mixture was stirred for 14 h at room temperature, then refluxed in 40 mL of toluene for 8 h. The compound 6c was obtained as light brown powder. Yield 1.40 g (48%), m.p. 168–175 °C. NMR 1H (DMSO-d6): 2.43–2.50 (m, 2H), 2.72–2.76 (m, 1H), 6.93 * (dd, J = 8.4, J = 2.6, 1H), 7.11 (dd, J = 8.4, J = 2.6, 1H), 7.31 * (s, 1H), 7.36 (s, 1H), 7.46 * (d, J = 8.4, 1H), 7.57 (d, J = 8.4, 1H), 10.65 * (s, 1H), 11.01 (s, 1H); *—major diastereomer, major:minor = 5:2 (by 1H NMR). NMR 13C (DMSO-d6): 15.71, 20.65, 27.19, 111.31, 112.26, 114.26, 117.68, 124.64, 131.51, 143.86, 172.45.
(1R*,2R*)-6′,7′-Dimethyl-2’-oxo-1’,2’-dihydrospiro[cyclopropane-1,3’-indole]-3-carbonitrile (6d)
The diazomethane solution was obtained from 5.30 g (60 mmol) of N-nitrosomethylurea, 10.00 g (180 mmol) of KOH in 27 mL of water and 80 mL of diethyl ether. After filtration through anhydrous sodium sulfate pad, the diazomethane solution was added to 1.05 g (5 mmol) of 2-(6,7-dimethyl-2-oxo-2,3-dihydro-1H-indol-3-ylidene)acetonitrile 3d. The reaction mixture was stirred for 12 h at room temperature, then refluxed in 25 mL of toluene for 8 h. The obtained mixture of compounds 6d and 1,3-dimethylated compound 6j was separated by column chromatography (eluent petroleum ether/ethyl acetate 8:1). Compound 6d was obtained as beige powder, single diastereomer, Rf 0.1, yield 0.378 g (34%), m.p. 145–147 °C. NMR 1H (CDCl3): 1.87 (dd, J = 6.8, J = 5.1, 1H), 2.12 (dd, J = 9.5, J = 4.9, 1H), 2.23 (s, 3H), 2.32 (s, 3H), 2.44 (dd, J = 9.5, J = 7.0, 1H), 6.94 (s, 2H), 9.87 (s, 1H). NMR 13C (CDCl3): 12.80, 14.39, 19.49, 20.53, 32.19, 117.62, 119.49, 121.35, 123.46, 133.36, 137.34, 138.30, 140.17, 175.87. Elemental analysis: found for C13H12N2O (%): C 73.57, H 5.93, N 12.98, calculated (%): C 73.56, H 5.70, N 13.20. Compound 6j was obtained as byproduct, white powder, Rf 0.25, yield 0.30 g (25%), m.p.145–147 °C. The spectral data of this compound were the same as for compound 6j obtained from 3j.
(1R*,2R*)-1′-Methyl-2’-oxo-1’,2’-dihydrospiro[cyclopropane-1,3′-indole]-3-carbonitrile (
6e) [
38]
The diazomethane solution was obtained from 7.5 g (85 mmol) of N-nitrosomethylurea, 15.2 g (270 mmol) of KOH in 41 mL of water and 124 mL of diethyl ether. After filtration through anhydrous sodium sulfate pad, the diazomethane solution was added to 1.50 g (8.2 mmol) of 2-(1-methyl-2-oxo-2,3-dihydro-1H-indol-3-ylidene)acetonitrile 3e. The reaction mixture was stirred for 12 h at room temperature, then refluxed in 30 mL of toluene for 8 h. The compound 6e was obtained as beige powder, single diastereoisomer after column chromatography. Yield 0.94 g (58%), m.p. 129–131 °C. NMR 1H (DMSO-d6): 1.91 (dd, J = 5.1, J = 6.8, 1H), 2.14 (dd, J = 5.1, J = 9.5, 1H), 2.45 (dd, J = 7.1, J = 9.46, 1H), 3.31 (s, 3H), 6.98 (d, J = 7.7, 1H), 7.16 (t, J = 7.5, 1H), 7.22 (d, J = 6.8, 1H), 7.40 (t, J = 7.5, 1H). NMR 13C (DMSO-d6): 14.81, 21.34, 26.88, 31.73, 108.71, 116.91, 118.88, 120.93, 120.90, 128.85, 144.14, 172.96. Elemental analysis: found for C12H10N2O (%): C 72.74, H 5.17, N 14.25, calculated (%): C 72.71, H 5.08, N 14.13.
(1R*,2R*)-1’-Methyl-5’-methoxy-2’-oxo-1’,2’-dihydrospiro[cyclopropane-1,3′-indol]-3-carbonitrile (
6g) [
39]
The diazomethane solution was obtained from 6.53 g (74 mmol) of
N-nitrosomethylurea, 12.40 g (220 mmol) of KOH in 31 mL of water and 100 mL of diethyl ether. After filtration through anhydrous sodium sulfate pad, the diazomethane solution was added to 1.40 g (6.5 mmol) of 2-(5-methoxy-1-methyl-2-oxo-2,3-dihydro-
1H-indol-3-ylidene)acetonitrile
3g. The reaction mixture was stirred for 12 h at room temperature, then refluxed in 30 mL of toluene for 8 h. The compound
6g was obtained as beige powder, single isomer after column chromatography, m.p. = 164–165 °C (m.p.
lit. = 164–165 °C [
39]), yield 1.02 g (68%). NMR
1H (CDCl
3): 1.88 (dd, J = 5.0, J = 6.8, 1H), 2.14 (dd, J = 4.9, J = 9.4, 1H), 2.46 (dd, J = 6.9, J = 9.4, 1H), 3.29 (s, 3H), 3.82 (s, 3H), 6.82 (d, J = 2.3, 1H), 6.87 (d, J = 8.6, 1H), 6.92 (dd, J = 2.3, J = 8.5, 1H). NMR
13C (CDCl
3): 14.69, 21.22, 26.75, 30.81, 31.60, 55.81, 108.24, 109.08, 113.22, 116.93, 137.50, 156.03, 172.51. HRMS-ESI: 251.0791, calculated for C
13H
12N
2O
2Na (M+Na): 251.0791.
The diazomethane solution was obtained from 3.00 g (34 mmol) of N-nitrosomethylurea, 5.75 g (102 mmol) of KOH in 16 mL of water and 46 mL of diethyl ether. After filtration through anhydrous sodium sulfate pad, the diazomethane solution was added to 0.80 g (3 mmol) of 2-(5-bromo-1-methyl-2-oxo-2,3-dihydro-1H-indol-3-ylidene)acetonitrile 3i. The reaction mixture was stirred for 12 h at room temperature, then refluxed in 10 mL of toluene for 8 h. The compound 6i was obtained as mixture of two diastereomers. Yield 0.30 g (36%), white powder, m.p. 205–207 °C. NMR 1H (CDCl3): major diastereomer: 1.91 (dd, J = 7.1, J = 5.2, 1H), 2.15 (dd, J = 9.5, J = 5.1, 1H), 2.48 (dd, J = 9.5, J = 7.1, 1H), 3.28 (s, 3H), 6.84 (d, J = 8.3, 1H), 7.30 (d, J = 1.8, 1H), 7.50 (dd, J = 8.3, J = 1.9, 1H); minor diastereomer: 2.01 (dd, J = 9.2, J = 5.2, 1H), 2.20 (dd, J = 7.5, J = 5.2, 1H), 2.36 (dd, J = 9.2, J = 7.5, 1H), 3.31 (s, 3H), 6.83 (d, J = 8.1, 1H), 6.96 (d, J = 1.8, 1H), 7.46 (dd, J = 8.3, J = 1.8, 1H); major:minor = 4.5:1 (by 1H NMR). NMR 13C (CDCl3) major: 15.06, 21.54, 26.89, 31.41, 109.96, 115.44, 116.34, 122.24, 124.03, 126.11, 131.63, 143.12, 172.32. Elemental analysis: found for C12H9N2OBr (%): C 52.04, H 3.25, N 10.06, calculated (%): C 52.01, H 3.27, N 10.11.
1′,6′,7′-Trimethyl-2′-oxo-1′,2′-dihydrospiro[cyclopropane-1,3′-indole]-3-carbonitrile (6j)
The diazomethane solution was obtained from 2.57 g (27 mmol) of N-nitrosomethylurea, 4.80 g (80 mmol) of KOH in 13 mL of water and 38 mL of diethyl ether. After filtration through anhydrous sodium sulfate pad, the diazomethane solution was added to 0.56 g (2.5 mmol) of 2-(1,6,7-trimethyl-2-oxo-2,3-dihydro-1H-indol-3-ylidene)acetonitrile 3j. The reaction mixture was stirred for 12 h at room temperature, then refluxed in 15 mL of toluene for 8 h. The compound 6j was obtained as mixture of diastereomers. Yield 0.57 g (93%), dark orange oil. NMR 1H (CDCl3): major diastereomer 1.79–1.82 (m, 1H), 2.04–2.10 (m, 1H), 2.33 (s, 3H), 2.35–2.41 (m, 1H), 2.50 (s, 3H), 3.57 (s, 3H), 6.90 (d, J = 7.7, 1H), 6.95 (d, J = 7.7, 1H); minor diastereomer, 1.87–1.91 (m, 1H), 2.06–2.11 (m, 1H), 2.21–2.25 (m, 1H), 2.29 (s, 3H), 2.48 (s, 3H), 3.58 (s, 3H), 6.53 (d, J = 7.5, 1H), 6.84 (d, J = 7.5, 1H); major:minor = 3:1 (by 1H NMR). NMR 13C (DMSO-d6): 13.96, 15.09, 20.84, 21.26, 30.87, 38.65, 117.94, 119.70, 124.18, 129.69, 130.91, 138.62, 142.12, 173.98. Elemental analysis: found for major isomer C14H14N2O (%): C 74.40, H 6.36, N 12.42, calculated (%): C 74.31, H 6.24, N 12.38.
3.1.7. General Procedure for Synthesis of N-methyl-substituted Compounds 6g, 6i,6j
Acetonitrile 6 was dissolved in dry THF, and then, sodium hydride (60% suspension in oil) was added. The mixture was stirred about 30 min before addition of excess MeI. The reaction mixture was stirred at room temperature for 48 h. Organic solvent was evaporated, oil residue was washed with ice-water, extracted by ethyl acetate and dried under MgSO4. The following compounds were obtained using this procedure:
To the solution of 5′-methoxy-2’-oxo-1’,2’-dihydrospiro[cyclopropane-1,3′-indol]-3-carbonitrile, 6b (1.00 g, 4.7 mmol) in 40 mL of THF 0.14 g (6 mmol) of sodium hydride was added. The reaction mixture was stirred for 5 min, and 2 mL (32 mmol) of MeI was added. Compound (6g) was obtained as beige powder, yield 1.01 g (93%). Spectral data are the same as for compound major isomer obtained using general procedure 3.1.6.
To the solution of 5’-bromo-2’-oxo-1’,2’-dihydrospiro[cyclopropane-1,3’-indole]-3-carbonitrile, 6c (1.28 g, 4.9 mmol) in 40 mL of THF 0.13 g (5.4 mmol) of sodium hydride was added. The reaction mixture was stirred for 5 min, and 1 mL (16 mmol) of MeI was added. Compound (6i) was obtained as beige powder, yield 1.01 g (74%). Spectral data are the same as for compounds obtained using the general procedure in 3.1.6.
1′,6′,7′-Trimethyl-2′-oxo-1′,2′-dihydrospiro[cyclopropane-1,3′-indole]-3-carbonitrile (6j)
To the solution of 6′,7′-dimethyl-2’-oxo-1’,2’-dihydrospiro[cyclopropane-1,3’-indole]-3-carbonitrile, 6d (1.80 g, 8.5 mmol) in 25 mL of THF 0.22 g (9 mmol) of sodium hydride was added. The reaction mixture was stirred for 5 min, and 2 mL (32 mmol) of MeI was added. Compound (6j) was obtained as dark orange oil, yield 1.20 g (62%). Spectral data are the same as for major isomer obtained using general procedure 3.1.6 from 3j.
3.1.8. General Procedure for Selective Reduction of Nitrile Group
Method A:
The acetonitrile 4 was dissolved in glacial acetic acid and hydrogenated at room temperature and atmospheric pressure in the presence of acetic anhydride. The platinum catalyst was filtered off, and the reaction mixture was evaporated to dry residue. The solid was washed with NaHCO3 and extracted with dichloromethane.
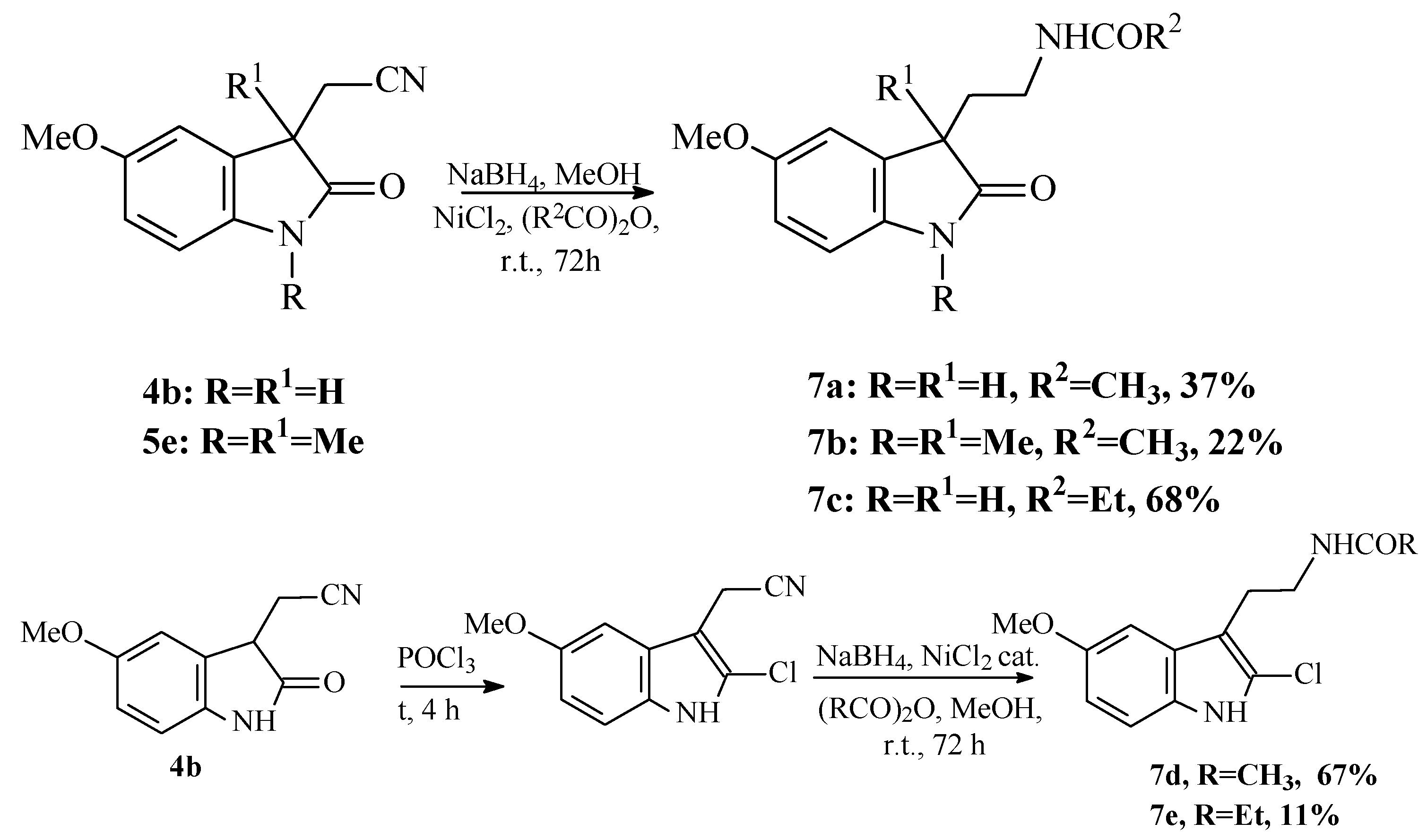
Method B:
The acetonitrile 4 was dissolved in methanol, and then, acetic anhydride and anhydrous NiCl2 were added. The reaction mixture was cooled, NaBH4 was added slowly portionwise. Reaction mixture was vigorously stirred for 4 days, the solvent was evaporated, dry residue washed with saturated solution of K2CO3 and extracted with dichloromethane.
The following compounds were synthesized according to these procedures:
Method A:
First, 1.00 g (4 mmol) of (5-methoxy-2-oxoindolin-3-yl)acetonitrile 4b was hydrogenated under vigorous stirring in 15 mL of AcOH in presence of 0.5 mL Ac2O with 50 mg (0.22 mmol) PtO2 until 0.222 l of H2 reacted (~4 h). Yield 1.00 g (80%), m.p. = 138–146 °C. NMR 1H (CDCl3): 1.95 (s, 3H), 1.98–2.09 (m, 1H), 2.17–2.24 (m, 1H), 3.41–3.51 (m, 2H), 3.77 (c, 3H, CH3O), 6.58 (br. s, 1H, NH), 6.72–6.75 (dd, J = 10.6, J = 6.4, 1H), 6.79–6.81 (d, J = 8.4, 1H), 6.9 (s, 1H), 9.04 (s,1H, NH). NMR 13C (CDCl3): 23.04 (CH3), 29.94, 36.96, 44.75, 55.76 (OCH3), 110.38, 111.09, 112.75, 130.59, 135.01, 155.76 (CAr-OCH3), 170.82 (C=O), 180.47 (C=O). IR, cm−1: 3298–3200 (NH), 1650 broad (C=O), 1697 (C=O). MS-EI, m/z: 248 (35%), 206 (7%, M+-CH3CO), 189 (45%), 176 (100%, M+-CH3CONHCH2), 163 (17%), 117 (23%), 83 (38%). Elemental analysis: found for C13H16N2O3 (%): C 62.80, H 6.46, N 11.22, calculated (%): C 62.89, H 6.50, N 11.28.
Method B:
To the solution of 0.436 g (2.2 mmol) of (5-methoxy-2-oxoindolin-3-yl)acetonitrile 4b in 10 mL of MeOH, 0.63 mL (6.7 mmol) of Ac2O, 0.018 g (0.46 mmol) of freshly prepared anhydrous NiCl2 and finally 0.44 g (11 mmol) NaBH4 were added at 0–10 °C. The reaction mixture was stirred at room temperature for 4 d, and twice per day, 0.4 g of NaBH4 was added additionally. Yield 0.20g (37%). The analytical data of obtained compound 7a were identical to compound prepared according to method A.
Method B:
To the solution of 0.124 г (0.54 mmol) 2-(1,3-dimethyl-5-methoxy-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile (5e), 0.0145 g (0.12 mmol) freshly prepared NiCl2, 0.16 mL (0.17 mmol) Ac2O in 10 mL MeOH 0.13 g (0.34 mmol) NaBH4 were added with cooling (10–15 °C). The reaction mixture was stirred at room temperature for 4 days, the additional 0.13 g (0.34 mmol) of NaBH4 was added each day twice per day. Yield 22% (0.033 g). M.p. = 125–130 °C.
NMR 1H (CDCl3): 1.30 (s, 3H), 1.84 (s, 3H), 1.94–2.24 (m, 2H), 3.15 (s, 3H), 3.17–3.21 (m, 2H), 3.79 (s, 3H, OCH3), 6.71 (d, J = 8.8, 1H), 6.74–6.82 (m, 4H). MS-EI (70 eV), m/z: 276 (5%), 245 (2%,), 218 (5%), 191 (50%), 174 (10%), 132 (15%), 118 (20%), 84 (100%), 72 (60%), 43 (80%). HRMS-ESI: 277.1546, calculated for C15H21N2O3 (M+H): 277.1547.
Method B:
To the solution of 0.211 g (1.00 mmol) of (5-methoxy-2-oxoindolin-3-yl)acetonitrile 4b in 10 mL of MeOH, 0.300 mL (1.0 mmol) of propionic anhydride, 0.027 g (0.21 mmol) of freshly prepared anhydrous NiCl2 and finally 0.200 g (5.3 mmol) NaBH4 were added at 0–10 °C. The reaction mixture was stirred at room temperature for 4 d and twice per day 0.100 g of NaBH4 were added additionally. Yield 0.177 g (68%).
NMR 1H (CDCl3): 1.26 (s, 3H), 2.04–2.08 (m, 3H), 2.18–2.28 (m, 2H), 3.20–3.27 (m, 1H), 3.73 (s, 3H), 6.68 (m, 2H), 6.93 (s, 1H). HRMS-ESI: 263.1389, calculated for C14H19N2O3 (M+H): 263.1390.
Method B:
To the solution of 0.109 g (5.40 mmol) of (5-methoxy-2-chloroindolin-3-yl)acetonitrile [
20] in 10 mL of MeOH, 0.14 mL (5.4 mmol) of propionic anhydride, 0.016 g (0.12 mmol) of freshly prepared anhydrous NiCl
2 and finally 0.110 g (3 mmol) NaBH
4 were added at 0–10 °C. The reaction mixture was stirred at room temperature for 4 d, and twice per day, 0.100 g of NaBH
4 was added additionally. Yield 0.017 g (11%).
NMR 1H (CDCl3): 1.25–1.27 (m, 3H), 2.12–2.18 (m, 2H), 3.02–3.04 (m, 2H), 3.68–3.73 (m, 2H), 3.84 (s, 3H), 6.83 (d, J = 7.1, 1H), 6.99 (d, J = 7.2, 1H), 7.15 (s, 1H). HRMS-ESI: 281.1053, calculated for C14H18ClN2O3 (M+H): 281.1051.
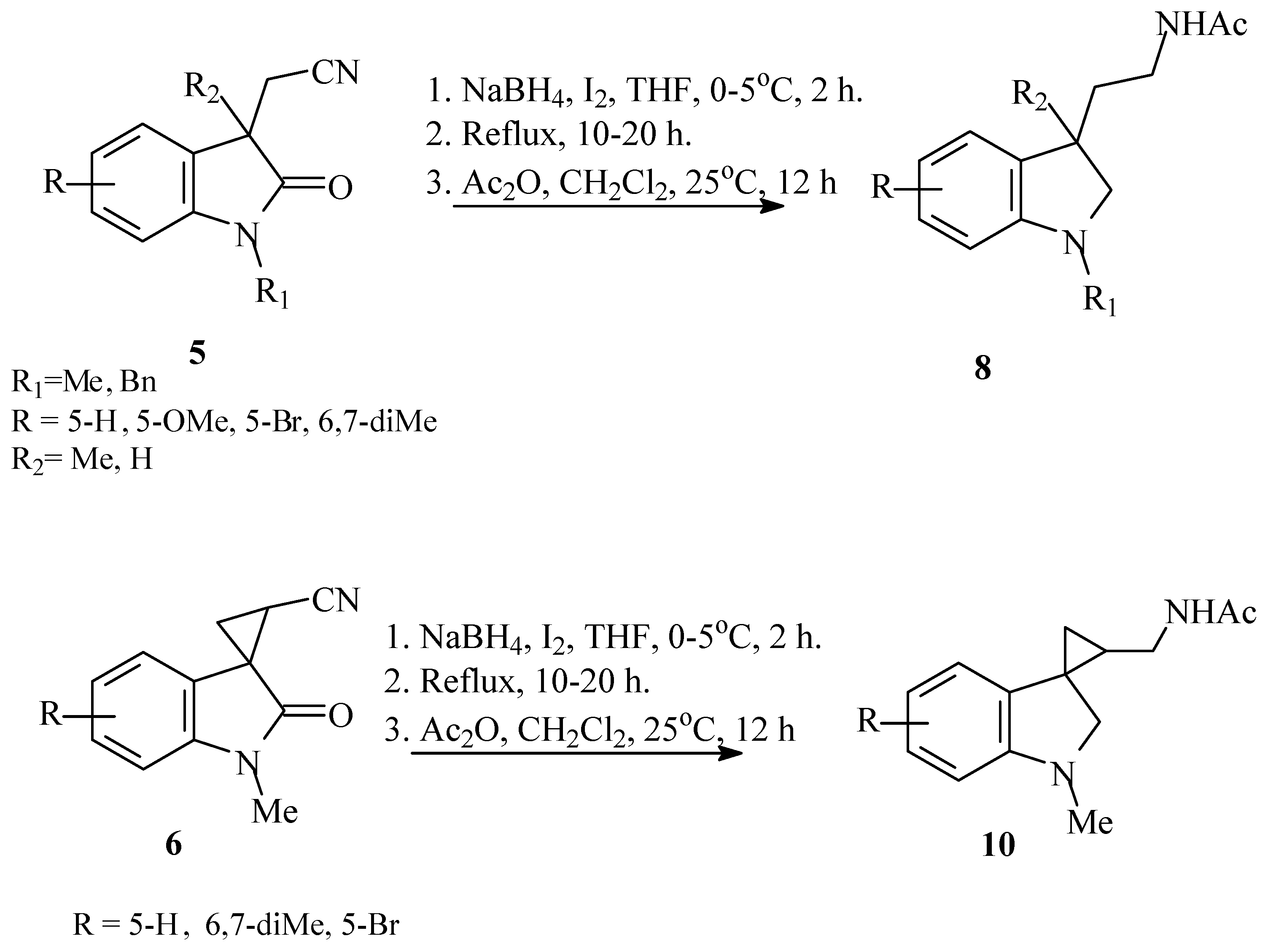
3.1.9. General Procedure for Synthesis of Melatonin Analogues 8,10
The acetonitriles (4–6) were dissolved in anhydrous THF, and NaBH4 was added at 0 °C in argon atmosphere. The solution of I2 in THF was added dropwise during 2–4 h at 0–5 °C. The reaction mixture was stirred at reflux until the solution color turned to light yellow (~2 h). The solvent was evaporated; dry residue was washed with 3N HCl, extracted with dichloromethane and dried under Na2SO4. To the dry dichloromethane fraction, Ac2O was added and reaction mixture was stirred for 14 h at room temperature. The reaction mixture was evaporated, and to the dry residue, EtOAc was added. The organic fraction was washed with saturated solution of sodium hydrocarbonate, or diluted solution of potassium carbonate was added. (Note: avoid the water treatment stage for 5-acetamido derivatives because of its good water solubility.) After additional extraction with dichloromethane, chloroform or ethyl acetate organic fraction was washed with water, dried with Na2SO4, and solvent was evaporated. Residue was purified using column chromatography (eluent ethyl acetate:chloroform 1:1). The following compounds were obtained using this general procedure:
The solution of 1.780 g (7.7 mmol) of iodine in 7 mL of THF was added portionwise to the mixture of 0.700 g (3.5 mmol) of 2-(1,3-dimethyl-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 5a and 0.600 g (16 mmol) of sodium borohydride in 30 mL of THF. After stirring with reflux for 5 h, the reaction was terminated by addition of 2 mL of acetic acid. Then, 2 mL of acetic anhydride was added to the reaction mixture and stirred for 14 h. The compound 8a was obtained after purification with column chromatography (Rf 0.28 in ethyl acetate/CH2Cl2 1:1) as light yellow oil that turned green on TLC. Yield 39% (0.360 g). 1H NMR (CDCl3): 1.24 (s, 3H), 1.64–1.79 (m, 2H), 1.76 (s, 3H), 2.66 (s, 3H), 2.91 (d, J = 8.9, 1H), 3.07–3.12 (m, 2H), 3.20 (d, J = 8.8, 1H), 6.41 (d, J = 7.8, 1H), 6.62 (t, J = 6.7, 1H), 6.90 (d, J = 6.6, 1H), 7.01 (t, J = 7.6, 1H). 13C (CDCl3): 22.68 (CH3), 25.53, 35.57, 35.80, 39.70, 42.32, 67.56, 107.26, 117.82, 121.78, 127.45, 136.62, 151.61, 169.74. HRMS-ESI: 255.1468, calculated for C14H20N2O(M+Na): 255.1468.
The solution of 1.840 g (7.2 mmol) of iodine in 10 mL of THF was added portionwise to the mixture of 1.000 g (3.6 mmol) of 2-(1-benzyl-3-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 5b and 0.680 g (18 mmol) of sodium borohydride in 40 mL of THF. After stirring with reflux for 10 h, the reaction was terminated by addition of 10 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 1 mL (10 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 8b was obtained as red brown oil after purification with column chromatography (Rf 0.32 in ethyl acetate). Yield 51% (0.555 g).
1H NMR (CDCl3): 1.35 (s, 3H), 1.76 (s, 3H), 1.78–1.89 (m, 2H), 3.05 (d, J = 9.1, 1H), 3.18–3.23 (m, 2H), 3.29 (d, J = 9.0, 1H), 4.13 (d, J = 14.7, 1H), 4.39 (d, J = 14.7, 1H), 5.91 (br. s, 1H), 6.57 (d, J = 7.8, 1H), 6.75 (t, J = 7.3, 1H), 7.04 (d, J = 7.3, 1H), 7.12 (t, J = 7.7, 1H), 7.28–7.37 (m, 5H). 13C NMR (CDCl3): 22.83, 26.18, 36.10, 40.32, 42.44, 53.29, 65.39, 107.22, 117.95, 122.24, 127.07, 127.72, 128.42, 136.38, 139.96, 151.35, 169.89. HRMS-ESI: 331.1781, calculated for C20H25N2ONa(M+Na): 331.1781.
The solution of 0.620 g (2.4 mmol) of iodine in 5 mL of THF was added portionwise to the mixture of 0.300 g (12 mmol) of 2-(3-ethyl-1-methyl-5-methoxy-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 5g and 0.213 g (5.6 mmol) of sodium borohydride in 20 mL of THF. After stirring with reflux for 12 h, the reaction was terminated by addition of 10 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 0.8 mL (8 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 8c was obtained as light red oil after purification with column chromatography (Rf 0.21 in ethyl acetate). Yield 31% (0.105 g). 1H NMR (CDCl3): 0.8 (t, J = 7.5, 1H), 1.63–1.80 (m, 2H), 1.84 (s, 3H), 1.92–2.11 (m, 2H), 2.72 (s, 3H), 2.96–3.06 (m, 4H), 3.75 (s, 3H), 5.73 (br.s, 1H), 6.45 (d, J = 8.5, 1H), 6.59 (d, J = 2.5, 1H), 6.69 (dd, J = 8.5, J = 2.5, 1H). 13C NMR (CDCl3): 8.75, 23.11, 26.20, 31.67, 35.85, 36.16, 46.65, 55.77, 66.32, 108.31, 110.42, 112.11, 137.09, 147.86, 153.29, 169.83. HRMS-ESI: 247.1805, calculated for C15H23N2O(M+H): 247.1805.
The solution of 5.89 g (23 mmol) of iodine in 7 mL of THF was added portionwise to the mixture of 1.74 g (7.7 mmol) of 2-(1-methyl-3-cyanomethyl-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 5d and 1.36 g (69 mmol) of sodium borohydride in 30 mL of THF. After stirring with reflux for 27 h, the reaction was terminated by addition of 10 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 1 mL (10 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 8d was obtained as light yellow oil after purification with column chromatography (Rf 0.53 in ethyl acetate). Yield 66% (1.56 g).
1H NMR (CDCl3): 1.72–1.77 (m, 2H), 1.85-1.92 (m, 2H), 2.09 (s, 3H), 2.72–2.81 (m, 2H), 2.77 (s, 6H), 3.16–3.21 (m, 4H), 6.47 (d, J = 7.8, 1H), 6.67 (t, J = 7.8, 1H), 6.94 (d, J = 7.3, 1H), 7.07 (t, J = 7.3, 1H). 13C NMR (CDCl3): 22.19, 29.79, 33.21, 34.95, 35.78, 43.72, 61.28, 108.12, 115.09, 122.05, 128.18, 138.59, 151.26, 166.26. HRMS-ESI: 304.2020, calculated for C17H26N3O2 (M+H): 304.2020.
The solution of 1.87 g (7.4 mmol) of iodine in 5 mL of THF was added portionwise to the mixture of 0.85 g (3.7 mmol) of 2-(1,3-dimethyl-5-methoxy-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 5e and 0.70 g (18 mmol) of sodium borohydride in 30 mL of THF. After stirring with reflux for 18 h, the reaction was terminated by addition of 10 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 1 mL (10 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 8e was obtained as light red oil after purification with column chromatography (Rf 0.31 in ethyl acetate). Yield 72% (0.70 g). 1H NMR (CDCl3): 1.32 (s, 3H), 1.70–1.82 (m, 2H), 1.84 (s, 3H), 2.71 (br.s, 3H), 2.90–2.95 (m, 1H), 3.02–3.10 (m, 1H), 3.26–3.34 (m, 2H); 3.75 (br. s, 3H), 5.98 (br. s, 1H), 6.47 (d, J = 7.7, 1H), 6.62 (s, 1H), 6.68 (d, J = 7.2, 1H). 13C NMR (CDCl3): 21.49 (CH3), 22.39, 30.34, 35.87, 36.62, 42.58, 55.31, 68.35, 108.09, 109.32, 111.89, 138.54, 146.17, 153.20, 166.26. HRMS-ESI: 263.1754, calculated for C15H23N2O2(M+H): 263.1754.
The solution of 0.762 g (3.0 mmol) of iodine in 7 mL of THF was added portionwise to the mixture of 0.45 g (1.5 mmol) of 2-(1-benzyl-3-methyl-5-methoxy-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 5f and 0.27 g (6.9 mmol) of sodium borohydride in 40 mL of THF. After stirring with reflux for 2 h, the reaction was terminated by addition of 3 mL of AcOH. Then, 2 mL (20 mmol) of acetic anhydride was added to the reaction mixture and stirred for 14 h. The compound 8f was obtained as reddish oil after purification with column chromatography (Rf 0.52 in ethyl acetate:CH2Cl2). Yield 77% (0.39 g). 1H NMR (CDCl3): 1.29 (s, 3H), 1.68–1.83 (m, 2H), 1.71 (s, 3H), 2.93–2.90 (m, 1H), 3.05–3.12 (m, 1H), 3.18–3.26 (m, 2H), 3.73 (s, 3H), 3.78 (s, 3H), 3.97 (d, J = 14.2, 1H), 4.30 (d, J = 14.3, 1H), 6.01 (br.s, 1H), 6.47–6.51 (m, 1H), 6.61–6.68 (m, 2H), 7.21–7.34 (m, 5H). 13C NMR (CDCl3): 22.85, 26.03, 36.09, 40.25, 42.71, 54.82, 55.74, 66.18, 108.26, 109.69, 112.05, 127.10, 127.95, 128.40, 134.27, 138.00, 138.27, 145.61, 153.29, 169.86. HRMS-ESI: 361.1886, calculated for C21H26N2O2Na(M+Na): 361.1886.
The solution of 1.120 g (4.4 mmol) of iodine in 10 mL of THF was added portionwise to the mixture of 0.471 g (2.2 mmol) of nitrile 3e and 0.390 g (10 mmol) of sodium borohydride in 20 mL of THF. After stirring with reflux for 5 h, the reaction was terminated by addition of 2 mL of acetic acid and 0.40 mL (4 mmol) of acetic anhydride was added to the reaction mixture and stirred for 14h. The compounds 8g and 9g were obtained as light red oils after purification with column chromatography.
Compound 8g, yield 14% (0.148 g). 1H NMR (CDCl3): 1.85 (s, 3H), 1.89–1.98 (m, 2H), 2.69 (s, 3H), 3.10-3.19 (m, 2H), 3.35–3.40 (m, 3H), 3.63 (s, 3H), 5.25 (s, 1H), 6.58 (d, J = 8.01, 1H), 6.66 (s, 1H), 6.77 (d, J = 8.8, 1H). HRMS-ESI: 249.1598, calculated: 249.1597 for C14H21N2O2 (M+H).
Compound 9g, yield 34% (0.186 g). 1H NMR (CDCl3): 1.94 (s, 3H), 2.93 (t, J = 6.7, 2H), 3.57 (q, J = 6.5, 2H), 3.74 (s, 3H), 3.87 (s, 3H), 5.59 (s, 1H), 6.92 (dd, J = 8.8, J = 2.4, 1H), 7.03 (d, J = 2.4, 1H), 7.21 (d, J = 8.8, 1H). 13C NMR (CDCl3): 23.13, 24.93, 32.56, 39.77, 55.77, 100.49, 109.87, 111.65, 112.86, 127.18, 127.85, 132.27, 153.55, 170.18. HRMS-ESI: 247.1442, calculated: 247.1441 for C14H19N2O2 (M+H).
The solution of 1.770 g (7 mmol) of iodine in 10 mL of THF was added portionwise to the mixture of 1.010 g (3.5 mmol) of 2-(1-benzyl-5-methoxy-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 4h and 0.600 g (16 mmol) of sodium borohydride in 20 mL of THF. After stirring with reflux for 10 h, the reaction was terminated by addition of 15 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 0.5 mL (5 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 8h was obtained as light yellow oil after purification with column chromatography. Yield 11% (0.125 g). 1H NMR (CDCl3): 1.72–1.74 (m, 4H), 1.94–2.05 (m, 2H), 3.01–3.12 (m, 3H), 3.20-3.40 (m, 2H), 3. (s, 3H), 4.22 (d, J = 14.9, 1H), 4.28 (d, J = 14.9, 1H), 6.56 (d, J = 6.8, 1H), 6.73 (t, J = 7.3, 1H), 7.13 (m, 5H), 7.66 (d, J = 7.8, 1H). HRMS-ESI: 325.1911, calculated for C20H25N2O2 (M+H): 325.1911.
The solution of 4.400 g (17.3 mmol) of iodine in 10 mL of THF was added portionwise to the mixture of 1.200 g (8.6 mmol) of 2-(1-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 4e and 1.520 g (40 mmol) of sodium borohydride in 20 mL of THF. After stirring with reflux for 10 h, the reaction was terminated by addition of 10 mL of 3N HCl. After extraction with methylene dichloride, 1.20 mL (13 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 8i was obtained as yellow oil after purification with column chromatography together with 9i as byproduct. For compound 8i: Yield 12% (0.225 g). NMR 1H (CDCl3): 1.27 (s, 3H), 1.64–1.77 (m, 2H), 1.87–2.06 (m, 3H), 3.23–3.26 (m, 5H), 3.66–3.75 (m, 2H), 7.26–7.29 (m, 1H), 7.48–7.51 (m, 1H), 7.59–7.63 (m, 1H), 7.82 (d, J = 8.1, 1H). MS: 216 (M+, 10%), 157 (80%), 144 (100%), 115 (20%), 89 (7%), 77 (10%). HRMS-ESI: 233.1647, calculated for C14H21N2O (M+H): 233.1648. For compound 9i: 2.01 (s, 3H), 3.01 (t, J=6.8, 2H), 3.61-3.66 (m, 2H), 3.78 (s, 3H), 6.03 (br. s 1H), 6.96 (s, 1H), 7.13 (t, J=7.9, 1H), 7.25 (d, J=8.7, 1H), 7.33 (d, J=8.2, 1H), 7.59 (d, J=7.8, 1H).
The solution of 1.710 g (6 mmol) of iodine in 5 mL of THF was added portionwise to the mixture of 0.870 g (3.1 mmol) of 2-(5-bromo-1,3-dimethyl-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile (5j) and 0.600 g (15 mmol) of sodium borohydride in 40 mL of THF. After stirring with reflux for 15 h, the reaction was terminated by addition of 10 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 0.80 mL (8 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 8j was obtained as yellow oil after purification with column chromatography (Rf 0.37 in ethyl acetate). Yield 21% (0.198 g). 1H NMR (CDCl3): 1.29 (s, 3H), 1.74 (s, 3H), 1.75–1.78 (m, 1H), 1.82–1.84 (m, 1H), 2.71 (s, 3H), 3.01 (d, J = 9.1, 1H), 3.11–3.17 (m, 2H), 3.33 (d, J = 9.4, 1H), 6.22 (br.s, 1H), 6.38 (d, J = 8.3, 1H), 7.02 (d, J = 1.9, 1H), 7.13 (dd, J = 8.3, J = 1.9, 1H). 13C NMR (CDCl3): 21.84, 25.71, 29.22, 29.34, 42.73, 60.46, 109.83, 121.11, 125.23, 130.38, 139.71, 149.51, 170.50. HRMS-ESI: 311.0753, calculated for C14H20N2OBr(M+H): 311.0753.
N-[2-(1,3,6,7-Tetramethyl-2,3-dihydro-1H-indol-3-yl)ethyl]acetamide (8k)
The solution of 1.81 g (7 mmol) of iodine in 7 mL of THF was added portionwise to the mixture of 0.814 g (3.6 mmol) of 2-(1,3,6,7-tetramethyl-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 5k and 0.81 g (21 mmol) of sodium borohydride in 30 mL of THF. After stirring with reflux for 15 h, the reaction was terminated by addition of 10 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 1 mL (10 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 8k was obtained as light orange oil after purification with column chromatography (Rf 0.36 in ethyl acetate). Yield 50% (0.47 g). 1H NMR (CDCl3): 1.27 (s, 3H), 1.70–1.77 (m, 2H), 1.84 (s, 3H), 2.20 (s, 3H), 2.21 (s, 3H), 2.89 (s, 3H), 3.02 (d, J = 10.1, 1H), 3.11–3.16 (m, 2H), 3.40 (d, J = 10.2, 1H), 6.72 (d, J = 7.7, 1H), 6.75 (d, J = 7.5, 1H). HRMS-ESI: 261.1962, calculated for C16H25N2O(M+H): 261.1961.
The solution of 0.526 g (2 mmol) of iodine in 2 mL of THF was added portionwise to the mixture of 0.296 g (1.38 mmol) of 2-(1,6,7-trimethyl-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 4j and 0.157 g (4 mmol) of sodium borohydride in 10 mL of THF. After stirring with reflux for 8 h, the reaction was terminated by addition of 15 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 0.3 mL (3 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 8l was obtained as light-brown oil after purification with column chromatography. Yield 24% (0.082 g). The product of aromatization, indol 9l, was also isolated after column chromatography For compound 8l: 1H NMR (DMSO-d6): 1.62–1.67 (m, 1H), 1.77–1.83 (m, 2H), 1.80 (s, 3H), 2.33 (s, 3H), 2.45 (s, 3H), 2.65 (s, 3H), 2.85–2.89 (m, 1H), 3.03–3.07 (m, 1H), 3.24–3.29 (m, 2H), 6.48 (d, J = 7.7, 1H), 6.69 (d, J = 7.2, 1H). 13C NMR (DMSO-d6): 13.47, 19.47, 23.13, 25.44, 30.03, 36.37, 36.56, 63.24, 110.68, 116.19, 116.54, 125.71, 126.37, 129.07, 129.93, 134.17, 169.16. HRMS-ESI: 247.1806, calculated for C15H23N2O (M+H): 247.1805. For compound 9l: 1H NMR (CDCl3): 1.94 (s, 3H), 2.40 (s, 3H), 2.65 (s, 3H), 2.88 (t, J = 6.8, 2H), 3.52-3.55 (m, 2H), 4.00 (s, 3H), 5.79 (br.s, 1H), 6.70 (s, 1H), 6.92 (d, J = 8.0, 1H), 7.28 (d, J = 8.1, 1H). 13C NMR (CDCl3): 14.36, 20.86, 23.14, 24.83, 37.22, 39.76, 110.55, 115.78, 119.35, 122.23, 127.44, 128.58, 130.06, 136.38, 170.04.
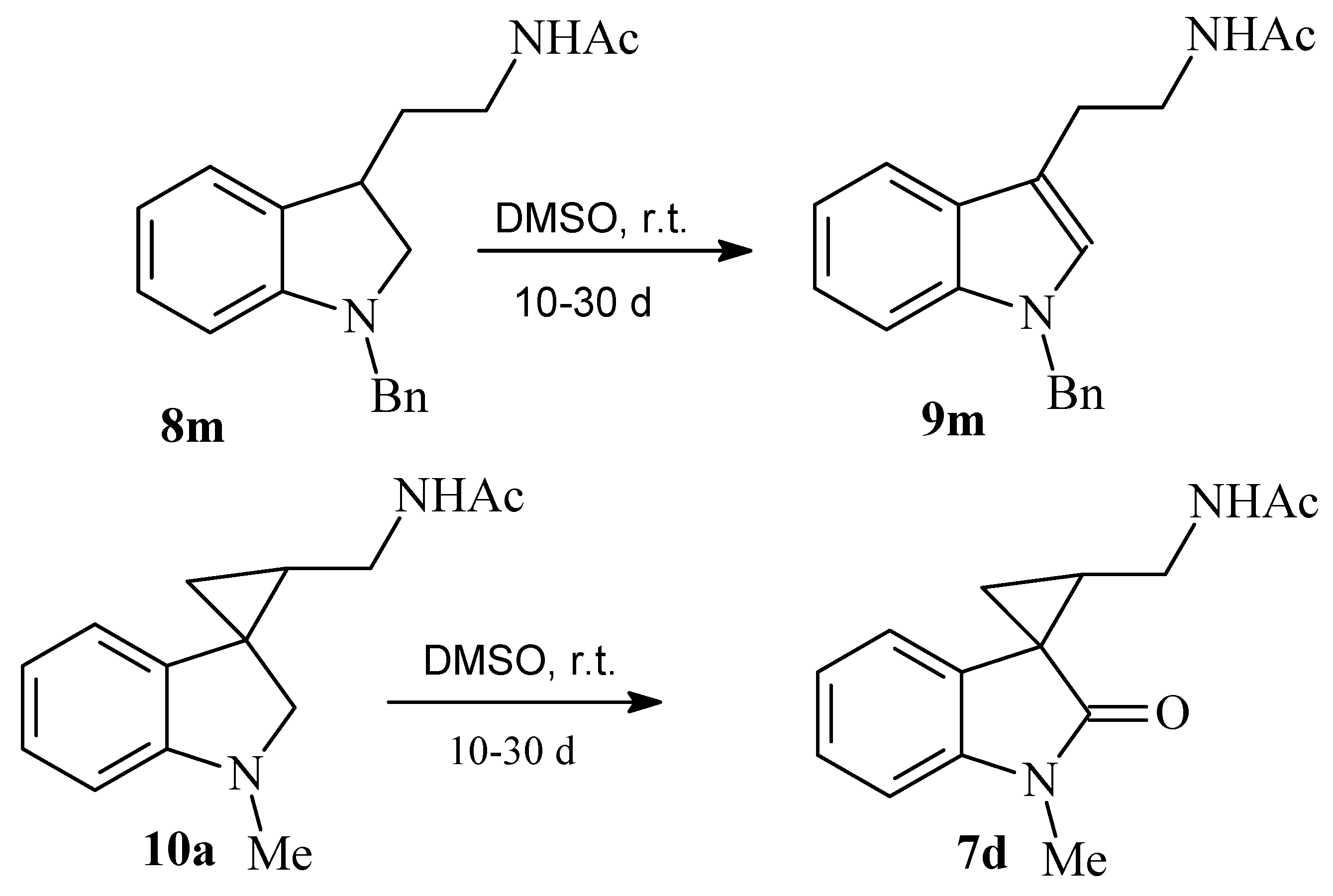
The solution of 3.200 g (14 mmol) of iodine in 10 mL of THF was added portionwise to the mixture of 1.100 g (4.2 mmol) of 2-(1-benzyl-2-oxo-2,3-dihydro-1H-indol-3-yl)acetonitrile 4f and 1.110 g (29 mmol) of sodium borohydride in 20 mL of THF. After stirring with reflux for 4 h, the reaction was terminated by addition of 7 mL of acetic acid; then, 1 mL (10 mmol) of acetic anhydride was added, and the reaction was stirred for 14 h. The compounds 8m and 9m were obtained as light yellow oil after purification with column chromatography (Rf 0.27 in EtOAc/chloroform 1:1). Yield 10% (0.130 g). For compound 8m: 1H NMR (CDCl3): 1.52–1.63 (m, 1H), 1.75 (s, 3H), 1.80–1.88 (m, 1H), 2.84–2.88 (m, 2H), 3.10–3.16 (m, 3H), 3.30–3.36 (m, 2H), 4.04 (d, J = 14.9, 1H), 4.10 (d, J = 14.9, 1H), 5.99–6.02 (br.s, 1H), 6.38 (d, J = 7.8, 1H), 6.55 (t, J = 7.3, 1H), 7.10–7.15 (m, 6H). MS-EI: 294 (M+, 7%), 203 (5%), 144 (30%), 130 (5%), 91 (100%), 77 (10%). HRMS-ESI: 309.1961, calculated for C20H25N2O (M+H): 309.1961. For compound 9m: 1H NMR (CDCl3): 1.75 (s, 3H), 2.82 (t, J = 7.1, 2H), 3.34 (t, J = 6.8, 2H), 5.08 (s, 1H), 6.24 (br.s., 1H), 6.80 (s, 1H), 6.91–6.99 (m, 5H), 7.4 (t, J = 7.1, 1H), 7.17–7.20 (m, 3H), 7.48 (d, J = 7.8, 1H).
The solution of 2.200 g (8.6 mmol) of iodine in 7 mL of THF was added portionwise to the mixture of 0.800 g (4 mmol) of 1′-methyl-2′-oxo-1′,2′-dihydrospiro[cyclopropane-1,3′-indole]-3-carbonitrile 6e and 0.912 g (24 mmol) of sodium borohydride in 30 mL of THF. After stirring with reflux for 10 h, the reaction was terminated by addition of 10 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 0.6 mL (5.5 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 10a was obtained as light red oil after purification with column chromatography (Rf 0.28 in ethyl acetate). Yield 76% (0.710 g). 1H NMR (CDCl3) (major diastereomer): 0.86-0.97 (m, 2H), 1.21-1.29 (m, 1H), 2.04 (s, 3H), 2.60 (s, 3H), 2.84–2.90 (m, 1H), 3.04 (d, J = 8.5, 1H), 3.15 (d, J = 8.5, 1H), 3.31–3.38 (m, 1H), 6.37 (d, J = 7.8, 1H), 6.51 (t, J = 7.5, 1H), 6.62 (d, J = 7.2, 1H), 6.92 (t, J = 7.6, 1H). 13C NMR (CDCl3): 15.08, 21.28, 22.09, 35.32, 37.82, 39.68, 64.85, 106.68, 117.14, 120.33, 126.68, 129.78, 153.96, 169.85. HRMS-ESI: 231.1491, calculated for C14H19N2O4(M+H): 231.1491.
The solution of 0.92 g (3.6 mmol) of iodine in 5 mL of THF was added portionwise to the mixture of 0.50 g (18 mmol) of 5′-bromo-1′-methyl-2′-oxo-1′,2′-dihydrospiro[cyclopropane-1,3′-indole]-3-carbonitrile 6i and 0.42 g (11 mmol) of sodium borohydride in 25 mL of THF. After stirring with reflux for 15 h, the reaction was terminated by addition of 10 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 0.80 mL (8 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 10b was obtained as light red oil after purification with column chromatography (Rf 0.18 in ethyl acetate). Yield 37% (0.21 g). 1H NMR (CDCl3): 1.08 (dd, J = 5.8, J = 5.8, 1H), 1.39–1.43 (m, 1H), 2.02 (s, 3H), 2.72 (s, 3H), 2.81–2.85 (m, 1H), 3.16 (d, J = 8.6, 1H), 3.33 (d, J = 8.5, 1H), 3.65–3.72 (m, 2H), 5.62 (s, 1H), 6.34 (d, J = 8.2, 1H), 6.77 (d, J = 2.0, 1H), 7.15 (dd, J = 8.2, J = 2.0, 1H). 13C NMR (CDCl3): 14.07, 20.93, 23.01, 27.75, 28.52, 35.81, 38.34, 60.30, 108.39, 109.39, 123.63, 129.96, 132.82, 153.83, 170.35. HRMS-ESI: 309.0597, calculated for C14H18N2OBr(M+H): 309.0597.
N-{1′,6′,7′-Trimethyl-1′,2′-dihydrospiro[cyclopropan-1,3′-indol]-3-ylmethyl}acetamide (10c)
The solution of 3.080 g (12 mmol) of iodine in 20 mL of THF was added portionwise to the mixture of 1.370 g (6 mmol) of 1′,6′,7′-trimethyl-2′-oxo-1′,2′-dihydrospiro[cyclopropane-1,3′-indole]-3-carbonitrile 6j and 1.160 g (36 mmol) of sodium borohydride in 20 mL of THF. After stirring with reflux for 24 h, the reaction was terminated by addition of 10 mL of 3N HCl. After extraction with 10 mL of methylene dichloride, 0.90 mL (9 mmol) of acetic anhydride was added to the organic fraction and stirred for 14 h. The compound 10c was obtained as yellow oil after purification with column chromatography (Rf 0.36 in ethyl acetate). Yield 35% (0.538 g). 1H NMR (CDCl3): 0.83–0.90 (m, 1H), 1.17–1.26 (m, 2H), 2.21 (s, 6H), 2.23 (s, 3H), 2.80-2.89 (m, 1H), 2.87 (s, 3H), 3.15 (d, J = 9.8, 1H), 3.39 (d, J = 9.8, 1H), 3.47–3.52 (m, 1H), 6.30 (d, J = 6.9, 1H), 6.65 (d, J = 7.1, 1H). 13C NMR (CDCl3): 13.78, 19.57, 20.23, 21.38, 22.06, 27.43, 29.71, 36.11, 41.50, 72.73, 115.53, 118.75, 121.77, 127.34, 129.07, 136.28, 142.49, 173.79. HRMS-ESI: 259.1805, calculated for C16H23N2O(M+H): 259.1805.


 and
and 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
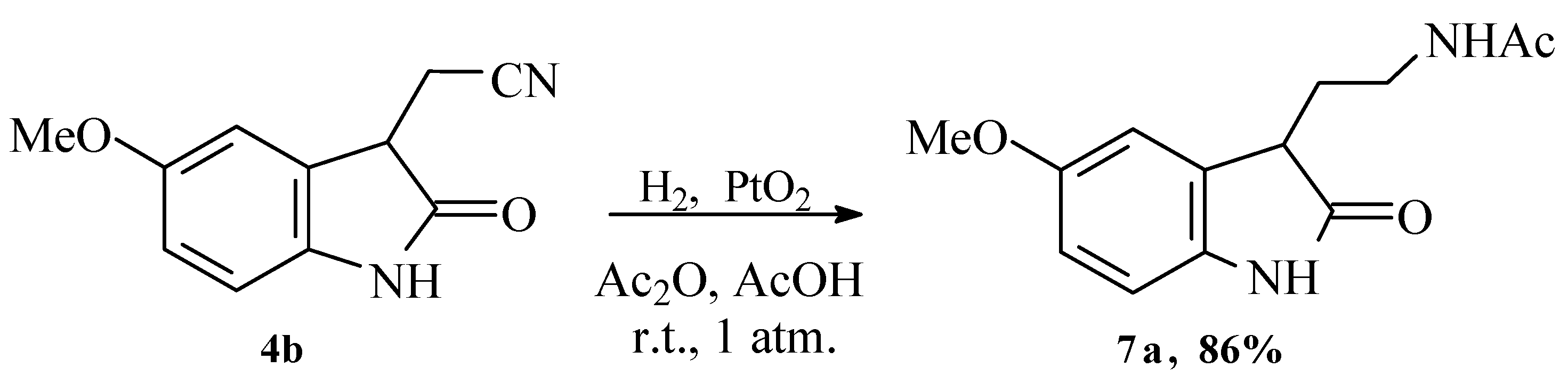{kind=link}
{kind=link}
{kind=link}
{kind=link}
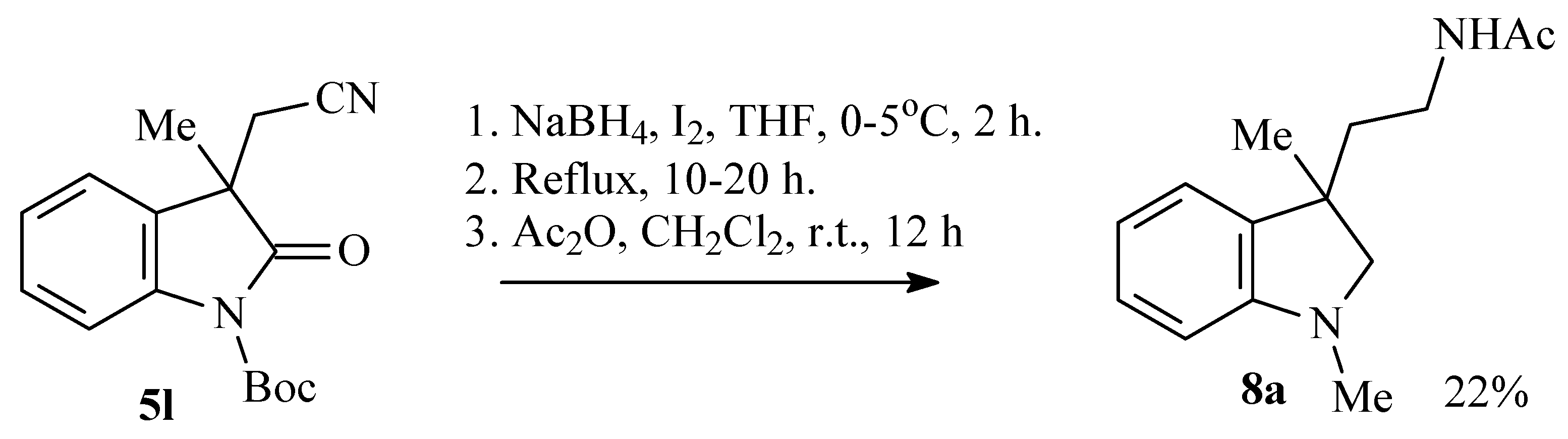{kind=link}
{kind=link}



