
Determination of Genotoxic Impurity N-Nitroso-N-methyl-4-aminobutyric Acid in Four Sartan Substances through Using Liquid Chromatography–Tandem Mass Spectrometry

Abstract
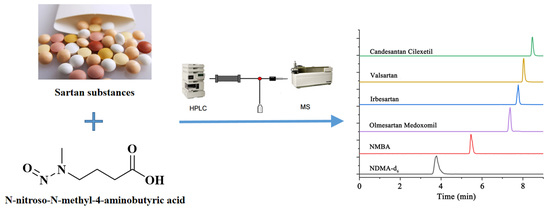
:
1. Introduction
2. Results and Discussion
2.1. Method Development
2.1.1. Optimization of Chromatographic Conditions
2.1.2. Optimization of Mass Spectrometric Conditions
2.1.3. Optimization of the Quantitative Analysis Method
2.2. Method Validation
2.2.1. Specificity
2.2.2. Linearity, LOQ and LOD
2.2.3. Accuracy
2.2.4. Precision
2.2.5. Stability
2.3. Detection of Actual Samples
3. Experimental
3.1. Chemicals and Materials
3.2. LC-MS/MS Determination of NMBA
3.3. Preparation of Standard and Sample Solutions
3.4. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APCI | Atmospheric pressure chemical ionization |
| ESI | Electron spray ion |
| LC-MS/MS | Liquid Chromatography–Tandem Mass Spectrometry |
| LOD | Limit of detection |
| LOQ | Limit of quantitation |
| NDBA | N-nitrosodibutylamine |
| NDEA | N-nitrosodiethylamine |
| NDMA | N-nitrosodimethylamine |
| NMBA | N-nitroso-N-methyl-4-aminobutyric acid |
| RSD | relative standard deviation |
References
- Li, K.; Ricker, K.; Tsai, F.C.; Hsieh, C.J.; Osborne, G.; Sun, M.; Sandy, M.S. Estimated cancer risks associated with nitrosamine contamination in commonly used medications. Int. J. Environ. Res. Public Health 2021, 18, 9465. [Google Scholar] [CrossRef] [PubMed]
- Dobo, K.L.; Kenyon, M.O.; Dirat, O.; Engel, M.; Fleetwood, A.; Martin, M.; Kalgutkar, A.S. Practical and science-based strategy for establishing acceptable intakes for drug product N-nitrosamine impurities. Chem. Res. Toxicol. 2022, 35, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Konstantinou, E.; Fotopoulou, F.; Drosos, A.; Dimakopoulou, N.; Zagoriti, Z.; Niarchos, A.; Makrynioti, D.; Kouretas, D.; Farsalinos, K.; Lagoumintzis, G.; et al. Tobacco-specific nitrosamines: A literature review. Food Chem. Toxicol. 2018, 118, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, M.A.; de Lima, P.C.; Novotny, T.S.; Santana, D.S.; Lima, M.E.D.; Dantas, A.S.C.L.; dos Santos, L.O.; de Souza, M.N.; Maranho, R.L.D.N.; Ochs, S.D.M. Development and Validation of Methods Based on High-Performance Liquid Chromatography-Tandem Mass Spectrometry for Determining N-nitrosamines Impurities in Sartan Pharmaceutical Products for Monitoring Program. 2022. Available online: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4057039 (accessed on 10 October 2022). [CrossRef]
- U.S. Food and Drug Administration. FDA Updates and Press Announcements on Angiotensin II Receptor Blocker (ARB) Recalls (Valsartan, Losartan, and Irbesartan). 28 February 2019. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-updates-and-press-announcements-angiotensin-ii-receptor-blocker-arb-recalls-valsartan-losartan (accessed on 10 October 2022).
- U.S. Food and Drug Administration. FDA Provides Update on Its Ongoing Investigation into ARB Drug Products; Reports on Finding of a New Nitrosamine Impurity in Certain Lots of Losartan and Product Recall. 1 March 2019. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm632425.htm (accessed on 10 October 2022).
- Lijinsky, W.; Reuber, M.D.; Saavedra, J.E.; Singer, G.M. Carcinogenesis in F344 rats by N-nitrosomethyl-N-propylamine derivatives. J. Natl. Cancer Inst. 1983, 70, 959–963. [Google Scholar] [PubMed]
- U.S. Food & Drug Administration. Development and Validation of a RapidFire-MS/MS Method for Screening of Nitrosamine Carcinogen Impurities N-Nitrosodimethylamine (NDMA), N-Nitrosodiethylamine (NDEA), N-Nitrosoethylisopropylamine (NEIPA), N-Nitrosodiisopropylamine (NDIPA), N-Nitrosodibutylamine (NDBA) and N-Nitroso-N-methyl-4-aminobutyric acid (NMBA) in ARB Drugs [EB/OL]. 2 June 2019. Available online: https://www.fda.gov/media/125477/download (accessed on 10 October 2022).
- Xu, W.F.; Jin, P.F.; Xu, S.; Zhang, S.S.; Wu, X.J. Determination of trace impurity NMBA in potassium losartan and its compound preparations by LC-Q-TOF-MS. Chin. J. Pharm. Anal. 2019, 39, 2047–2050. [Google Scholar]
- Zou, Y.; Sun, L.P.; Li, X.D.; Hamada, N. Determination of N-Nitroso-N-Methyl-4-Aminobutyric acid in losartan potassium using high performance liquid chromatography triple quadrupole mass spectrometry. Chin. Pharm. J. 2020, 55, 228–232. [Google Scholar]
- Chidella, K.S.; Dasari, V.B.; Anireddy, J. Ultra-sensitive LC-MS/MS method for the trace level quantification of six potential genotoxic nitrosamine impurities in telmisartan. Am. J. Anal. Chem. 2021, 12, 227–240. [Google Scholar] [CrossRef]
- Ripollés, C.; Pitarch, E.; Sancho, J.V.; López, F.J.; Hernández, F. Determination of eight nitrosamines in water at the ng/L levels by liquid chromatography coupled to atmospheric pressure chemical ionization tandem mass spectrometry. Anal. Chim. Acta 2011, 702, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Chang, C.C.; Wang, L.J.; Chen, W.C.; Fan, S.Y.; Zang, C.Z.; Hsu, Y.H.; Lin, M.C.; Tseng, S.H.; Wang, D.Y. A multi-analyte LC-MS/MS method for screening and quantification of nitrosamines in sartans. J. Food Drug Anal. 2020, 28, 98–107. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Precursor (m/z) | Product (m/z) | Dwell Time (ms) | Declustering Potential (V) | Collision Energy (V) |
|---|---|---|---|---|---|
| NDMA-d6 | 81.0 | 46.2 ① | 200 | 55 | 25 |
| NMBA | 147.3 | 117.0 ① | 200 | 31 | 10 |
| 87.0 ② | 200 | 16 | |||
| Candesartan cilexetil | 611.4 | 441.3 ① | 200 | 80 | 16 |
| 423.1 ② | 200 | 24 | |||
| Olmesartan medoxomi | 559.4 | 541.1 ① | 200 | 71 | 19 |
| 207.1 ② | 200 | 36 | |||
| Irbesartan | 429.9 | 207.1 ① | 200 | 161 | 32 |
| 402.2 ② | 200 | 21 | |||
| Valsartan | 436.3 | 235.2 ① | 200 | 85 | 24 |
| 291.3 ② | 200 | 24 |
| Method | Linearity Range (ng/mL) | Regression Equation | R2 | Matrix | Average Recovery (%) | RSD % |
|---|---|---|---|---|---|---|
| Internal standard method | 3~30 | y = 0.0169x + 0.0018 | 0.9996 | Candesartan cilexetil | 105.3 | 4.0 |
| Olmesartan medoxomi | 103.6 | 2.9 | ||||
| Irbesartan | 98.9 | 3.6 | ||||
| Valsartan | 96.5 | 1.8 | ||||
| External standard method | 3~30 | y = 2641.3x + 245.03 | 0.9998 | Candesartan cilexetil | 112.8 | 7.2 |
| Olmesartan medoxomi | 114.6 | 6.9 | ||||
| Irbesartan | 102.4 | 5.6 | ||||
| Valsartan | 115.4 | 6.8 |
| Linearity Range (ng/mL) | Regression Equation | R2 | LOD (ng/mL) | LOQ (ng/mL) | |
|---|---|---|---|---|---|
| K1-K5 | 3~24 | y = 0.0178x + 0.0039 | 0.9990 | 3.0 | 0.9 |
| K1-K7 | 3~45 | y = 0.0166x − 0.0020 | 0.9994 |
| Matrix | Spiked Concentration (ng/mL) | Average Detected Concentration (ng/mL) | Average Recovery (%) | RSD (%) |
|---|---|---|---|---|
| Candesartan cilexetil | 3.0035 | 3.2643 | 108.7 | 3.7 |
| 30.0350 | 32.9359 | 109.7 | 2.6 | |
| Olmesartan medoxomi | 3.0035 | 2.8315 | 94.3 | 3.0 |
| 30.0350 | 30.3470 | 101.0 | 3.8 | |
| Irbesartan | 3.0035 | 2.6994 | 89.9 | 3.8 |
| 30.0350 | 30.6522 | 102.1 | 4.9 | |
| Valsartan | 3.0035 | 3.4747 | 115.7 | 3.8 |
| 30.0350 | 30.3651 | 101.1 | 5.2 |
| Matrix | Spiked Concentration (ng/mL) | Intra-Day (n = 6) | Inter-Day (n = 18) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | Average Recovery (%) | RSD (%) | |||||
| Average Recovery (%) | RSD (%) | Average Recovery (%) | RSD (%) | Average Recovery (%) | RSD (%) | ||||
| Candesartan cilexetil | 30.0350 | 102.3 | 2.3 | 105.7 | 2.2 | 98.5 | 3.8 | 102.2 | 4.0 |
| Olmesartan medoxomi | 104.5 | 2.9 | 109.2 | 3.5 | 103.9 | 2.3 | 105.8 | 3.6 | |
| Irbesartan | 95.7 | 3.9 | 97.5 | 4.5 | 102.5 | 3.8 | 98.6 | 5.0 | |
| Valsartan | 101.3 | 3.4 | 102.6 | 3.4 | 107.0 | 4.5 | 103.6 | 4.3 | |
| Time (h) | Spiked Concentration (ng/mL) | Average Detected Concentration (ng/mL) | Average Recovery (%) | RSD (%) |
|---|---|---|---|---|
| 0 | 30.0350 | 29.8781 | 99.5 | 3.6 |
| 2 | 29.4553 | 98.1 | ||
| 4 | 29.2079 | 97.3 | ||
| 6 | 28.8273 | 96.0 | ||
| 9 | 31.7491 | 105.7 | ||
| 12 | 31.2361 | 104.0 | ||
| 24 | 30.3306 | 101.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, B.; Guo, D.; Mai, B.; Fan, J. Determination of Genotoxic Impurity N-Nitroso-N-methyl-4-aminobutyric Acid in Four Sartan Substances through Using Liquid Chromatography–Tandem Mass Spectrometry. Molecules 2022, 27, 7498. https://doi.org/10.3390/molecules27217498
Xie B, Guo D, Mai B, Fan J. Determination of Genotoxic Impurity N-Nitroso-N-methyl-4-aminobutyric Acid in Four Sartan Substances through Using Liquid Chromatography–Tandem Mass Spectrometry. Molecules. 2022; 27(21):7498. https://doi.org/10.3390/molecules27217498
Chicago/Turabian StyleXie, Bin, Dong Guo, Binliang Mai, and Jun Fan. 2022. "Determination of Genotoxic Impurity N-Nitroso-N-methyl-4-aminobutyric Acid in Four Sartan Substances through Using Liquid Chromatography–Tandem Mass Spectrometry" Molecules 27, no. 21: 7498. https://doi.org/10.3390/molecules27217498
APA StyleXie, B., Guo, D., Mai, B., & Fan, J. (2022). Determination of Genotoxic Impurity N-Nitroso-N-methyl-4-aminobutyric Acid in Four Sartan Substances through Using Liquid Chromatography–Tandem Mass Spectrometry. Molecules, 27(21), 7498. https://doi.org/10.3390/molecules27217498