

Bispyrrolidinoindoline Epi(poly)thiodioxopiperazines (BPI-ETPs) and Simplified Mimetics: Structural Characterization, Bioactivities, and Total Synthesis †

Abstract
:
1. Introduction
2. Isolation, Structural Elucidation, and Biological Activities of Tryptophan-Derived BPI-ETP Alkaloids
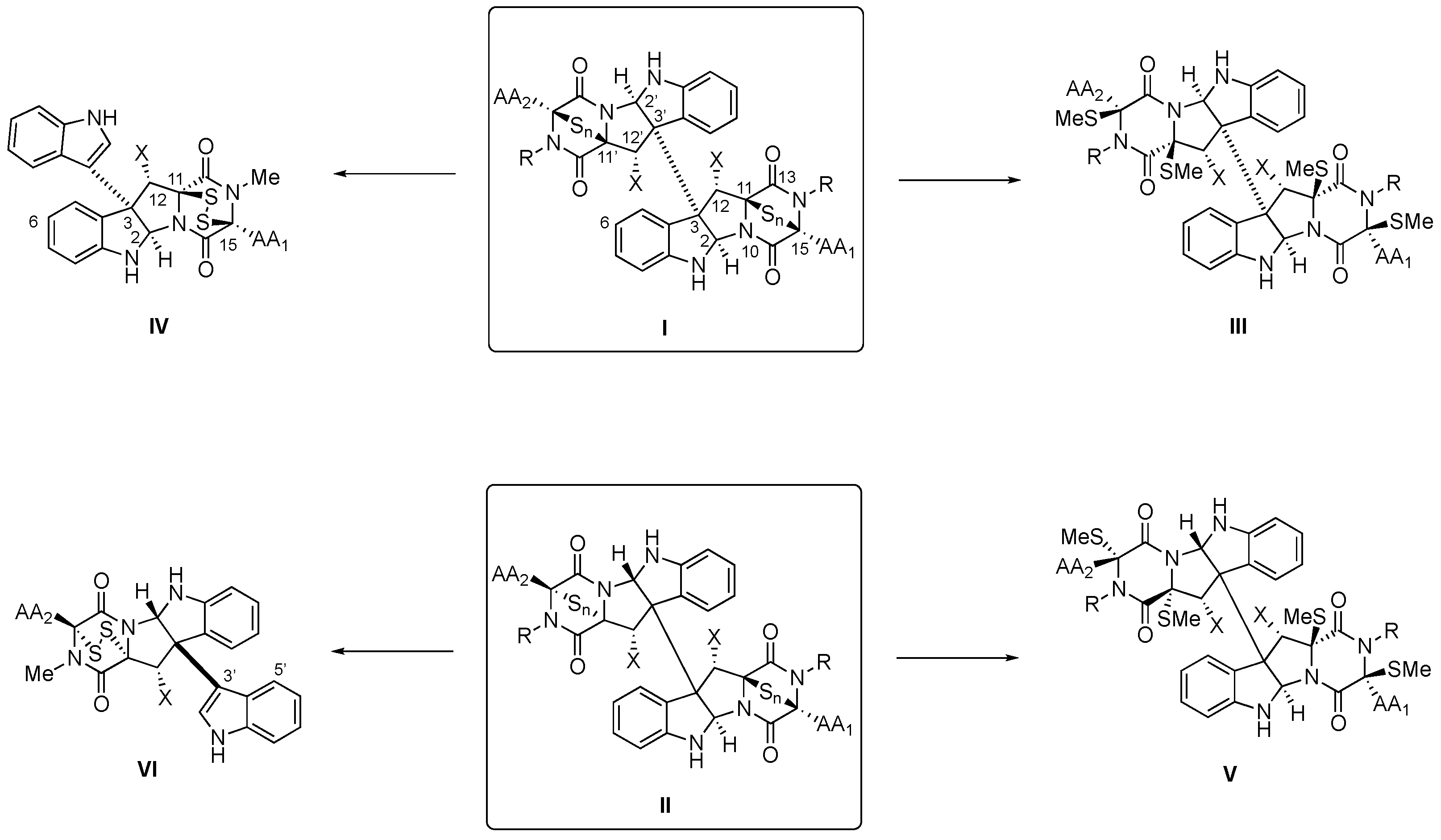
2.1. Structural Elucidation
2.2. BPI-ETP Alkaloids
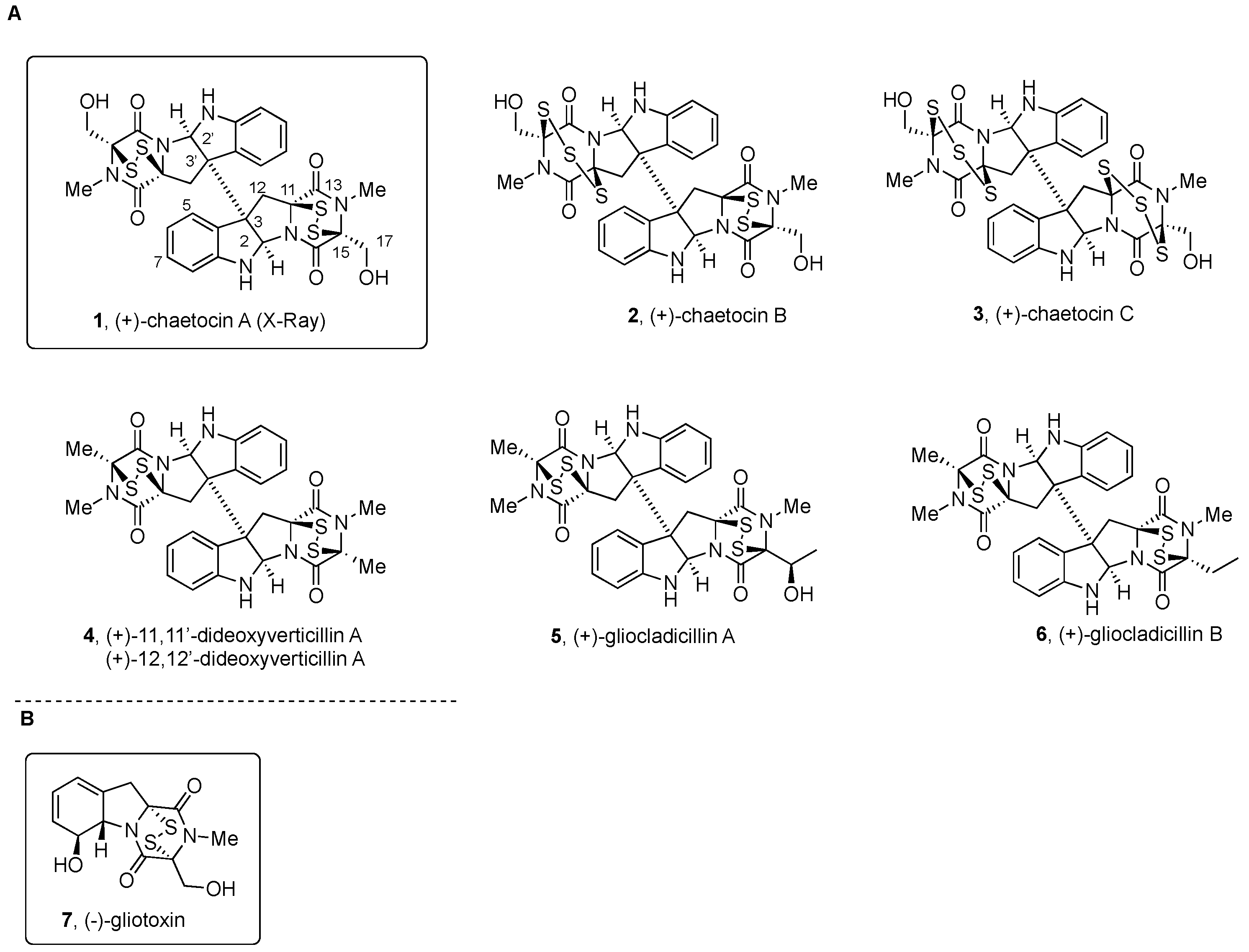
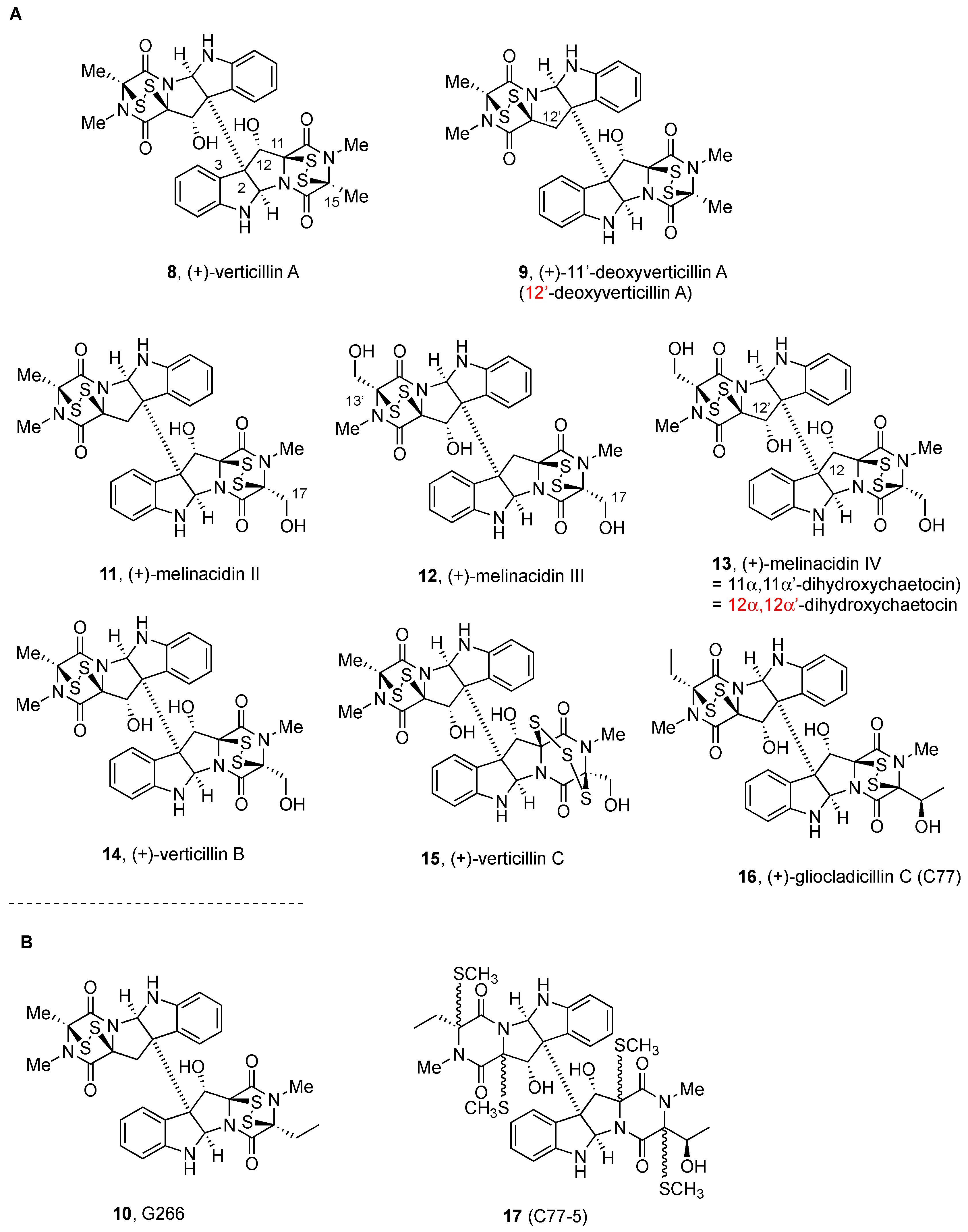
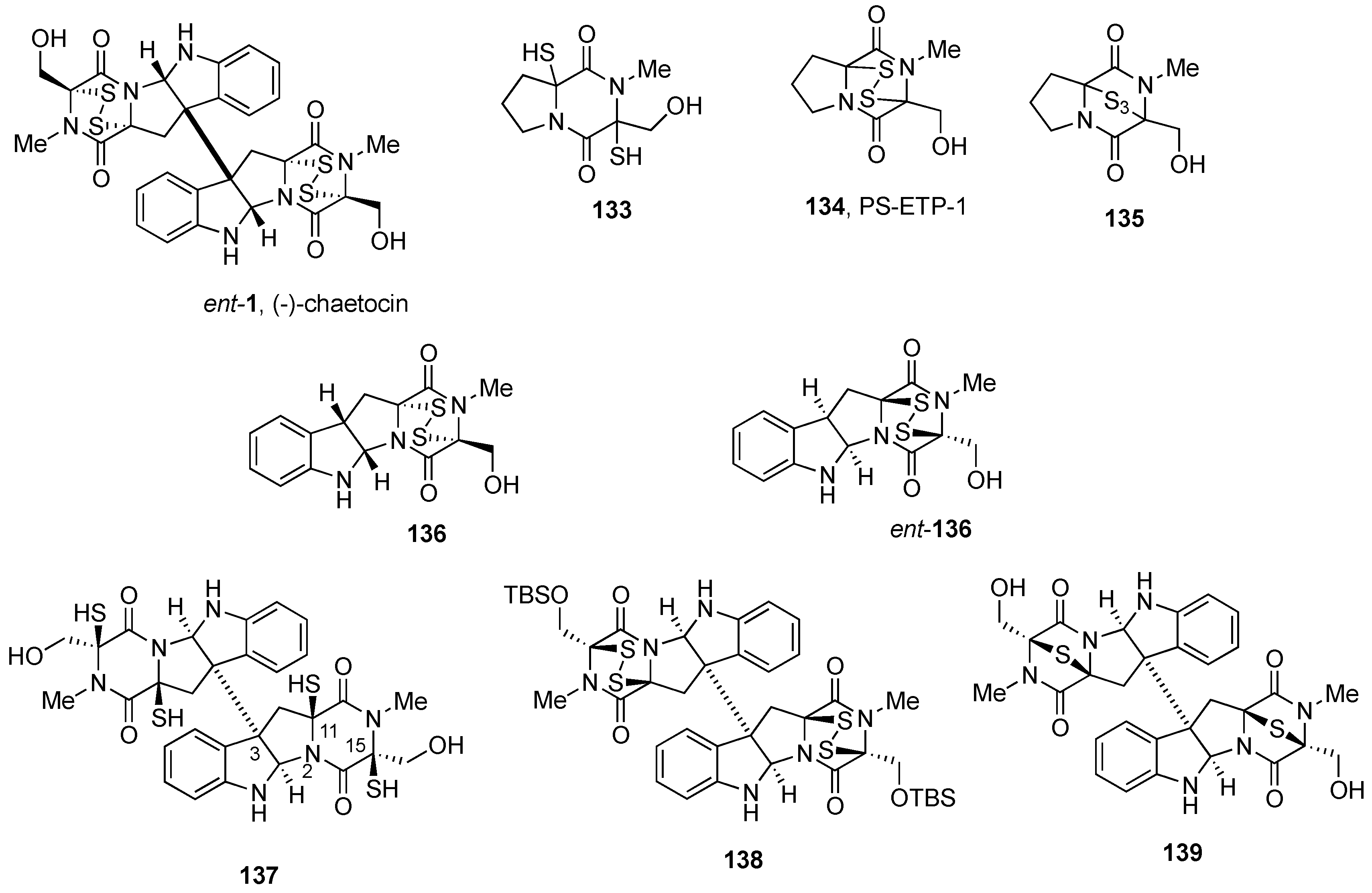
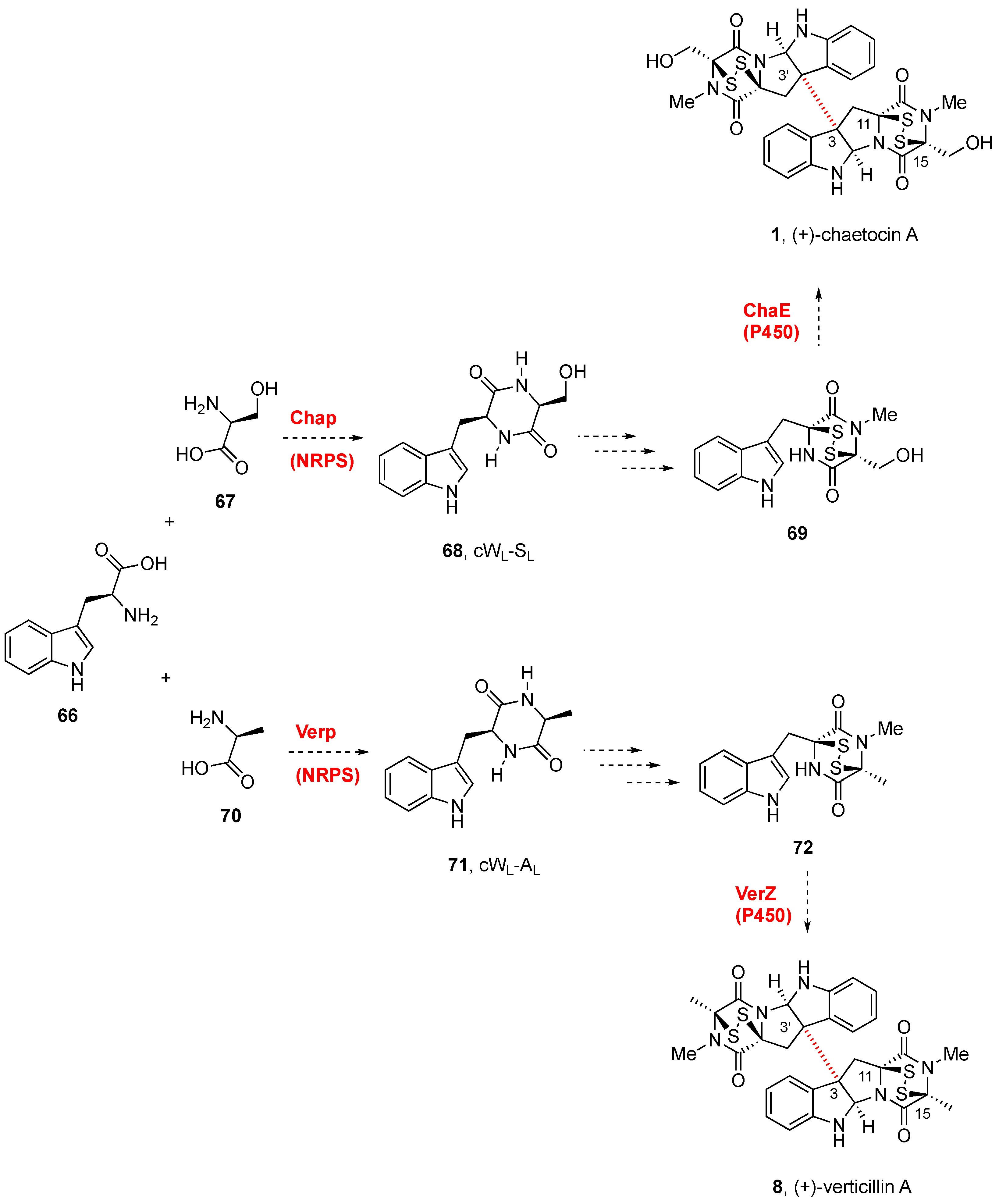
2.2.1. Chaetocin and Analogues



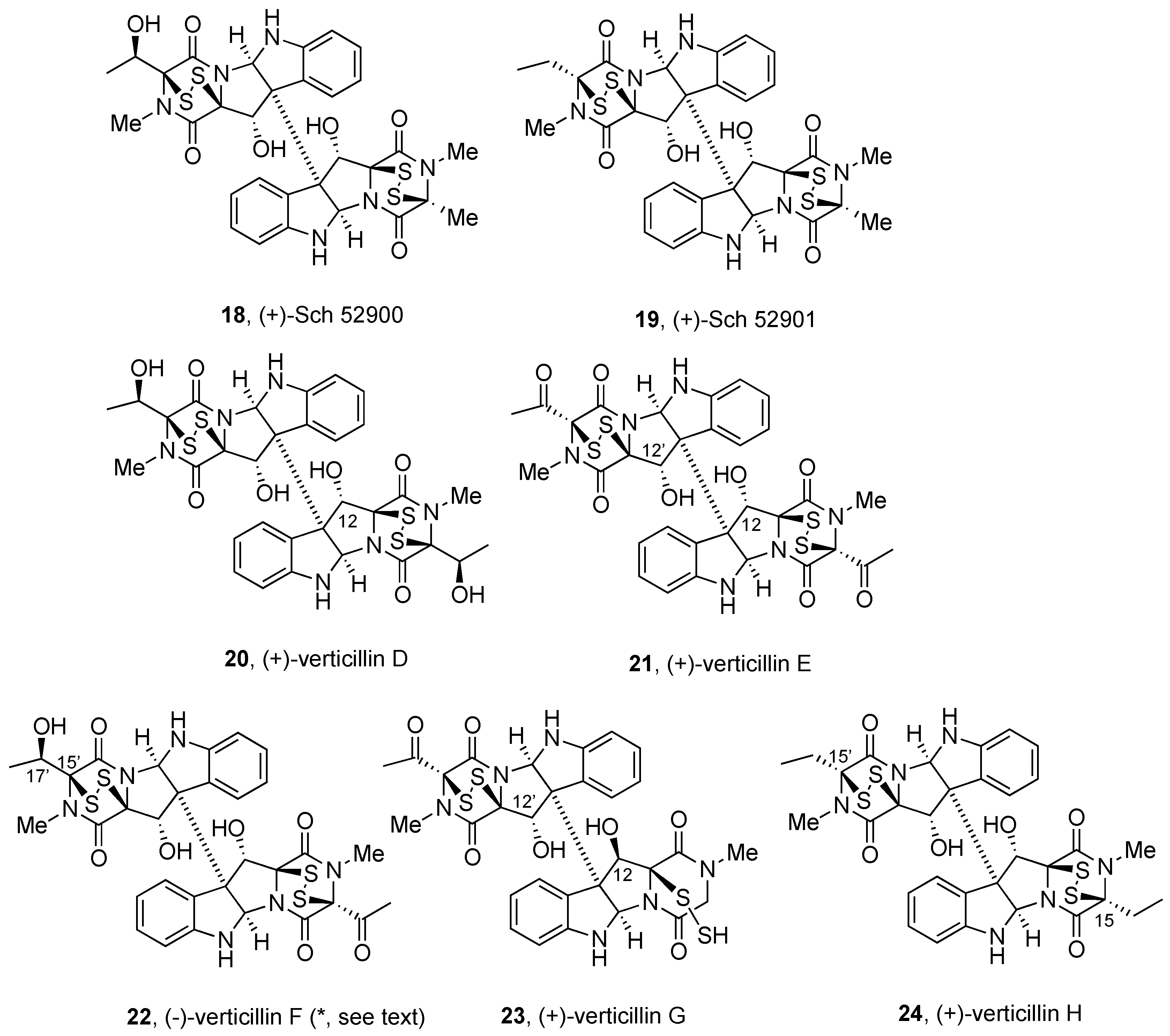
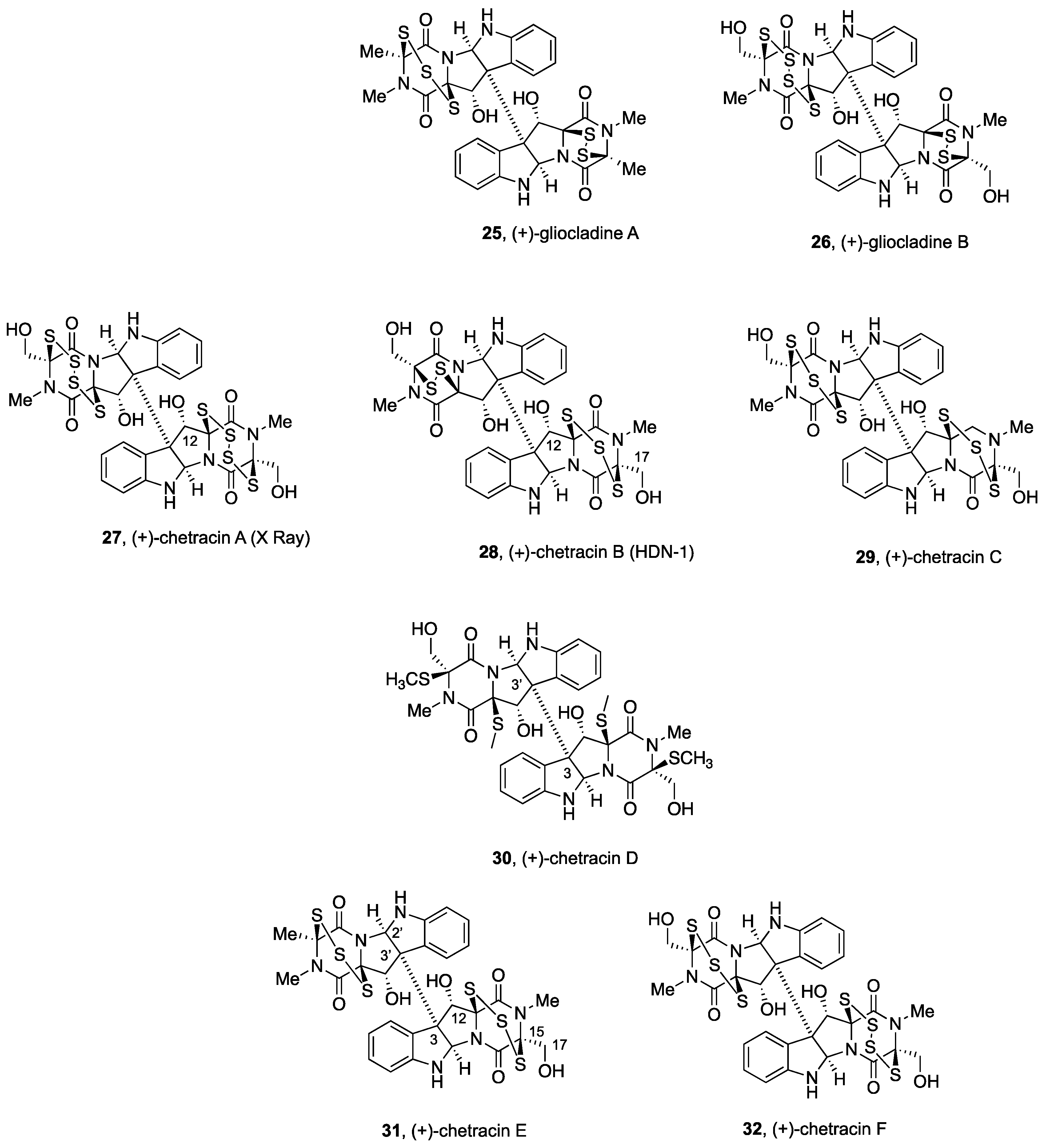
2.2.2. Gliocladines and Chetracins

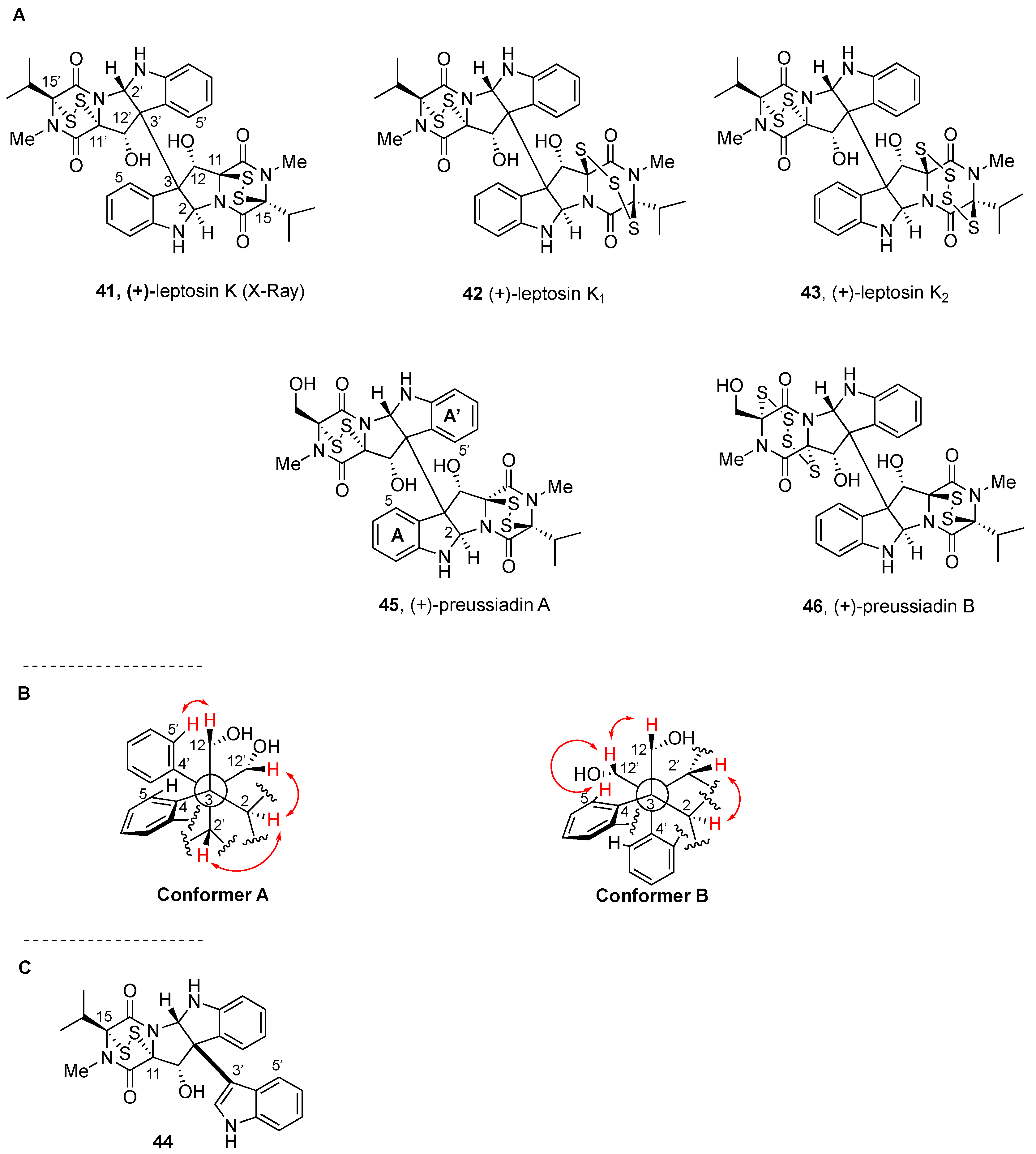
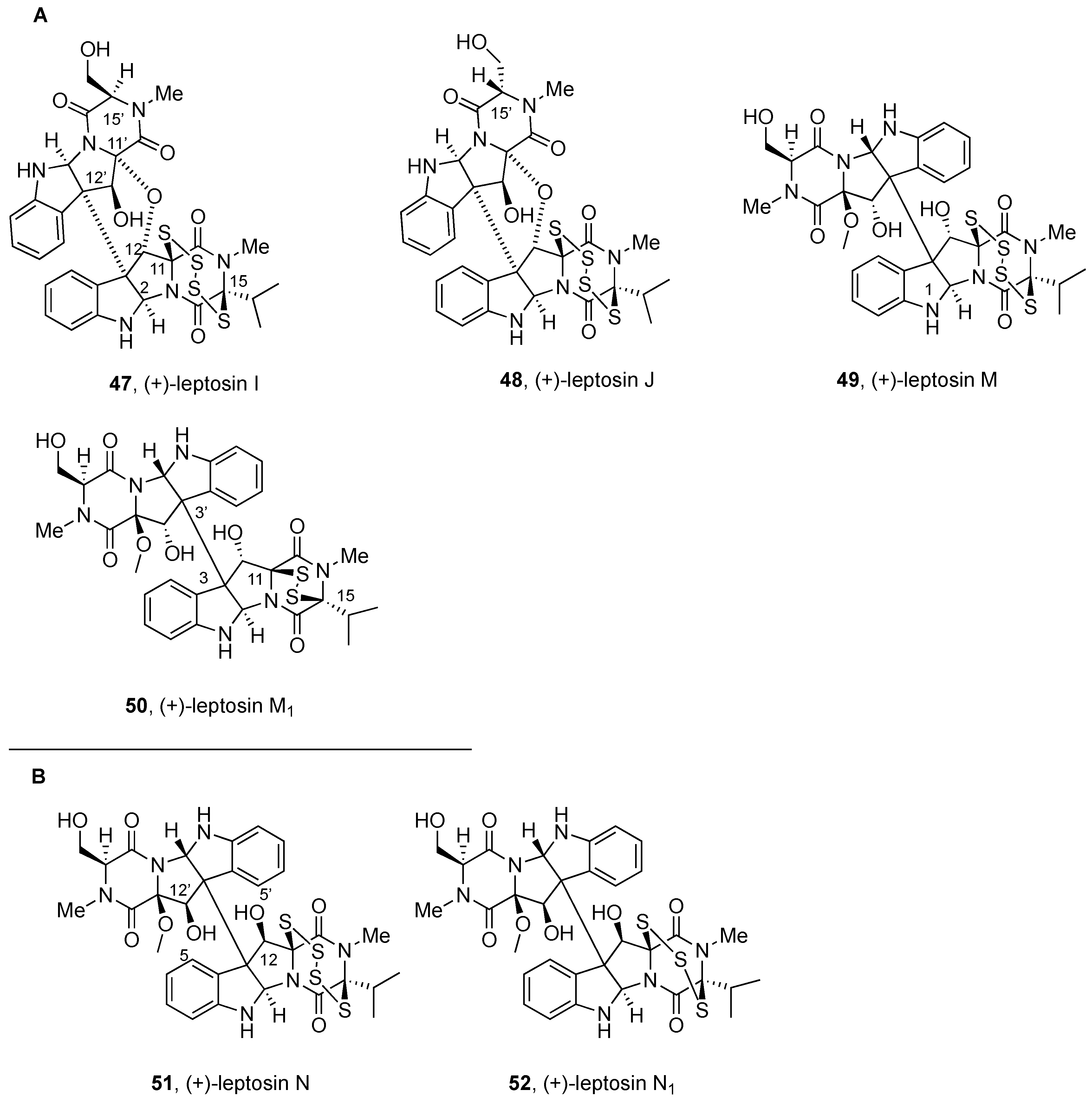
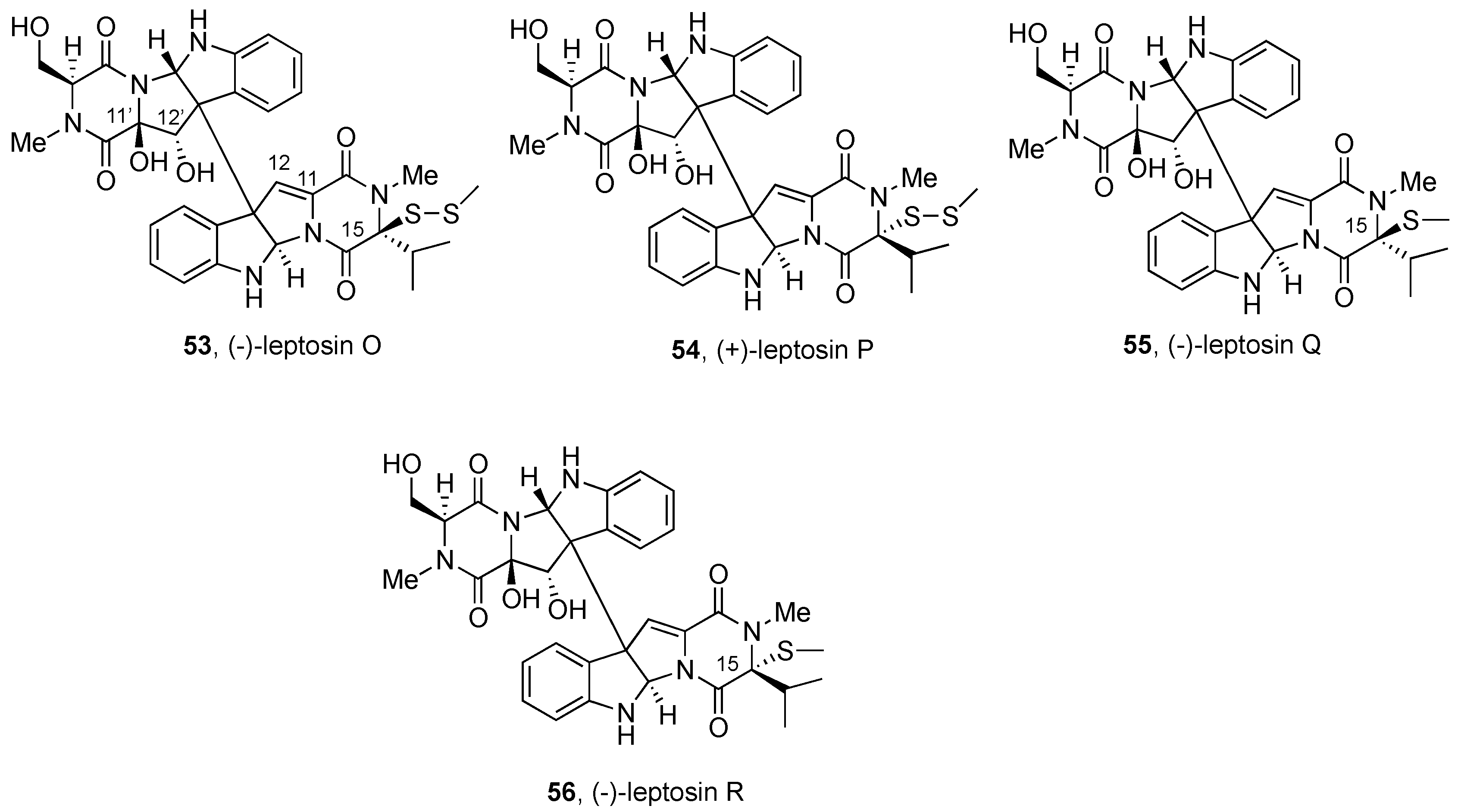
2.2.3. Leptosins and Preussiadins




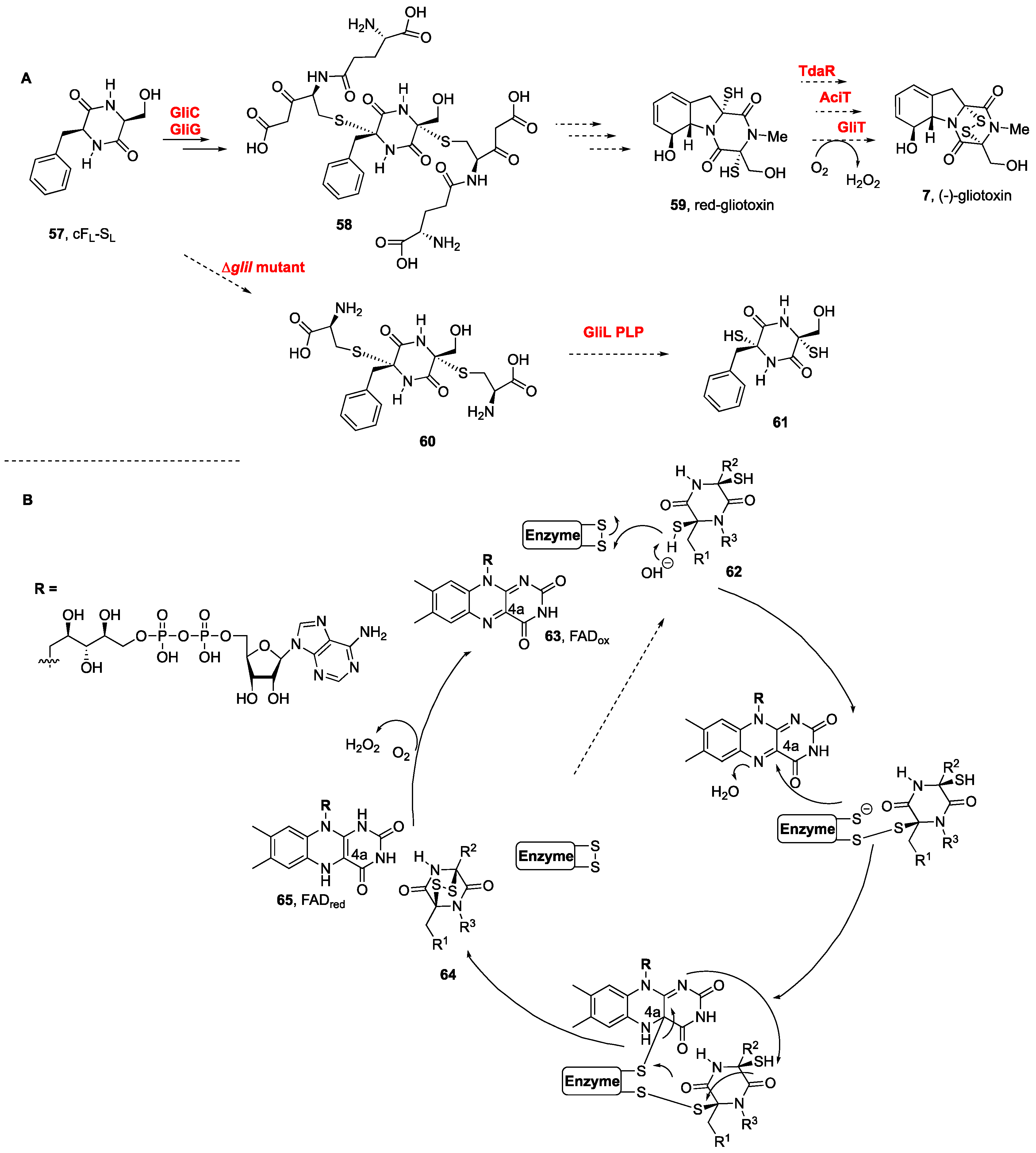
3. Biogenesis of BPI-ETPs
4. Total Synthesis of Tryptophan-Derived BPI-ETP Alkaloids
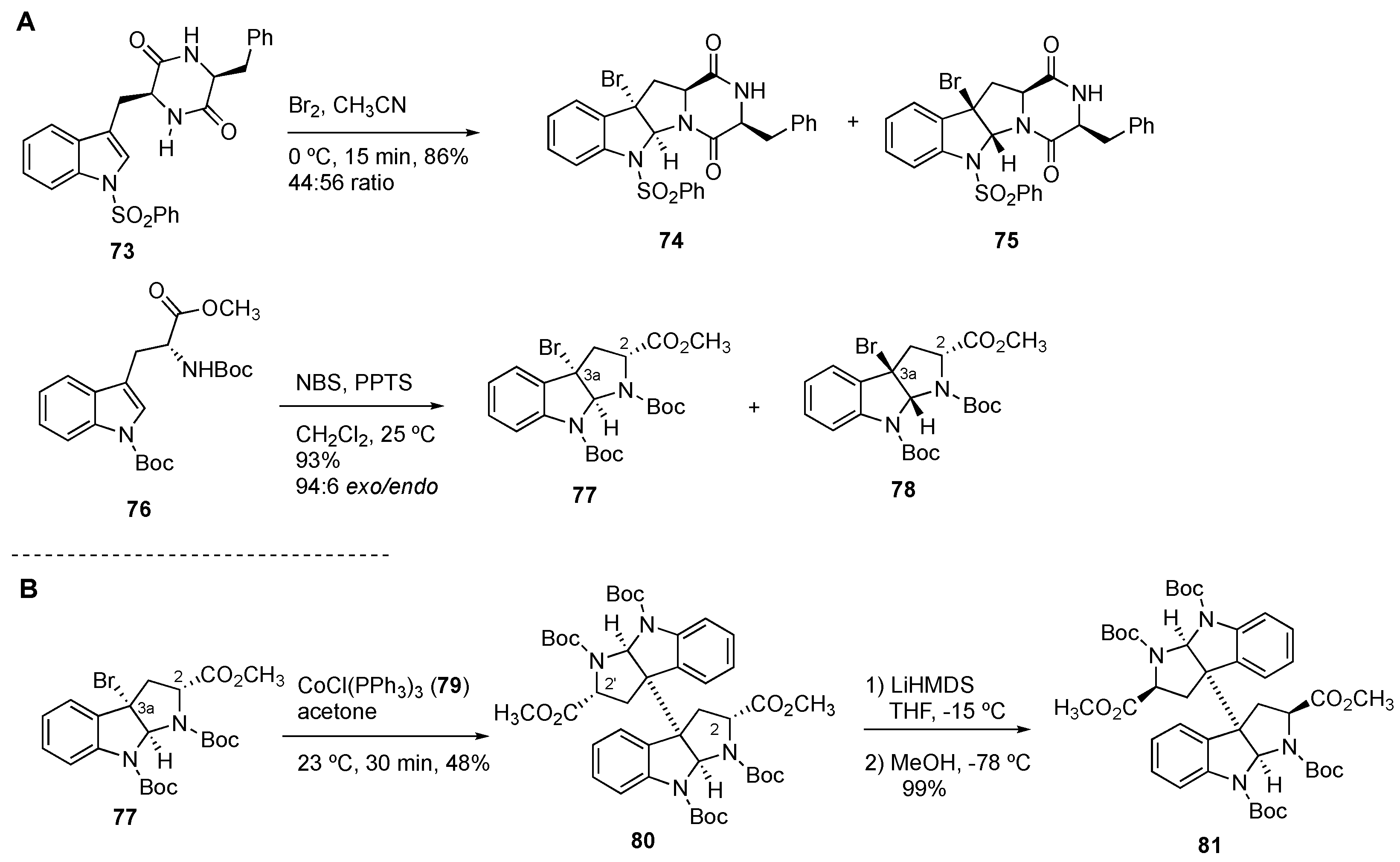
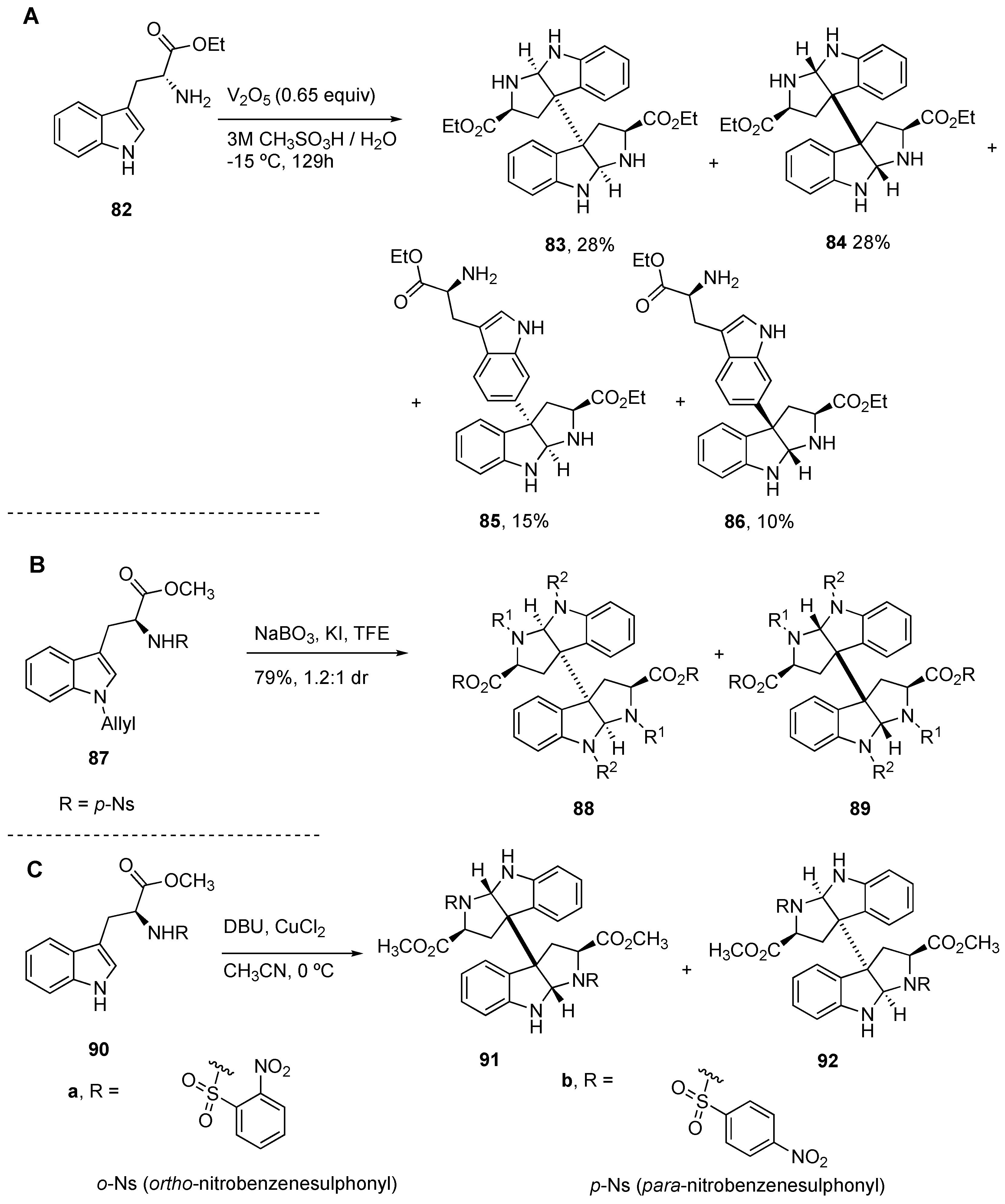
4.1. General Construction of Tryptophan-Derived Bis-Pyrrolidinoindolines
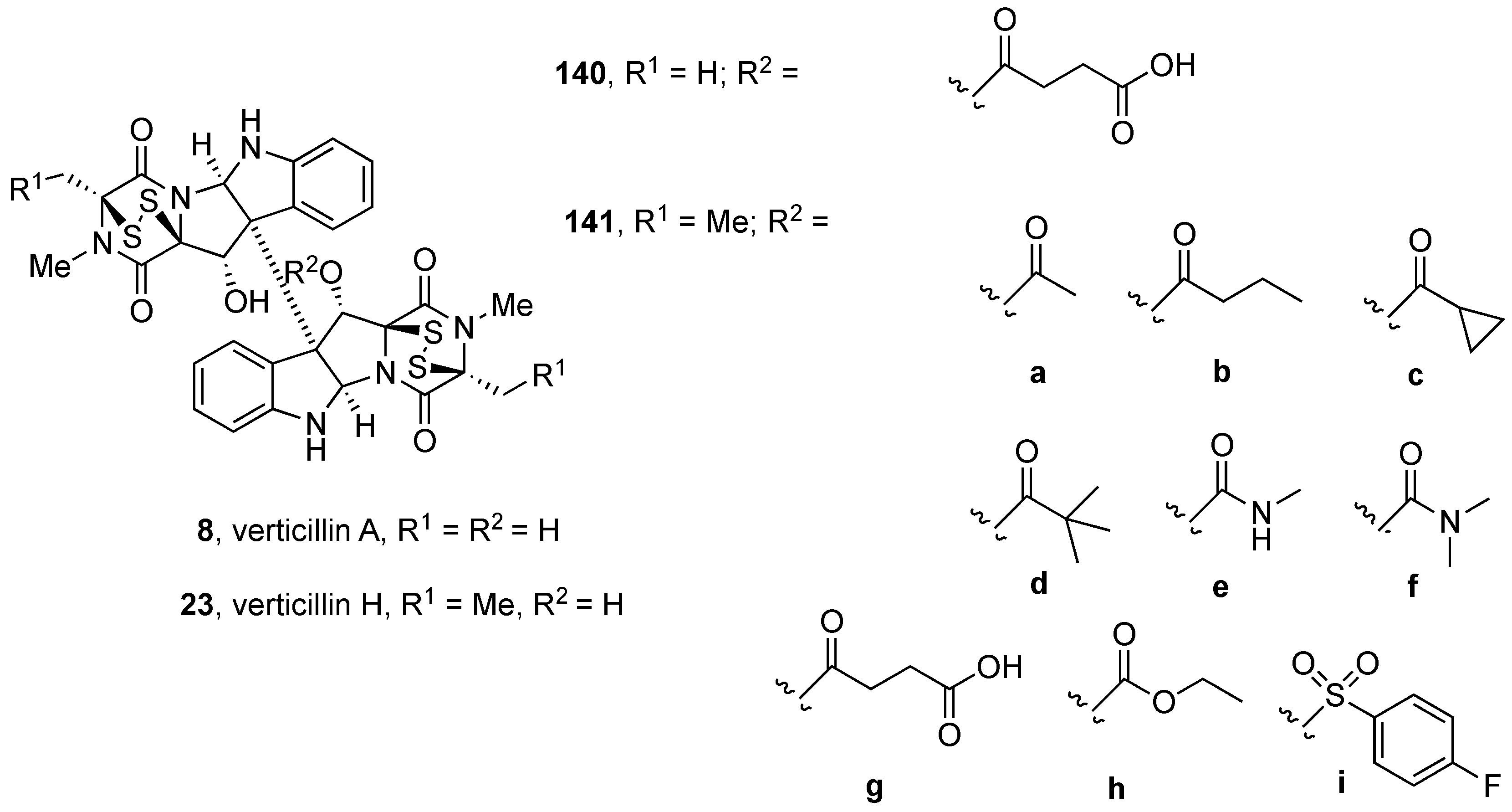
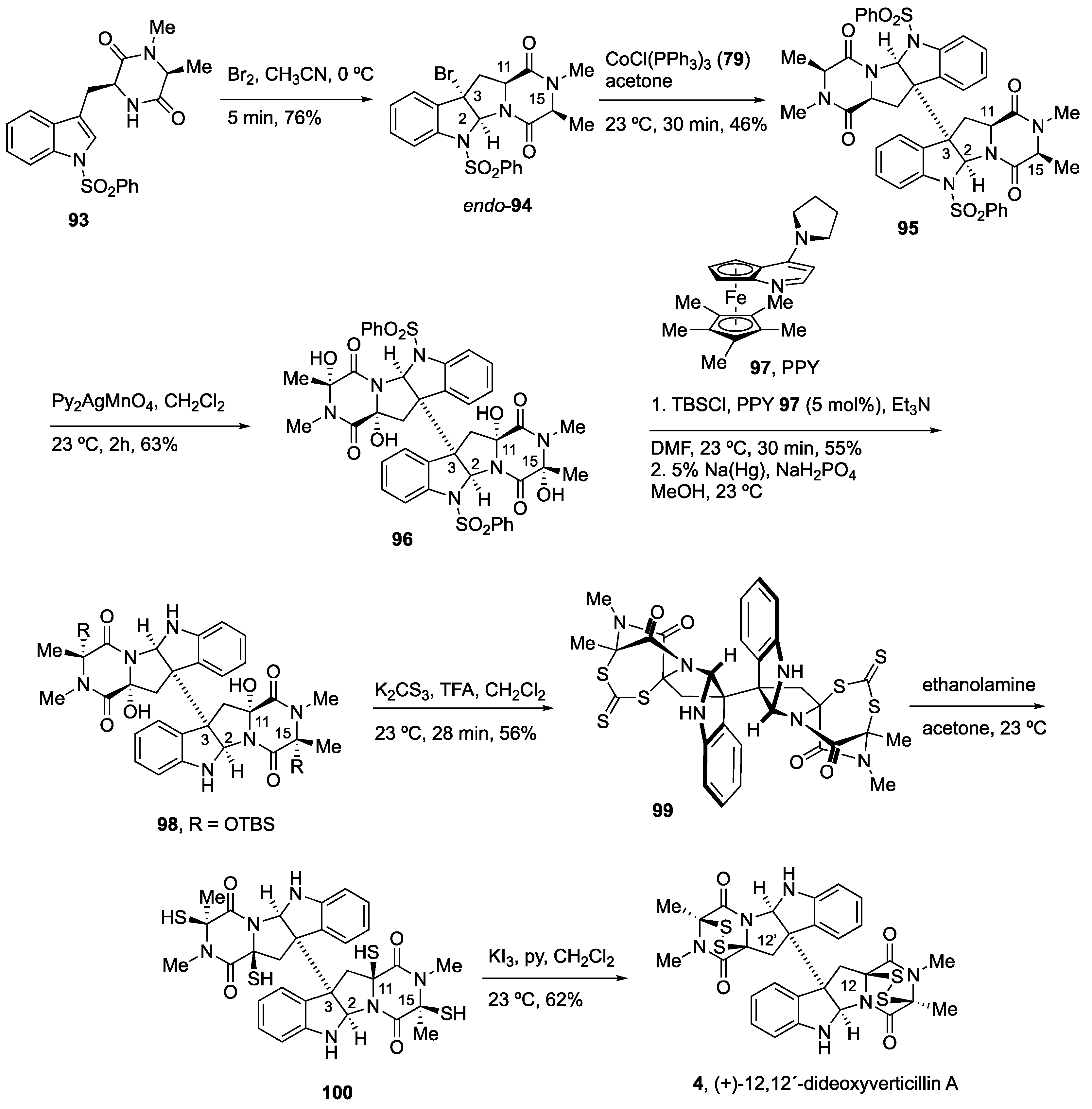
4.2. Synthesis of (+)-12,12’-Dideoxyverticillin A (4)
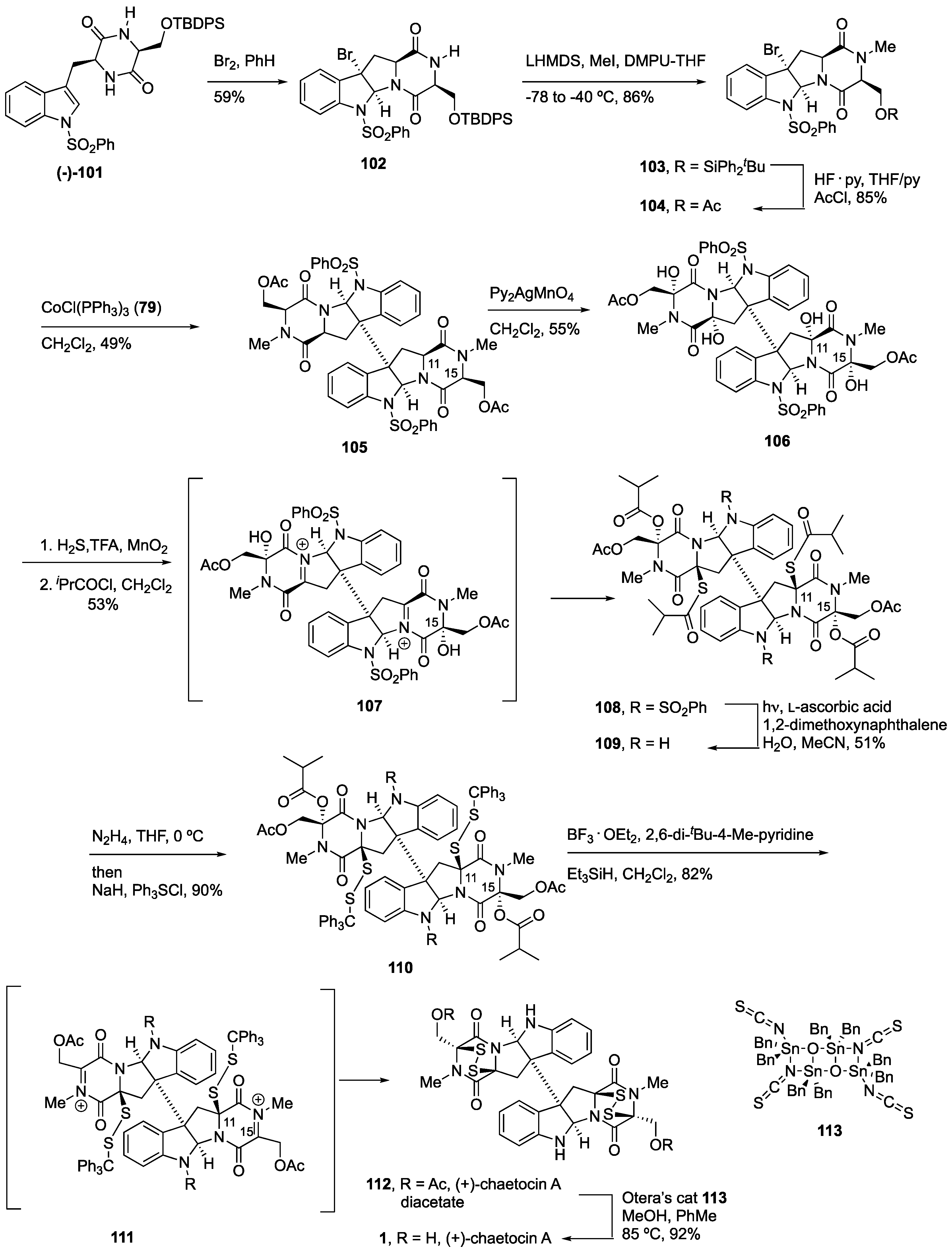
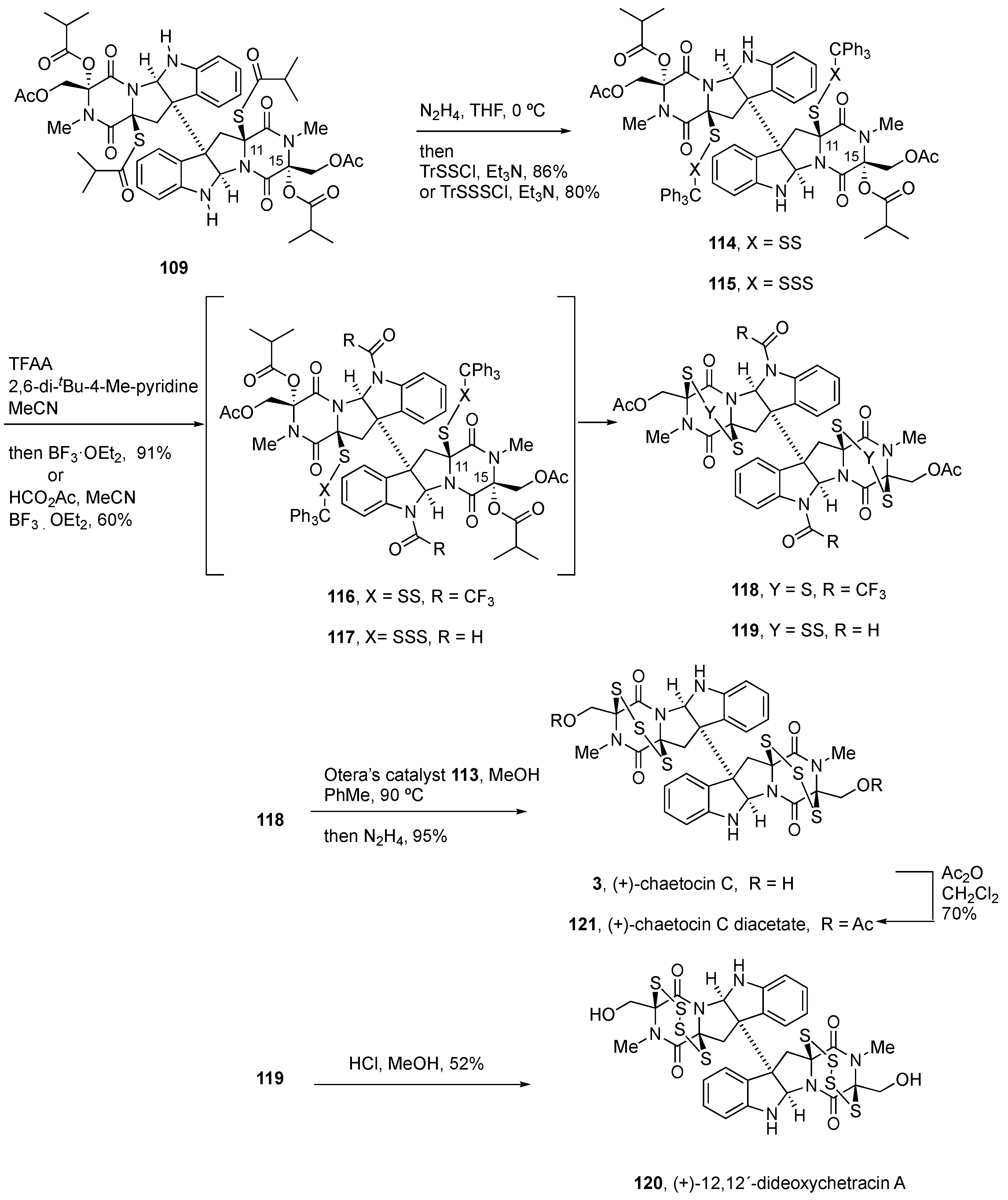
4.3. Synthesis of (+)-Chaetocin A (1) and (+)-Chaetocin C (3)
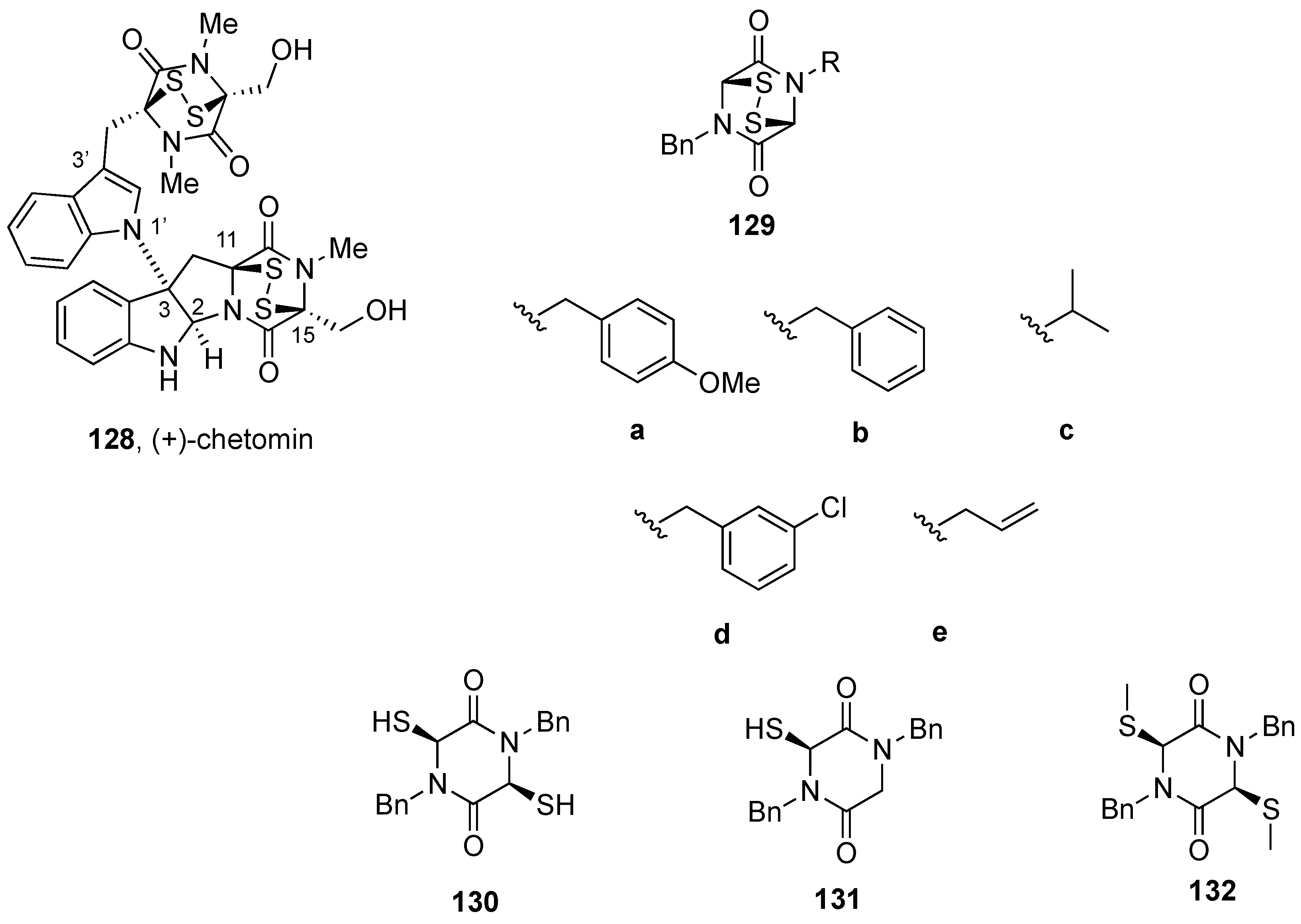
4.4. Synthesis of (+)-12,12´-Dideoxychetracin A (128)

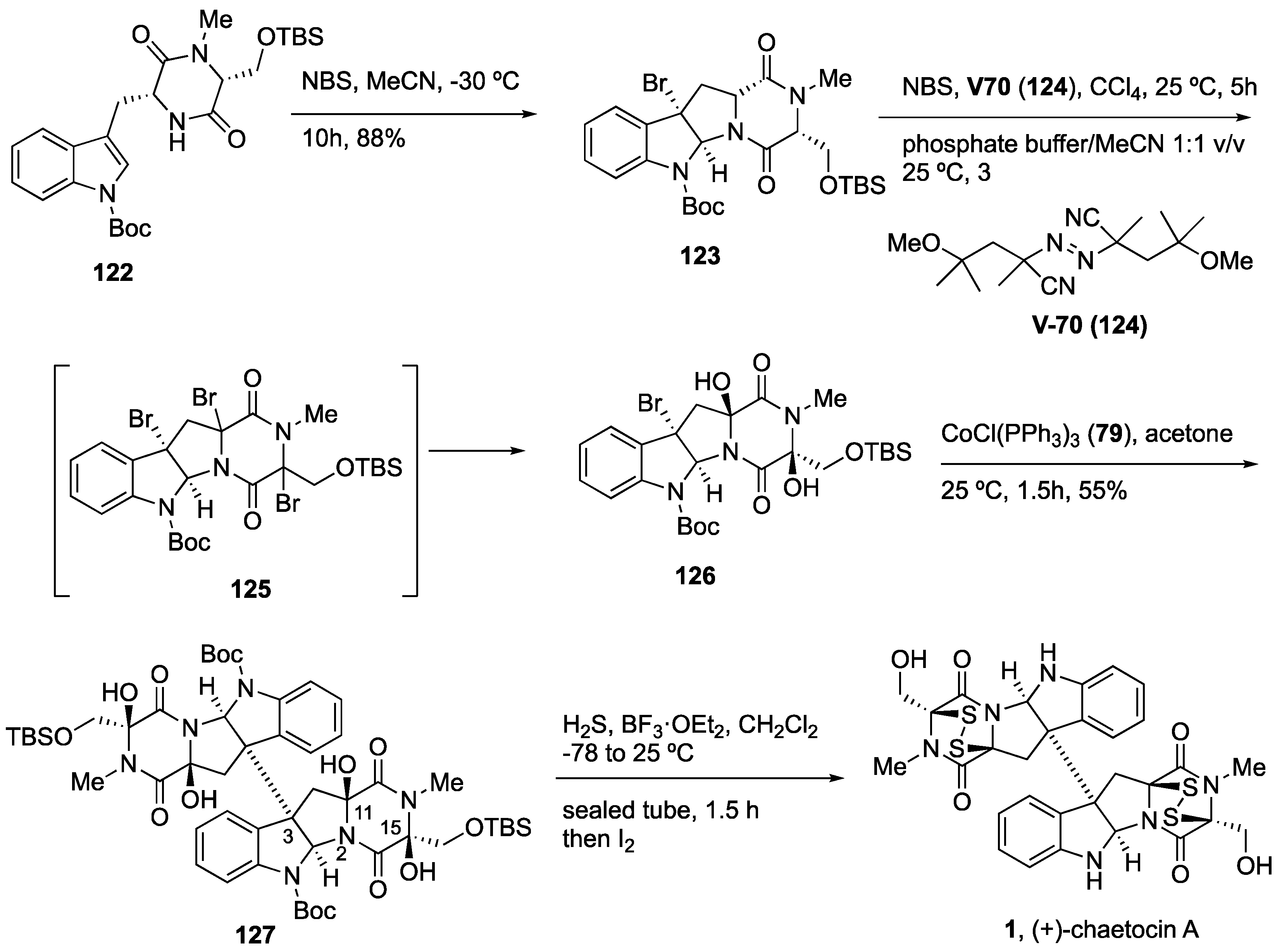
4.5. Synthesis of (+)-Chaetocin A (1)
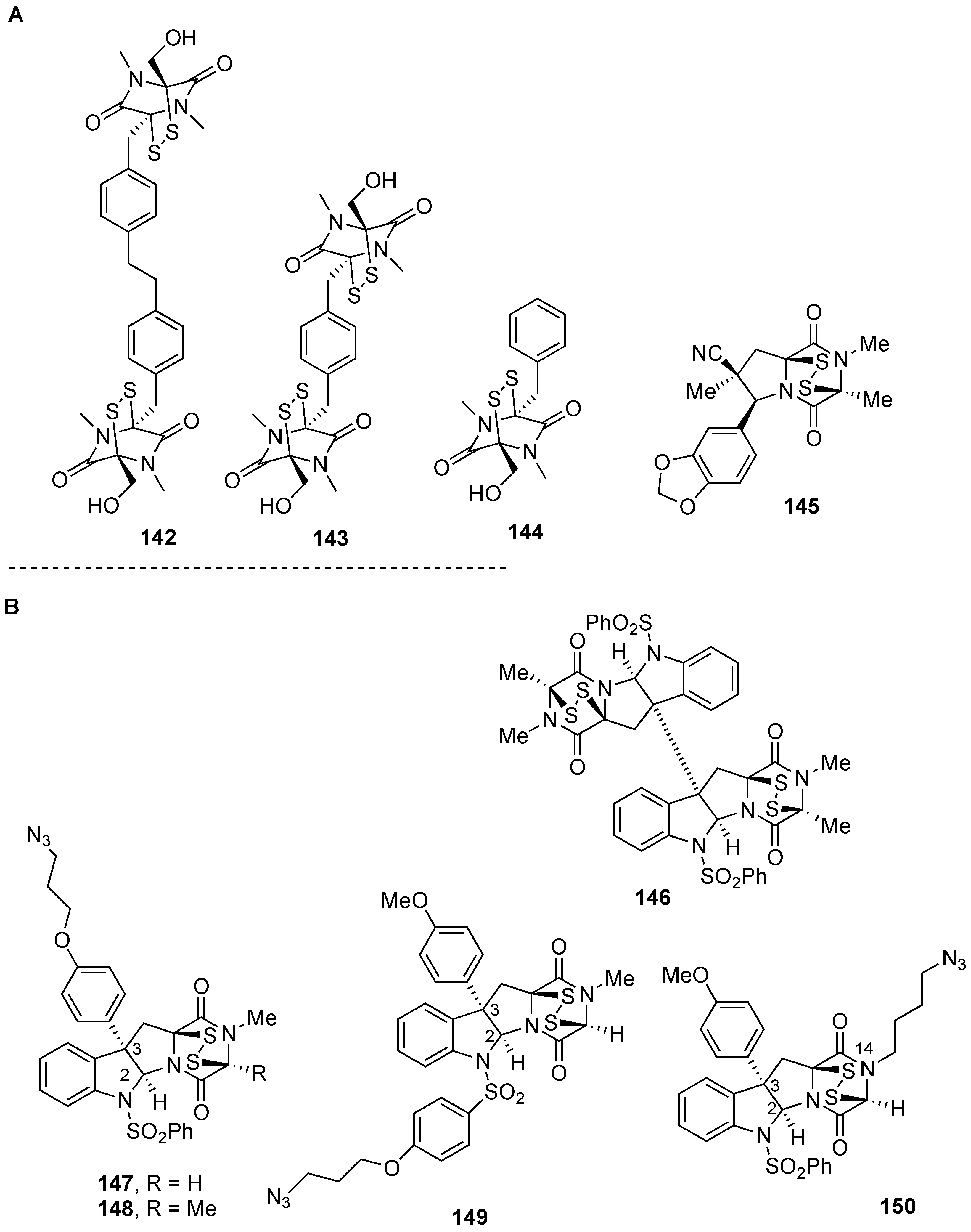
5. Biological Activities of Natural Product-Inspired (Dithio)dioxopiperazines
6. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, X.; Li, Y.; Zhang, X.; Lai, D.; Zhou, L. Structural Diversity and Biological Activities of the Cyclodipeptides from Fungi. Molecules 2017, 22, 2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojarska, J.; Mieczkowski, A.; Ziora, Z.M.; Skwarczynski, M.; Toth, I.; Shalash, A.O.; Parang, K.; El-Mowafi, S.A.; Mohammed, E.H.M.; Elnagdy, S.; et al. Cyclic Dipeptides: The Biological and Structural Landscape with Special Focus on the Anti-Cancer Proline-Based Scaffold. Biomolecules 2021, 11, 1515. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhou, A.; Xia, C. Strategies for total synthesis of bispyrrolidinoindoline alkaloids. Nat. Prod. Rep. 2022, 39, 1015–1044. [Google Scholar] [CrossRef] [PubMed]
- Barrow, C.J.; Cai, P.; Snyder, J.K.; Sedlock, D.M.; Sun, H.H.; Cooper, R. WIN 64821, a new competitive antagonist to substance P, isolated from an Aspergillus species: Structure determination and solution conformation. J. Org. Chem. 1993, 58, 6016–6021. [Google Scholar] [CrossRef]
- Borthwick, A.D. 2,5-Diketopiperazines: Synthesis, Reactions, Medicinal Chemistry, and Bioactive Natural Products. Chem. Rev. 2012, 112, 3641–3716. [Google Scholar] [CrossRef]
- Ciarkowski, J. CNDO/2 quantum-mechanical calculations of the conformational flexibility of the diketopiperazine skeleton. Biopolymers 1984, 23, 397–407. [Google Scholar] [CrossRef]
- Liskamp, R.M.J.; Rijkers, D.T.S.; Kruijtzer, J.A.W.; Kemmink, J. Peptides and Proteins as a Continuing Exciting Source of Inspiration for Peptidomimetics. ChemBioChem 2011, 12, 1626–1653. [Google Scholar] [CrossRef]
- Bai, W.-J.; Wang, X. Appreciation of symmetry in natural product synthesis. Nat. Prod. Rep. 2017, 34, 1345–1358. [Google Scholar] [CrossRef]
- Ma, Y.-M.; Liang, X.-A.; Kong, Y.; Jia, B. Structural Diversity and Biological Activities of Indole Diketopiperazine Alkaloids from Fungi. J. Agric. Food Chem. 2016, 64, 6659–6671. [Google Scholar] [CrossRef]
- Gardiner, D.M.; Waring, P.; Howlett, B.J. The epipolythiodioxopiperazine (ETP) class of fungal toxins: Distribution, mode of action, functions and biosynthesis. Microbiology 2005, 151, 1021–1032. [Google Scholar] [CrossRef]
- Jiang, C.-S.; Müller, W.E.G.; Schröder, H.C.; Guo, Y.-W. Disulfide- and Multisulfide-Containing Metabolites from Marine Organisms. Chem. Rev. 2011, 112, 2179–2207. [Google Scholar] [CrossRef] [PubMed]
- Welch, T.R.; Williams, R.M. Epidithiodioxopiperazines. Occurrence, synthesis and biogenesis. Nat. Prod. Rep. 2014, 31, 1376–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, N.G.M.; Pereira, R.B.; Andrade, P.B.; Valentão, P. Double the Chemistry, Double the Fun: Structural Diversity and Biological Activity of Marine-Derived Diketopiperazine Dimers. Mar. Drugs 2019, 17, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Li, H.Q.; Zong, S.C.; Gao, J.M.; Zhang, A.L. Chemical and Bioactive Diversities of the Genus Chaetomium Secondary Metabolites. Mini-Rev. Med. Chem. 2012, 12, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Morimoto, K.; Hamasaki, T. Flavoglaucin, a metabolite of Eurotium chevalieri, its antioxidation and synergism with tocopherol. J. Am. Oil Chem. Soc. 1984, 61, 1864–1868. [Google Scholar] [CrossRef]
- Slack, G.J.; Puniani, E.; Frisvad, J.C.; Samson, R.A.; Miller, J.D. Secondary metabolites from Eurotium species, Aspergillus calidoustus and A. insuetus common in Canadian homes with a review of their chemistry and biological activities. Mycol. Res. 2009, 113, 480–490. [Google Scholar] [CrossRef]
- Scott, T.A.; Piel, J. The hidden enzymology of bacterial natural product biosynthesis. Nat. Rev. Chem. 2019, 3, 404–425. [Google Scholar] [CrossRef]
- Giessen, T.W.; Marahiel, M.A. Rational and combinatorial tailoring of bioactive cyclic dipeptides. Front. Microbiol. 2015, 6, 785. [Google Scholar] [CrossRef]
- Hauser, D.; Weber, H.P.; Sigg, H.P. Isolierung und Strukturaufklärung von Chaetocin. Helv. Chim. Acta 1970, 53, 1061–1073. [Google Scholar] [CrossRef]
- Katagiri, K.; Sato, K.; Hakayama, S.; Matsushima, T.; Minato, H. Verticillin A, a new antibiotic from Verticillium sp. J. Antibiot. 1970, 23, 420–422. [Google Scholar] [CrossRef]
- Weindling, R.; Emerson, O.H. The isolation of a toxic substance from the culture filtrate of Trichoderma. Phytopathology 1936, 26, 1068–1070. [Google Scholar]
- Bell, M.R.; Johnson, J.R.; Wildi, B.S.; Woodward, R.B. THE STRUCTURE OF GLIOTOXIN. J. Am. Chem. Soc. 1958, 80, 1001. [Google Scholar] [CrossRef]
- Walsh, C.T. Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat. Prod. Rep. 2015, 33, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Moore, B.S. Enzymatic Cascade Reactions in Biosynthesis. Angew. Chem. Int. Ed. 2018, 58, 6846–6879. [Google Scholar] [CrossRef]
- Gondry, M.; Sauguet, L.; Belin, P.; Thai, R.; Amouroux, R.; Tellier, C.; Tuphile, K.; Jacquet, M.; Braud, S.; Courçon, M.; et al. Cyclodipeptide synthases are a family of tRNA-dependent peptide bond–forming enzymes. Nat. Chem. Biol. 2009, 5, 414–420. [Google Scholar] [CrossRef]
- Gondry, M.; Jacques, I.B.; Thai, R.; Babin, M.; Canu, N.; Seguin, J.; Belin, P.; Pernodet, J.-L.; Moutiez, M. A Comprehensive Overview of the Cyclodipeptide Synthase Family Enriched with the Characterization of 32 New Enzymes. Front. Microbiol. 2018, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Canu, N.; Moutiez, M.; Belin, P.; Gondry, M. Cyclodipeptide synthases: A promising biotechnological tool for the synthesis of diverse 2,5-diketopiperazines. Nat. Prod. Rep. 2019, 37, 312–321. [Google Scholar] [CrossRef]
- Shende, V.V.; Khatri, Y.; Newmister, S.A.; Sanders, J.N.; Lindovska, P.; Yu, F.; Doyon, T.J.; Kim, J.; Houk, K.N.; Movassaghi, M.; et al. Structure and Function of NzeB, a Versatile C–C and C–N Bond-Forming Diketopiperazine Dimerase. J. Am. Chem. Soc. 2020, 142, 17413–17424. [Google Scholar] [CrossRef]
- Yao, T.; Liu, J.; Liu, Z.; Li, T.; Li, H.; Che, Q.; Zhu, T.; Li, D.; Gu, Q.; Li, W. Genome mining of cyclodipeptide synthases unravels unusual tRNA-dependent diketopiperazine-terpene biosynthetic machinery. Nature Commun. 2018, 9, 4091. [Google Scholar] [CrossRef] [Green Version]
- Canu, N.; Belin, P.; Thai, R.; Correia, I.; Lequin, O.; Seguin, J.; Moutiez, M.; Gondry, M. Incorporation of Non-canonical Amino Acids into 2,5-Diketopiperazines by Cyclodipeptide Synthases. Angew. Chem. Int. Ed. 2018, 57, 3118–3122. [Google Scholar] [CrossRef]
- Borgman, P.; Lopez, R.D.; Lane, A.L. The expanding spectrum of diketopiperazine natural product biosynthetic pathways containing cyclodipeptide synthases. Org. Biomol. Chem. 2019, 17, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- García-Domínguez, P.; Areal, A.; Alvarez, R.; de Lera, A.R. Chemical synthesis in competition with global genome mining and heterologous expression for the preparation of dimeric tryptophan-derived 2,5-dioxopiperazines. Nat. Prod. Rep. 2022, 39, 1172–1225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, J.; Cheng, F.; Li, S. Cytochrome P450 enzymes in fungal natural product biosynthesis. Nat. Prod. Rep. 2021, 38, 1072–1099. [Google Scholar] [CrossRef]
- Liu, J.; Liu, A.; Hu, Y. Enzymatic dimerization in the biosynthetic pathway of microbial natural products. Nat. Prod. Rep. 2021, 38, 1469–1505. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fan, J.; Zhang, P.; Hu, Y.; Liu, X.; Li, S.-M.; Yin, W.-B. New insights into the disulfide bond formation enzymes in epidithiodiketopiperazine alkaloids. Chem. Sci. 2021, 12, 4132–4138. [Google Scholar] [CrossRef] [PubMed]
- Patteson, J.B.; Cai, W.; Johnson, R.A.; Maria, K.C.S.; Li, B. Identification of the Biosynthetic Pathway for the Antibiotic Bicyclomycin. Biochemistry 2017, 57, 61–65. [Google Scholar] [CrossRef]
- Meng, S.; Han, W.; Zhao, J.; Jian, X.; Pan, H.; Tang, G. A Six-Oxidase Cascade for Tandem C−H Bond Activation Revealed by Reconstitution of Bicyclomycin Biosynthesis. Angew. Chem. Int. Ed. 2017, 57, 719–723. [Google Scholar] [CrossRef]
- Giessen, T.W.; von Tesmar, A.M.; Marahiel, M.A. A tRNA-Dependent Two-Enzyme Pathway for the Generation of Singly and Doubly Methylated Ditryptophan 2,5-Diketopiperazines. Biochemistry 2013, 52, 4274–4283. [Google Scholar] [CrossRef]
- Yu, H.; Xie, X.; Li, S.-M. Coupling of Guanine with cyclo-l-Trp-l-Trp Mediated by a Cytochrome P450 Homologue from Streptomyces purpureus. Org. Lett. 2018, 20, 4921–4925. [Google Scholar] [CrossRef]
- Shi, J.; Xu, X.; Zhao, E.J.; Zhang, B.; Li, W.; Zhao, Y.; Jiao, R.H.; Tan, R.X.; Ge, H.M. Genome Mining and Enzymatic Total Biosynthesis of Purincyclamide. Org. Lett. 2019, 21, 6825–6829. [Google Scholar] [CrossRef]
- Yu, H.; Xie, X.; Li, S.-M. Coupling of cyclo-L-Trp-L-Trp with Hypoxanthine Increases the Structure Diversity of Guanitrypmycins. Org. Lett. 2019, 21, 9104–9108. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xie, X.; Li, S.-M. Guanitrypmycin Biosynthetic Pathways Imply Cytochrome P450 Mediated Regio- and Stereospecific Guaninyl-Transfer Reactions. Angew. Chem. Int. Ed. 2019, 58, 11534–11540. [Google Scholar] [CrossRef] [PubMed]
- Yet, L. Priviledged Structures in Drug Discovery: Medicinal Chemistry and Synthesis; John Wiley & Sons, Inc.: Hoboken, NY, USA, 2018. [Google Scholar]
- Cook, K.M.; Hilton, S.T.; Mecinović, J.; Motherwell, W.B.; Figg, W.D.; Schofield, C.J. Epidithiodiketopiperazines Block the Interaction between Hypoxia-inducible Factor-1α (HIF-1α) and p300 by a Zinc Ejection Mechanism. J. Biol. Chem. 2009, 284, 26831–26838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasa, E.; Hamashima, Y.; Sodeoka, M. Epipolithiodiketopiperazines Alkaloids: Total Synthesis and Biological Activities. Isr. J. Chem. 2011, 51, 420–433. [Google Scholar] [CrossRef]
- Jiang, C.-S.; Wang, L.; Guo, Y.-W. Epipolythiodioxopiperazines from Fungi: Chemistry and Bioactivities. Recent Advances in Medicinal Chemistry; Atta-ur-Rahman, Choudhary, M.I., Perry, G., Eds.; Bentham eBooks: Soest, The Netherlands, 2015; Volume 2, pp. 76–106. [Google Scholar] [CrossRef]
- Kim, J.; Movassaghi, M. Biogenetically inspired syntheses of alkaloid natural products. Chem. Soc. Rev. 2009, 38, 3035–3050. [Google Scholar] [CrossRef]
- Lathrop, S.P.; Kim, J.; Movassaghi, M. Radical-mediated Dimerization and Oxidation Reactions for the Synthesis of Complex Alkaloids. CHIMIA 2012, 66, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Movassaghi, M. Biogenetically-Inspired Total Synthesis of Epidithiodiketopiperazines and Related Alkaloids. Acc. Chem. Res. 2015, 48, 1159–1171. [Google Scholar] [CrossRef] [Green Version]
- Sil, B.C.; Hilton, S.T. 2-Epi-3,6-dithio-2,5-diketopiperazines (ETPs): An overview of synthetic approaches to the ETP core. In Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Cambridge, MA, USA, 2021; Volume 32, pp. 27–105. [Google Scholar]
- Zhao, P.; Xue, Y.; Li, J.; Li, X.; Zu, X.; Zhao, Z.; Quan, C.; Gao, W.; Feng, S. Non-lipopeptide fungi-derived peptide antibiotics developed since 2000. Biotechnol. Lett. 2019, 41, 651–673. [Google Scholar] [CrossRef]
- Minato, H.; Matsumoto, M.; Katayama, T. Verticillin A, a new antibiotic from Verticillium sp. J. Chem. Soc. D 1971, 44–45. [Google Scholar] [CrossRef]
- Safe, S.; Taylor, A. Sporidesmins. Part XI. The reaction of triphenylphosphine with epipolythiodioxopiperazines. J. Chem. Soc. C 1971, 1189–1192. [Google Scholar] [CrossRef]
- Herscheid, J.D.M.; Tijhuis, M.W.; Noordik, J.H.; Ottenheijm, H.C.J. Desulfurization of epidithiodioxopiperazines. A mechanistic and chiroptical study. J. Am. Chem. Soc. 1979, 101, 1159–1162. [Google Scholar] [CrossRef]
- Ferezou, J.P.; Quesneau-Thierry, A.; Cesario, M.; Pascard, C.; Barbier, M. Desulfurization of the epidithiopiperazinedione sirodesmin PL with triphenylphosphine: Retention of configuration at the bridgehead carbon atoms. J. Am. Chem. Soc. 1983, 105, 5402–5406. [Google Scholar] [CrossRef]
- Cherblanc, F.; Lo, Y.-P.; De Gussem, E.; Alcazar-Fuoli, L.; Bignell, E.; He, Y.; Chapman-Rothe, N.; Bultinck, P.; Herrebout, W.A.; Brown, R.; et al. On the Determination of the Stereochemistry of Semisynthetic Natural Product Analogues using Chiroptical Spectroscopy: Desulfurization of Epidithiodioxopiperazine Fungal Metabolites. Chem. –Eur. J. 2011, 17, 11868–11875. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, C.; Numata, A.; Ito, Y.; Matsumura, E.; Araki, H.; Iwaki, H.; Kushida, K. Leptosins, antitumour metabolites of a fungus isolated from a marine alga. J. Chem. Soc. Perkin Trans. 1 1994, 13, 1859–1864. [Google Scholar] [CrossRef]
- Li, Y.; Sun, K.-L.; Wang, Y.; Fu, P.; Liu, P.-P.; Wang, C.; Zhu, W.-M. A cytotoxic pyrrolidinoindoline diketopiperazine dimer from the algal fungus Eurotium herbariorum HT-2. Chin. Chem. Lett. 2013, 24, 1049–1052. [Google Scholar] [CrossRef]
- Weber, H.P. The molecular structure and absolute configuration of chaetocin. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1972, 28, 2945–2951. [Google Scholar] [CrossRef] [Green Version]
- Watts, K.R.; Ratnam, J.; Ang, K.-H.; Tenney, K.; Compton, J.E.; McKerrow, J.; Crews, P. Assessing the trypanocidal potential of natural and semi-synthetic diketopiperazines from two deep water marine-derived fungi. Bioorganic Med. Chem. 2010, 18, 2566–2574. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S.; Spilsbury, J.F. Gliotoxin from Aspergillus chevalieri (Mangin) Thom et Church. Nature 1965, 206, 619. [Google Scholar] [CrossRef]
- Saito, T.; Suzuki, Y.; Koyama, K.; Natori, S.; Iitaka, Y.; Kinosita, T. Chetracin A and Chaetocins B and C, Three New Epipolythiodioxo-piperazines from Chaetomium spp. Chem. Pharm. Bull. 1988, 36, 1942–1956. [Google Scholar] [CrossRef] [Green Version]
- Greiner, D.; Bonaldi, T.; Eskeland, R.; Roemer, E.; Imhof, A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat. Chem. Biol. 2005, 1, 143–145. [Google Scholar] [CrossRef]
- Copeland, R.A.; Solomon, M.E.; Richon, V.M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 2009, 8, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.C.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Li, L.; Yang, D.; Zeng, L.; Yewei, X.; Yu, B.; Liao, G.; Chen, J. Recent progress in histone methyltransferase (G9a) inhibitors as anticancer agents. Eur. J. Med. Chem. 2019, 179, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Milite, C.; Feoli, A.; Viviano, M.; Rescigno, D.; Mai, A.; Castellano, S.; Sbardella, G. Progress in the Development of Lysine Methyltransferase SETD8 Inhibitors. ChemMedChem 2016, 11, 1680–1685. [Google Scholar] [CrossRef]
- Song, X.; Zhao, Z.; Qi, X.; Tang, S.; Wang, Q.; Zhu, T.; Gu, Q.; Liu, M.; Li, J. Identification of epipolythiodioxopiperazines HDN-1 and chaetocin as novel inhibitor of heat shock protein 90. Oncotarget 2015, 6, 5263–5274. [Google Scholar] [CrossRef] [Green Version]
- Greiner, D.; Bonaldi, T.; Eskeland, R.; Roemer, E.; Imhof, A. Reply to "Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases". Nat. Chem. Biol. 2013, 9, 137. [Google Scholar] [CrossRef]
- Cherblanc, F.L.; Chapman, K.L.; Brown, R.; Fuchter, M.J. Chaetocin is a nonspecific inhibitor of histone lysine methyl-transferases. Nat. Chem. Biol. 2013, 9, 136–137. [Google Scholar] [CrossRef]
- Cherblanc, F.L.; Chapman, K.L.; Reid, J.; Borg, A.J.; Sundriyal, S.; Alcazar-Fuoli, L.; Bignell, E.; Demetriades, M.; Schofield, C.J.; DiMaggio, J.P.A.; et al. On the Histone Lysine Methyltransferase Activity of Fungal Metabolite Chaetocin. J. Med. Chem. 2013, 56, 8616–8625. [Google Scholar] [CrossRef]
- Argoudelis, A.D. Melinacidins II, III and IV, new “3,6-epidithiadiketopiperazines antibiotics”. J. Antibiot. 1972, 25, 171–178. [Google Scholar] [CrossRef]
- Argoudelis, A.D.; Mizsak, S.A. Melinacidins II, III and IV Structural Studies. J. Antibiot. 1977, 30, 468–473. [Google Scholar] [CrossRef]
- Hauser, D.; Loosli, H.R.; Niklaus, P. Isolierung von 11α,11′α-Dihydroxychaetocin aus Verticillium tenerum. Helv. Chim. Acta 1972, 55, 2182–2187. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Koyama, K.; Natori, S.; Iitaka, Y. Chetracin A, a new epipolythiodioxopiperazine having a tetrasulfide bridge from Chaetomium abuense and C. retardatum. Tetrahedron Lett. 1985, 26, 4731–4734. [Google Scholar] [CrossRef]
- Son, B.W.; Jensen, P.R.; Kauffman, C.A.; Fenical, W. New Cytotoxic Epidithiodioxopiperazines Related to Verticillin A From A Marine Isolate of the Fungus Penicillium. Nat. Prod. Lett. 1999, 13, 213–222. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Chen, Y.; Guo, X.-N.; Zhang, X.-W.; Zhao, W.-M.; Zhong, L.; Zhou, J.; Xi, Y.; Lin, L.-P.; Ding, J. 11,11′-Dideoxyverticillin: A natural compound possessing growth factor receptor tyrosine kinase-inhibitory effect with anti-tumor activity. Anti-Cancer Drugs 2005, 16, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, H.; Du, Z.; Liu, X.-Z.; Che, Y.; Ye, X. Ecology-based screen identifies new metabolites from a Cordyceps-colonizing fungus as cancer cell proliferation inhibitors and apoptosis inducers. Cell Prolif. 2009, 42, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.; Graf, T.N.; Ayers, S.; Adcock, A.F.; Kroll, D.J.; Yang, J.; Swanson, S.M.; Munoz-Acuna, U.; de Blanco, E.J.C.; Agrawal, R.; et al. Cytotoxic epipolythiodioxopiperazine alkaloids from filamentous fungi of the Bionectriaceae. J. Antibiot. 2012, 65, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.-Y.; He, H.-P.; Shen, Y.-M.; Zhang, K.-Q. Nematicidal Epipolysulfanyldioxopiperazines from Gliocladium roseum. J. Nat. Prod. 2005, 68, 1510–1513; [Google Scholar] [CrossRef]
- Paschall, A.V.; Yang, D.; Lu, C.; Choi, J.-H.; Li, X.; Liu, F.; Figueroa, M.; Oberlies, N.H.; Pearce, C.; Bollag, W.B.; et al. H3K9 Trimethylation Silences Fas Expression To Confer Colon Carcinoma Immune Escape and 5-Fluorouracil Chemoresistance. J. Immunol. 2015, 195, 1868–1882. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Paschall, A.V.; Shi, H.; Savage, N.; Waller, J.L.; Sabbatini, M.E.; Oberlies, N.H.; Pearce, C.; Liu, K. The MLL1-H3K4me3 Axis-Mediated PD-L1 Expression and Pancreatic Cancer Immune Evasion. JNCI: J. Natl. Cancer Inst. 2017, 109, djw283. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Liu, Q.; Yang, D.; Bollag, W.B.; Robertson, K.; Wu, P.; Liu, K. Verticillin A Overcomes Apoptosis Resistance in Human Colon Carcinoma through DNA Methylation-Dependent Upregulation of BNIP3. Cancer Res. 2011, 71, 6807–6816. [Google Scholar] [CrossRef]
- He, P.; Che, Y.-S.; He, Q.-J.; Chen, Y.; Ding, J. G226, a novel epipolythiodioxopiperazine derivative, induces autophagy and caspase-dependent apoptosis in human breast cancer cells in vitro. Acta Pharmacol. Sin. 2014, 35, 1055–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebead, G.A.; Overy, D.P.; Berrué, F.; Kerr, R.G. Westerdykella reniformis sp. nov., producing the antibiotic metabolites melinacidin IV and chetracin B. IMA Fungus 2012, 3, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Minato, H.; Matsumoto, M.; Katayama, T. Studies on the metabolites of Verticillium sp. Structures of verticillins A, B, and C. J. Chem. Soc. Perkin 1 1973, 17, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.K.; Gloer, J.B.; Wicklow, D.T. New Verticillin and Glisoprenin Analogues from Gliocladium catenulatum, a Myco-parasite of Aspergillus flavus Sclerotia. J. Nat. Prod. 1999, 62, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, C.; Minoura, K.; Yamada, T.; Numata, A.; Kushida, K.; Shingu, T.; Hagishita, S.; Nakai, H.; Sato, T.; Harada, H. Potent cytotoxic metabolites from a Leptosphaeria species. Structure determination and conformational analysis. Tetrahedron 1995, 51, 3483–3498. [Google Scholar] [CrossRef]
- Che, Y.; Liu, X.; Ye, X.; Guo, H.; Chen, Y.; Liu, S. Method for preparing gliocladicillin C and application thereof. CN Patent 101805699B, 29 May 2013. [Google Scholar]
- Li, E.; Hou, B.; Gao, Q.; Xu, Y.; Zhang, C.; Liu, X.; Jiang, X.; Che, Y. Disulfide Cleavage in a Dimeric Epipolythiodioxopiperazine Natural Product Diminishes Its Apoptosis-Inducing Effect but Enhances Autophagy in Tumor Cells. J. Nat. Prod. 2020, 83, 601–609. [Google Scholar] [CrossRef]
- Chu, M.; Truumees, I.; Rothofsky, M.L.; Patel, M.G.; Gentile, F.; Das, P.R.; Puar, M.S.; Lin, S.L. Inhibition of c-fos Pro-to-oncogene Induction by Sch 52900 and Sch 52901, Novel Diketopiperazines Produced by Gliocladium sp. J. Antibiot. 1995, 48, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Erkel, G.; Gehrt, A.; Anke, T.; Sterner, O. Induction of Differentiation in Acute Promyelocytic Leukemia Cells (HL-60) by the Verticillin Derivative Sch 52900. Z. Naturforsch. 2002, 57c, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Ebrahim, W.; Kjer, J.; El Amrani, M.; Wray, V.; Lin, W.; Ebel, R.; Lai, D.; Proksch, P. Pullularins E and F, Two New Peptides from the Endophytic Fungus Bionectria ochroleuca Isolated from the Mangrove Plant Sonneratia caseolaris. Mar. Drugs 2012, 10, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.-J.; Park, S.-H.; Koshino, H.; Kim, Y.-H.; Kim, W.-G. Verticillin G, a New Antibacterial Compound from Bionectra byssicola. J. Antibiot. 2007, 60, 61–64. [Google Scholar] [CrossRef]
- Li, L.; Li, D.; Luan, Y.; Gu, Q.; Zhu, T. Cytotoxic Metabolites from the Antarctic Psychrophilic Fungus Oidiodendron truncatum. J. Nat. Prod. 2012, 75, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Pickering, T.L.; Saunders, K.J.; Tobolsky, A.V. Disproportionation of organic polysulfides. J. Am. Chem. Soc. 1967, 89, 2364–2367. [Google Scholar] [CrossRef]
- Yu, G.; Wang, Y.; Yu, R.; Feng, Y.; Wang, L.; Che, Q.; Gu, Q.; Li, D.; Li, J.; Zhu, T. Chetracins E and F, cytotoxic epipolythiodioxopiperazines from the marine-derived fungus Acrostalagmus luteoalbus HDN13-530. RSC Adv. 2018, 8, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, C.; Numata, A.; Matsumura, E.; Minoura, K.; Eto, H.; Shingu, T.; Ito, T.; Hasegawa, T. Leptosins I and J, cytotoxic substances produced by a Leptosphaeria sp. Physico-chemical properties and structures. J. Antibiot. 1994, 47, 1242–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, C.; Takai, Y.; Kimura, Y.; Numata, A.; Shigematsu, N.; Tanaka, H. Cytotoxic metabolites from a fungal adherent of a marine alga. Phytochemistry 1995, 38, 155–158. [Google Scholar] [CrossRef]
- Du, L.; Robles, A.J.; King, J.B.; Mooberry, S.L.; Cichewicz, R.H. Cytotoxic Dimeric Epipolythiodiketopiperazines from the Ascomycetous Fungus Preussia typharum. J. Nat. Prod. 2014, 77, 1459–1466. [Google Scholar] [CrossRef]
- Yanagihara, M.; Sasaki-Takahashi, N.; Sugahara, T.; Yamamoto, S.; Shinomi, M.; Yamashita, I.; Hayashida, M.; Yamanoha, B.; Numata, A.; Yamori, T.; et al. Leptosins isolated from marine fungus Leptoshaeria species inhibit DNA topoisomerases I and/or II and induce apoptosis by inactivation of Akt/protein kinase B. Cancer Sci. 2005, 96, 816–824. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yamada, T.; Iwamoto, C.; Yamagaki, N.; Yamanouchi, T.; Minoura, K.; Yamori, T.; Uehara, Y.; Andoh, T.; Umemura, K.; Numata, A. Leptosins M–N1, cytotoxic metabolites from a Leptosphaeria species separated from a marine alga. Structure determination and biological activities. Tetrahedron 2002, 58, 479–487. [Google Scholar] [CrossRef]
- Yamada, T.; Iwamoto, C.; Yamagaki, N.; Yamanouchi, T.; Minoura, K.; Hagishita, S.; Numata, A. Leptosins O-S, Cytotoxic Metabolites of a Strain of Leptosphaeria sp. Isolated from a Marine Alga. Heterocycles 2004, 63, 641. [Google Scholar] [CrossRef]
- Wang, N.; Saidhareddy, P.; Jiang, X. Construction of sulfur-containing moieties in the total synthesis of natural products. Nat. Prod. Rep. 2019, 37, 246–275. [Google Scholar] [CrossRef]
- Hai, Y.; Wei, M.-Y.; Wang, C.-Y.; Gu, Y.-C.; Shao, C.-L. The intriguing chemistry and biology of sulfur-containing natural products from marine microorganisms (1987–2020). Mar. Life Sci. Technol. 2021, 3, 488–518. [Google Scholar] [CrossRef]
- Sun, C.; Tian, W.; Lin, Z.; Qu, X. Biosynthesis of pyrroloindoline-containing natural products. Nat. Prod. Rep. 2022, 39, 1721–1765. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, K.L.; Scharf, D.H.; Litomska, A.; Hertweck, C. Enzymatic Carbon–Sulfur Bond Formation in Natural Product Biosynthesis. Chem. Rev. 2017, 117, 5521–5577. [Google Scholar] [CrossRef] [PubMed]
- Scharf, D.H.; Groll, M.; Habel, A.; Heinekamp, T.; Hertweck, C.; Brakhage, A.A.; Huber, E.M. Flavoenzyme-Catalyzed Formation of Disulfide Bonds in Natural Products. Angew. Chem. Int. Ed. 2014, 53, 2221–2224. [Google Scholar] [CrossRef]
- Harken, L.; Li, S.-M. Modifications of diketopiperazines assembled by cyclodipeptide synthases with cytochrome P450 enzymes. App. Microbiol. Biotechnol. 2021, 105, 2277–2285. [Google Scholar] [CrossRef]
- Scherlach, K.; Hertweck, C. Mining and unearthing hidden biosynthetic potential. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Pedras, M.S.C.; Séguin-Swartz, G.; Abrams, S.R. Minor phytotoxins from the blackleg fungus Phoma lingam. Phytochemistry 1990, 29, 777–782. [Google Scholar] [CrossRef]
- Cramer, R.A.; Gamcsik, M.P.; Brooking, R.M.; Najvar, L.K.; Kirkpatrick, W.R.; Patterson, T.F.; Balibar, C.J.; Graybill, J.R.; Perfect, J.R.; Abraham, S.N.; et al. Disruption of a Nonribosomal Peptide Synthetase in Aspergillus fumigatus Eliminates Gliotoxin Production. Eukaryotic Cell 2006, 5, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Scharf, D.H.; Remme, N.; Habel, A.; Chankhamjon, P.; Scherlach, K.; Heinekamp, T.; Hortschansky, P.; Brakhage, A.A.; Hertweck, C. A Dedicated Glutathione S-Transferase Mediates Carbon–Sulfur Bond Formation in Gliotoxin Biosynthesis. J. Am. Chem. Soc. 2011, 133, 12322–12325. [Google Scholar] [CrossRef]
- Davis, C.; Carberry, S.; Schrettl, M.; Singh, I.; Stephens, J.C.; Barry, S.M.; Kavanagh, K.; Challis, G.L.; Brougham, D.; Doyle, S. The Role of Glutathione S-Transferase GliG in Gliotoxin Biosynthesis in Aspergillus fumigatus. Chem. Biol. 2011, 18, 542–552. [Google Scholar] [CrossRef]
- Scharf, D.H.; Chankhamjon, P.; Scherlach, K.; Heinekamp, T.; Willing, K.; Brakhage, A.A.; Hertweck, C. Epidithiodiketopiperazine Biosynthesis: A Four-Enzyme Cascade Converts Glutathione Conjugates into Transannular Disulfide Bridges. Angew. Chem. Int. Ed. 2013, 52, 11092–11095. [Google Scholar] [CrossRef] [PubMed]
- Gerken, T.; Walsh, C.T. Cloning and Sequencing of the Chaetocin Biosynthetic Gene Cluster. ChemBioChem 2013, 14, 2256–2258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, P.; Pan, Y.; Zhu, Y.; Liu, X.; Che, Y.; Liu, G. Identification and characterization of the verticillin biosynthetic gene cluster in Clonostachys rogersoniana. Fungal Genet. Biol. 2017, 103, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Hao, T.; Wang, Y.; Pan, Y.; Ren, F.; Liu, X.; Che, Y.; Liu, G. VerZ, a Zn(II)2Cys6 DNA-binding protein, regulates the biosynthesis of verticillin in Clonostachys rogersoniana. Microbiology 2017, 163, 1654–1663. [Google Scholar] [CrossRef]
- Zhu, S.; Ren, F.; Guo, Z.; Liu, J.; Liu, X.; Liu, G.; Che, Y. Rogersonins A and B, Imidazolone N-Oxide-Incorporating Indole Alkaloids from a verG Disruption Mutant of Clonostachys rogersoniana. J. Nat. Prod. 2019, 82, 462–468. [Google Scholar] [CrossRef]
- Wang, Y.; Ren, J.; Li, H.; Pan, Y.; Liu, X.; Che, Y.; Liu, G. The disruption of verM activates the production of gliocladiosin A and B in Clonostachys rogersoniana. Org. Biomol. Chem. 2019, 17, 6782–6785. [Google Scholar] [CrossRef]
- Movassaghi, M.; Schmidt, M.A.; Ashenhurst, J.A. Concise Total Synthesis of (+)-WIN 64821 and (−)-Ditryptophenaline. Angew. Chem. Int. Ed. 2008, 47, 1485–1487. [Google Scholar] [CrossRef]
- Movassaghi, M.; Ahmad, O.K.; Lathrop, S.P. Directed Heterodimerization: Stereocontrolled Assembly via Solvent-Caged Unsymmetrical Diazene Fragmentation. J. Am. Chem. Soc. 2011, 133, 13002–13005. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Balado, C.; Rodríguez-Graña, P.; de Lera, A.R. Stereocontrolled and Versatile Total Synthesis of Bispyrrolidinoindoline Diketopiperazine Alkaloids: Structural Revision of the Fungal Isolate (+)-Asperdimin. Chem.—Eur. J. 2009, 15, 9928–9937. [Google Scholar] [CrossRef]
- López, C.S.; Pérez-Balado, C.; Rodríguez-Graña, P.; de Lera, R. Mechanistic Insights into the Stereocontrolled Synthesis of Hexahydropyrrolo[2,3-b]indoles by Electrophilic Activation of Tryptophan Derivatives. Org. Lett. 2007, 10, 77–80. [Google Scholar] [CrossRef]
- Crich, D.; Banerjee, A. Chemistry of the Hexahydropyrrolo[2,3-b]indoles: Configuration, Conformation, Reactivity, and Applications in Synthesis. Acc. Chem. Res. 2007, 40, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Tadano, S.; Mukaeda, Y.; Ishikawa, H. Bio-Inspired Dimerization Reaction of Tryptophan Derivatives in Aqueous Acidic Media: Three-Step Syntheses of (+)-WIN 64821, (−)-Ditryptophenaline, and (+)-Naseseazine B. Angew. Chem. Int. Ed. 2013, 52, 7990–7994. [Google Scholar] [CrossRef] [PubMed]
- Tadano, S.; Sugimachi, Y.; Sumimoto, M.; Tsukamoto, S.; Ishikawa, H. Collective Synthesis and Biological Evaluation of Tryptophan-Based Dimeric Diketopiperazine Alkaloids. Chem.—Eur. J. 2015, 22, 1277–1291. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-K.; Yang, J.-S.; Dai, K.-L.; Zhang, F.-M.; Zhang, X.-M.; Tu, Y.-Q. Exploration of a KI-catalyzed oxidation system for direct construction of bispyrrolidino[2,3-b]indolines and the total synthesis of (+)-WIN 64821. Chem. Commun. 2019, 56, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Deng, X.; Tong, X.; Li, D.; Ding, M.; Zhou, A.; Xia, C. Copper-Mediated Dimerization to Access 3a,3a′-Bispyrrolidinoindoline: Diastereoselective Synthesis of (+)-WIN 64821 and (−)-Ditryptophenaline. Org. Lett. 2015, 17, 206–209. [Google Scholar] [CrossRef]
- Boyer, N.; Morrison, K.C.; Kim, J.; Hergenrother, P.J.; Movassaghi, M. Synthesis and anticancer activity of epipolythiodiketopiperazine alkaloids. Chem. Sci. 2013, 4, 1646–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, N.C.; Movassaghi, M. Concise total synthesis of (+)-gliocladins B and C. Chem. Sci. 2012, 3, 1798–1803. [Google Scholar] [CrossRef] [Green Version]
- Kieffer, M.E.; Chuang, K.; Reisman, S.E. A copper-catalyzed arylation of tryptamines for the direct synthesis of aryl pyrroloindolines. Chem. Sci. 2012, 3, 3170–3174. [Google Scholar] [CrossRef]
- Kieffer, M.E.; Chuang, K.V.; Reisman, S.E. Copper-Catalyzed Diastereoselective Arylation of Tryptophan Derivatives: Total Synthesis of (+)-Naseseazines A and B. J. Am. Chem. Soc. 2013, 135, 5557–5560. [Google Scholar] [CrossRef] [Green Version]
- Repka, L.M.; Reisman, S.E. Recent Developments in the Catalytic, Asymmetric Construction of Pyrroloindolines Bearing All-Carbon Quaternary Stereocenters. J. Org. Chem. 2013, 78, 12314–12320. [Google Scholar] [CrossRef]
- Kim, J.; Ashenhurst, J.A.; Movassaghi, M. Total Synthesis of (+)-11,11’-Dideoxyverticillin A. Science 2009, 324, 238–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Movassaghi, M. General Approach to Epipolythiodiketopiperazine Alkaloids: Total Synthesis of (+)-Chaetocin A and C and (+)-12,12’-Dideoxychetracin A. J. Am. Chem. Soc. 2010, 132, 14376–14378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasa, E.; Hamashima, Y.; Fujishiro, S.; Higuchi, E.; Ito, A.; Yoshida, M.; Sodeoka, M. Total Synthesis of (+)-Chaetocin and its Analogues: Their Histone Methyltransferase G9a Inhibitory Activity. J. Am. Chem. Soc. 2010, 132, 4078–4079. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, E.; Hamashima, Y.; Fujishiro, S.; Hashizume, D.; Sodeoka, M. Total syntheses of chaetocin and ent-chaetocin. Tetrahedron 2011, 67, 6587–6599. [Google Scholar] [CrossRef]
- Brewer, D.; McInnes, A.G.; Smith, D.G.; Taylor, A.; Walter, J.A.; Loosli, H.R.; Kis, Z.L. Sporidesmins. Part 16. The structure of chetomin, a toxic metabolite of Chaetomium cochliodes, by N-15 and C-13 Nuclear Magnetic Resonance spectroscopy. J. Chem. Soc., Perkin Trans. 1 1978, 1248–1251. [Google Scholar] [CrossRef]
- Kung, A.L.; Zabludoff, S.D.; France, D.S.; Freedman, S.J.; A Tanner, E.; Vieira, A.; Cornell-Kennon, S.; Lee, J.; Wang, B.; Wang, J.; et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 2004, 6, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Block, K.M.; Wang, H.; Szabó, L.Z.; Polaske, N.W.; Henchey, L.K.; Dubey, R.; Kushal, S.; László, C.F.; Makhoul, J.; Song, Z.; et al. Direct Inhibition of Hypoxia-Inducible Transcription Factor Complex with Designed Dimeric Epidithiodiketopiperazine. J. Am. Chem. Soc. 2009, 131, 18078–18088. [Google Scholar] [CrossRef] [Green Version]
- Dubey, R.; Levin, M.D.; Szabo, L.Z.; Laszlo, C.F.; Kushal, S.; Singh, J.B.; Oh, P.; Schnitzer, J.E.; Olenyuk, B.Z. Suppression of Tumor Growth by Designed Dimeric Epidithiodiketopiperazine Targeting Hypoxia-Inducible Transcription Factor Complex. J. Am. Chem. Soc. 2013, 135, 4537–4549. [Google Scholar] [CrossRef]
- Fujishiro, S.; Dodo, K.; Iwasa, E.; Teng, Y.; Sohtome, Y.; Hamashima, Y.; Ito, A.; Yoshida, M.; Sodeoka, M. Epidithiodiketopiperazine as a pharmacophore for protein lysine methyltransferase G9a inhibitors: Reducing cytotoxicity by structural simplification. Bioorg. Med. Chem. Lett. 2013, 23, 733–736. [Google Scholar] [CrossRef]
- Teng, Y.; Iuchi, K.; Iwasa, E.; Fujishiro, S.; Hamashima, Y.; Dodo, K.; Sodeoka, M. Unnatural enantiomer of chaetocin shows strong apoptosis-inducing activity through caspase-8/caspase-3 activation. Bioorg. Med. Chem. 2010, 20, 5085–5088. [Google Scholar] [CrossRef]
- Cherblanc, F.L.; Davidson, R.W.M.; Di Fruscia, P.; Srimongkolpithak, N.; Fuchter, M.J. Perspectives on natural product epigenetic modulators in chemical biology and medicine. Nat. Prod. Rep. 2013, 30, 605–624. [Google Scholar] [CrossRef] [PubMed]
- Cherblanc, F.L.; Lo, Y.-P.; Herrebout, W.A.; Bultinck, P.; Rzepa, H.S.; Fuchter, M.J. Mechanistic and Chiroptical Studies on the Desulfurization of Epidithiodioxopiperazines Reveal Universal Retention of Configuration at the Bridgehead Carbon Atoms. J. Org. Chem. 2013, 78, 11646–11655. [Google Scholar] [CrossRef] [PubMed]
- Waring, P.; Eichner, R.D.; Müllbacher, A. The chemistry and biology of the immunomodulating agent gliotoxin and related epipolythiodioxopiperazines. Med. Res. Rev. 1988, 8, 499–524. [Google Scholar] [CrossRef] [PubMed]
- Isham, C.R.; Tibodeau, J.D.; Jin, W.; Xu, R.; Timm, M.M.; Bible, K.C. Chaetocin: A promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress. Blood 2006, 109, 2579–2588. [Google Scholar] [CrossRef] [Green Version]
- Isham, C.R.; Tibodeau, J.D.; Bossou, A.R.; Merchan, J.R.; Bible, K.C. The anticancer effects of chaetocin are independent of programmed cell death and hypoxia, and are associated with inhibition of endothelial cell proliferation. Br. J. Cancer 2011, 106, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Tibodeau, J.D.; Benson, L.M.; Isham, C.R.; Owen, W.G.; Bible, K.C. The Anticancer Agent Chaetocin Is a Competitive Substrate and Inhibitor of Thioredoxin Reductase. Antioxid. Redox Signal. 2009, 11, 1097–1106. [Google Scholar] [CrossRef] [Green Version]
- Munday, R. Studies on the mechanism of toxicity of the mycotoxin, sporidesmin. I. Generation of superoxide radical by sporidesmin. Chem. Biol. Interact. 1982, 41, 361–374. [Google Scholar] [CrossRef]
- Bernardo, P.H.; Brasch, N.; Chai, C.; Waring, P. A Novel Redox Mechanism for the Glutathione-dependent Reversible Uptake of a Fungal Toxin in Cells. J. Biol. Chem. 2003, 278, 46549–46555. [Google Scholar] [CrossRef] [Green Version]
- Amrine, C.S.M.; Huntsman, A.C.; Doyle, M.G.; Burdette, J.E.; Pearce, C.J.; Fuchs, J.R.; Oberlies, N.H. Semisynthetic Derivatives of the Verticillin Class of Natural Products through Acylation of the C11 Hydroxy Group. ACS Med. Chem. Lett. 2021, 12, 625–630. [Google Scholar] [CrossRef]
- Baumann, M.; Dieskau, A.P.; Loertscher, B.M.; Walton, M.C.; Nam, S.; Xie, J.; Horne, D.; Overman, L.E. Tricyclic analogues of epidithiodioxopiperazine alkaloids with promising in vitro and in vivo antitumor activity. Chem. Sci. 2015, 6, 4451–4457. [Google Scholar] [CrossRef]
- Olsson, C.R.; Payette, J.N.; Cheah, J.H.; Movassaghi, M. Synthesis of Potent Cytotoxic Epidithiodiketopiperazines Designed for Derivatization. J. Org. Chem. 2020, 85, 4648–4662. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
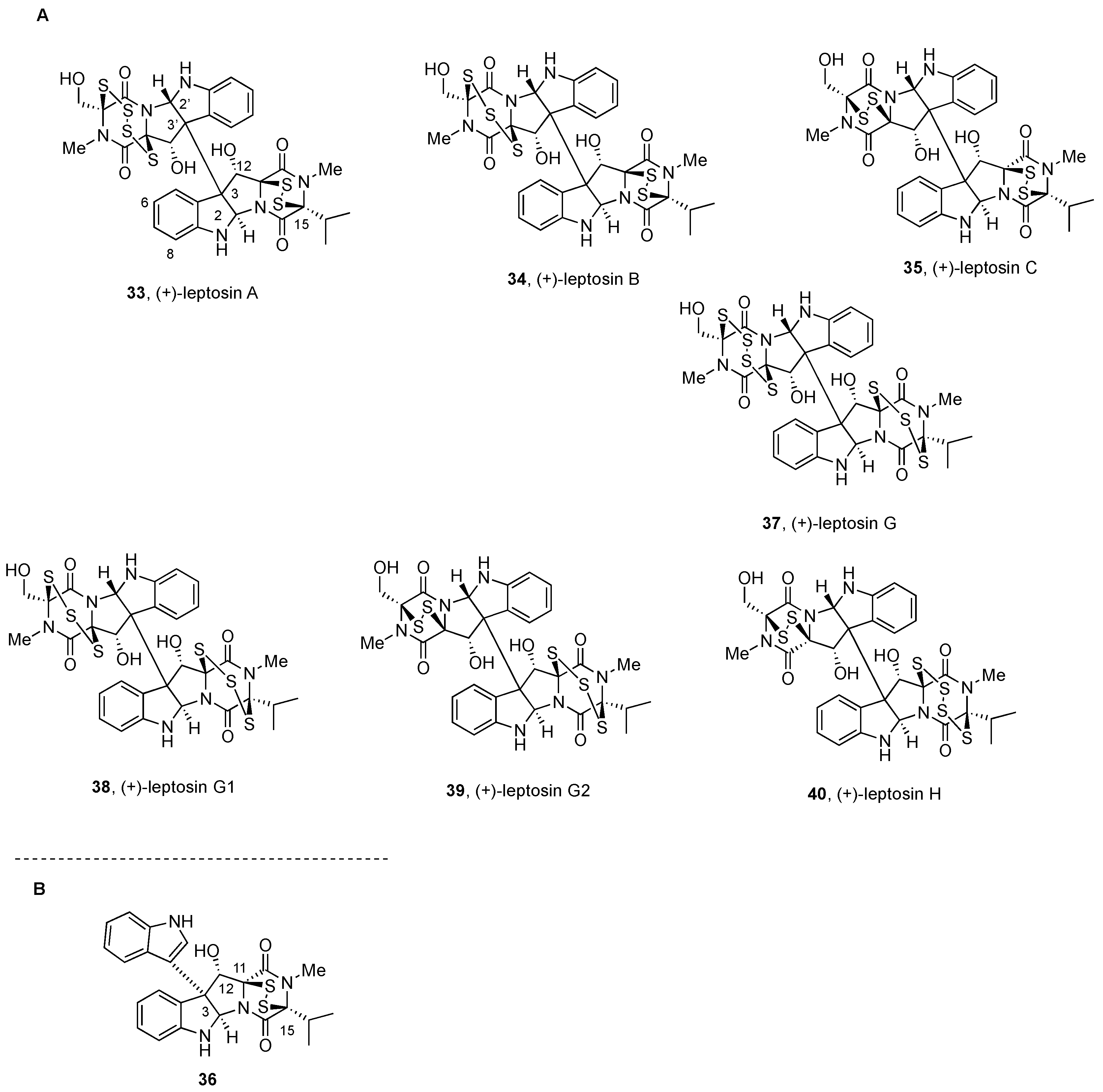{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, C.; García-Domínguez, P.; Álvarez, R.; de Lera, A.R. Bispyrrolidinoindoline Epi(poly)thiodioxopiperazines (BPI-ETPs) and Simplified Mimetics: Structural Characterization, Bioactivities, and Total Synthesis. Molecules 2022, 27, 7585. https://doi.org/10.3390/molecules27217585
Martínez C, García-Domínguez P, Álvarez R, de Lera AR. Bispyrrolidinoindoline Epi(poly)thiodioxopiperazines (BPI-ETPs) and Simplified Mimetics: Structural Characterization, Bioactivities, and Total Synthesis. Molecules. 2022; 27(21):7585. https://doi.org/10.3390/molecules27217585
Chicago/Turabian StyleMartínez, Claudio, Patricia García-Domínguez, Rosana Álvarez, and Angel R. de Lera. 2022. "Bispyrrolidinoindoline Epi(poly)thiodioxopiperazines (BPI-ETPs) and Simplified Mimetics: Structural Characterization, Bioactivities, and Total Synthesis" Molecules 27, no. 21: 7585. https://doi.org/10.3390/molecules27217585
APA StyleMartínez, C., García-Domínguez, P., Álvarez, R., & de Lera, A. R. (2022). Bispyrrolidinoindoline Epi(poly)thiodioxopiperazines (BPI-ETPs) and Simplified Mimetics: Structural Characterization, Bioactivities, and Total Synthesis. Molecules, 27(21), 7585. https://doi.org/10.3390/molecules27217585