


Electrochemistry of Tetrathiafulvalene Ligands Assembled on the Surface of Gold Nanoparticles
Abstract
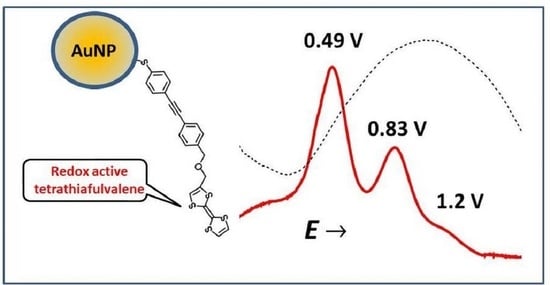
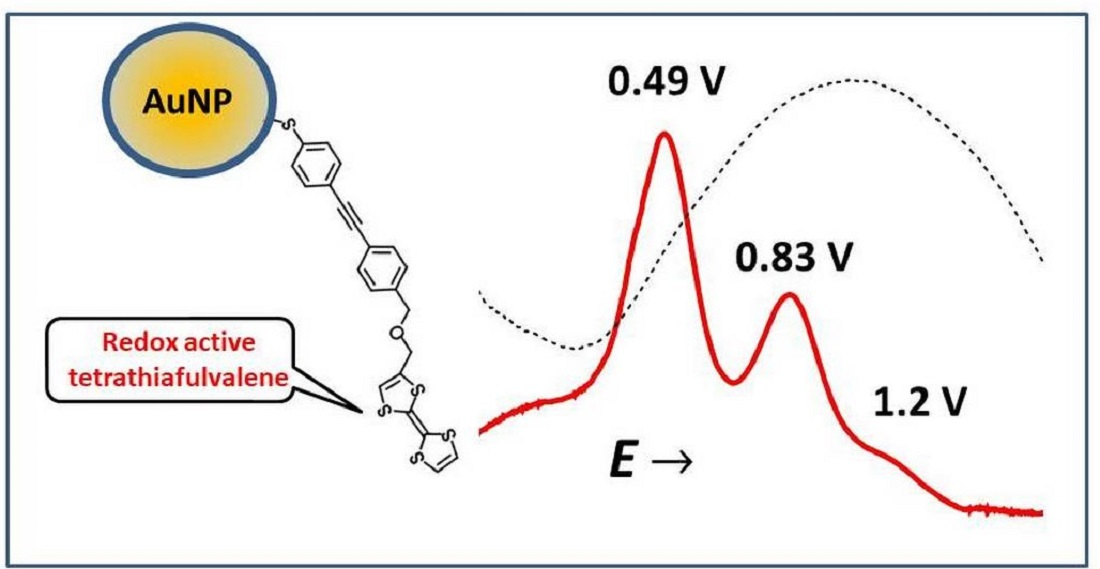
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
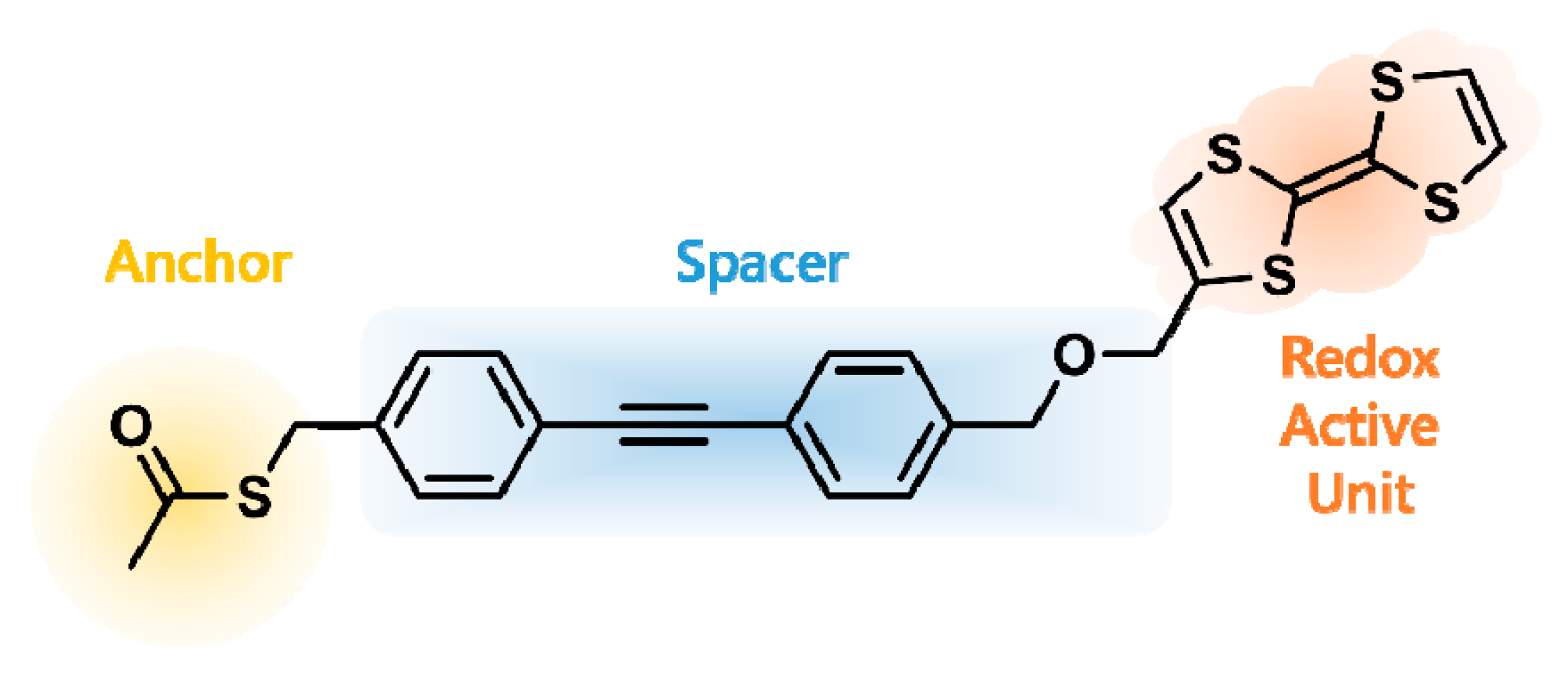
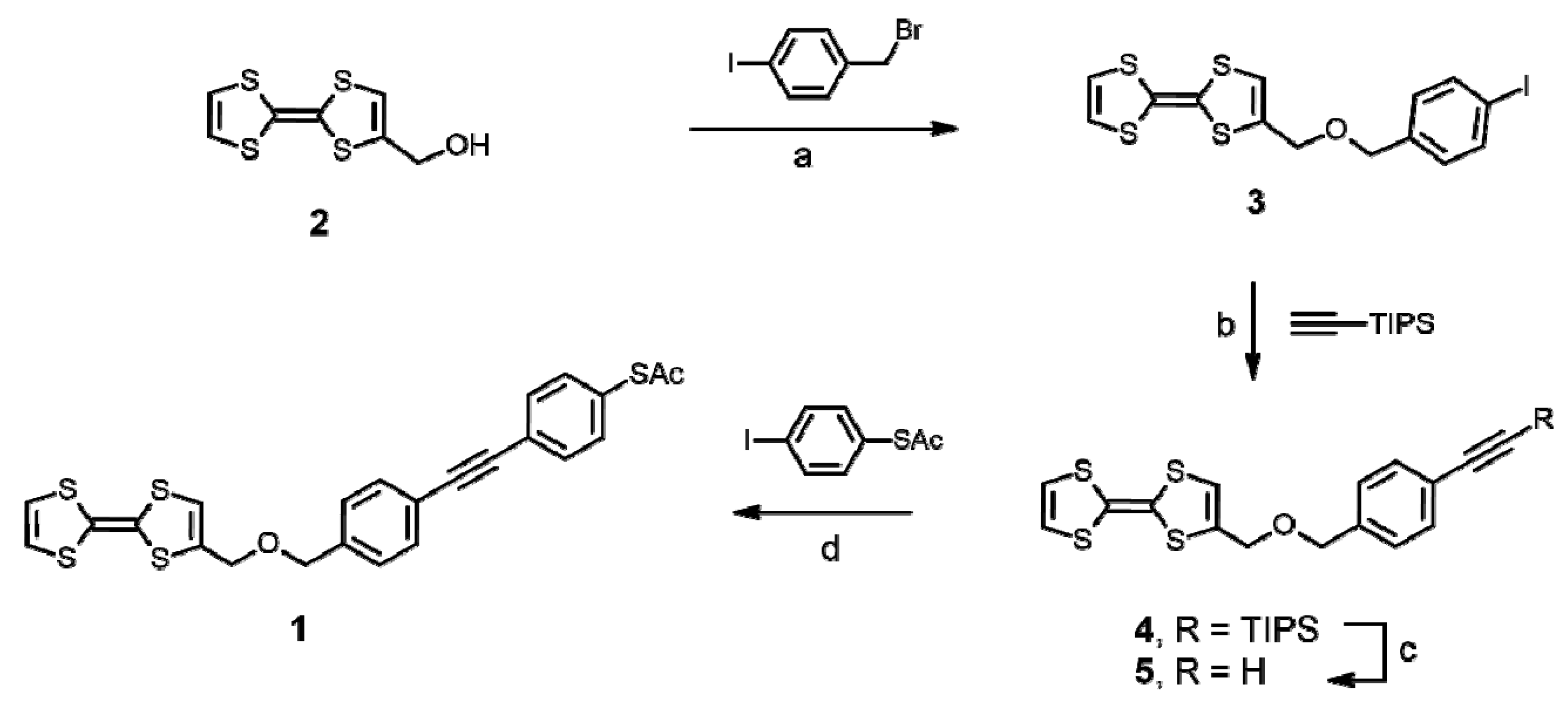
2.1. Synthesis of TTF Ligand 1
2.2. Surface Modification of AuNPs
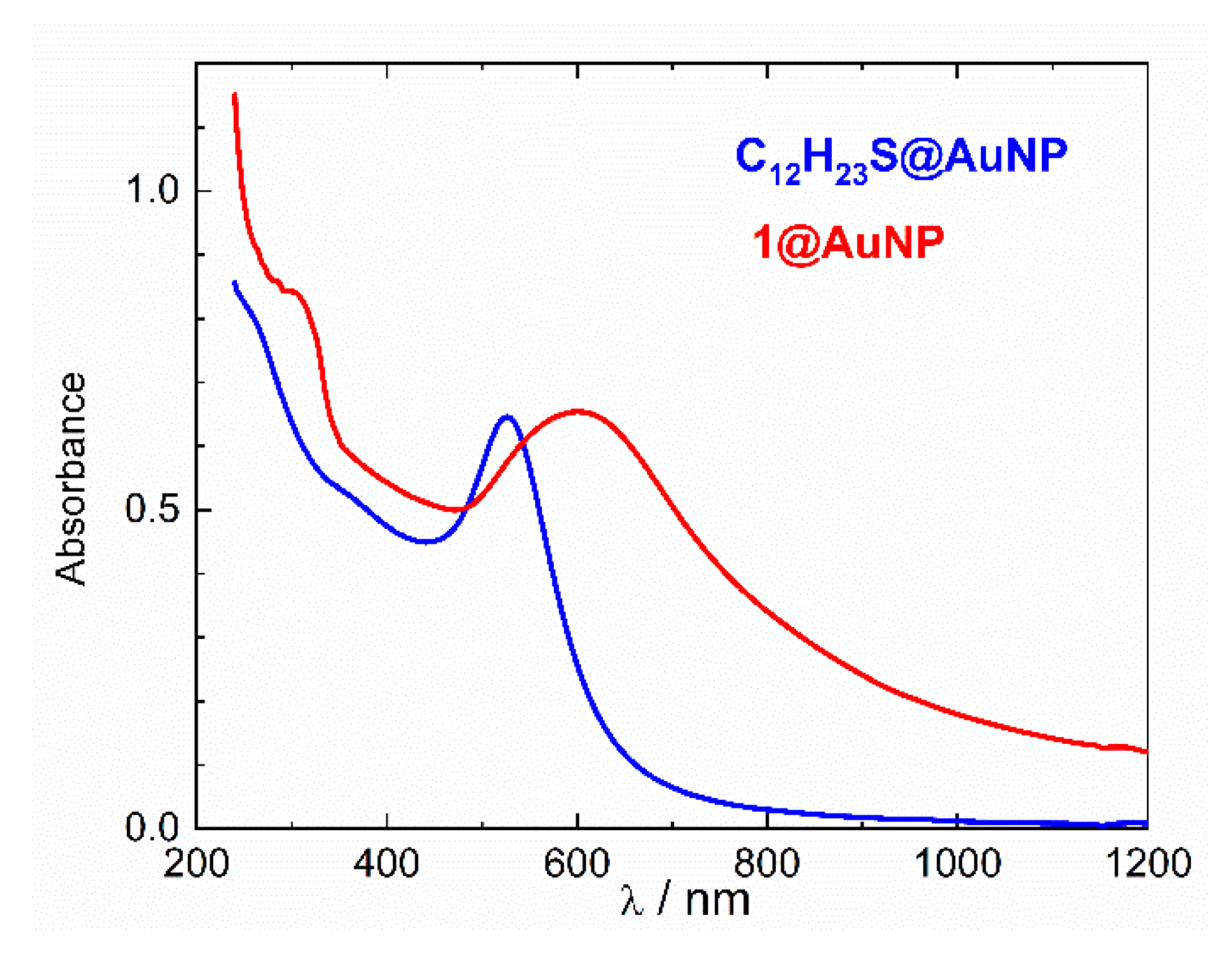
2.3. UV-Vis Spectroscopy
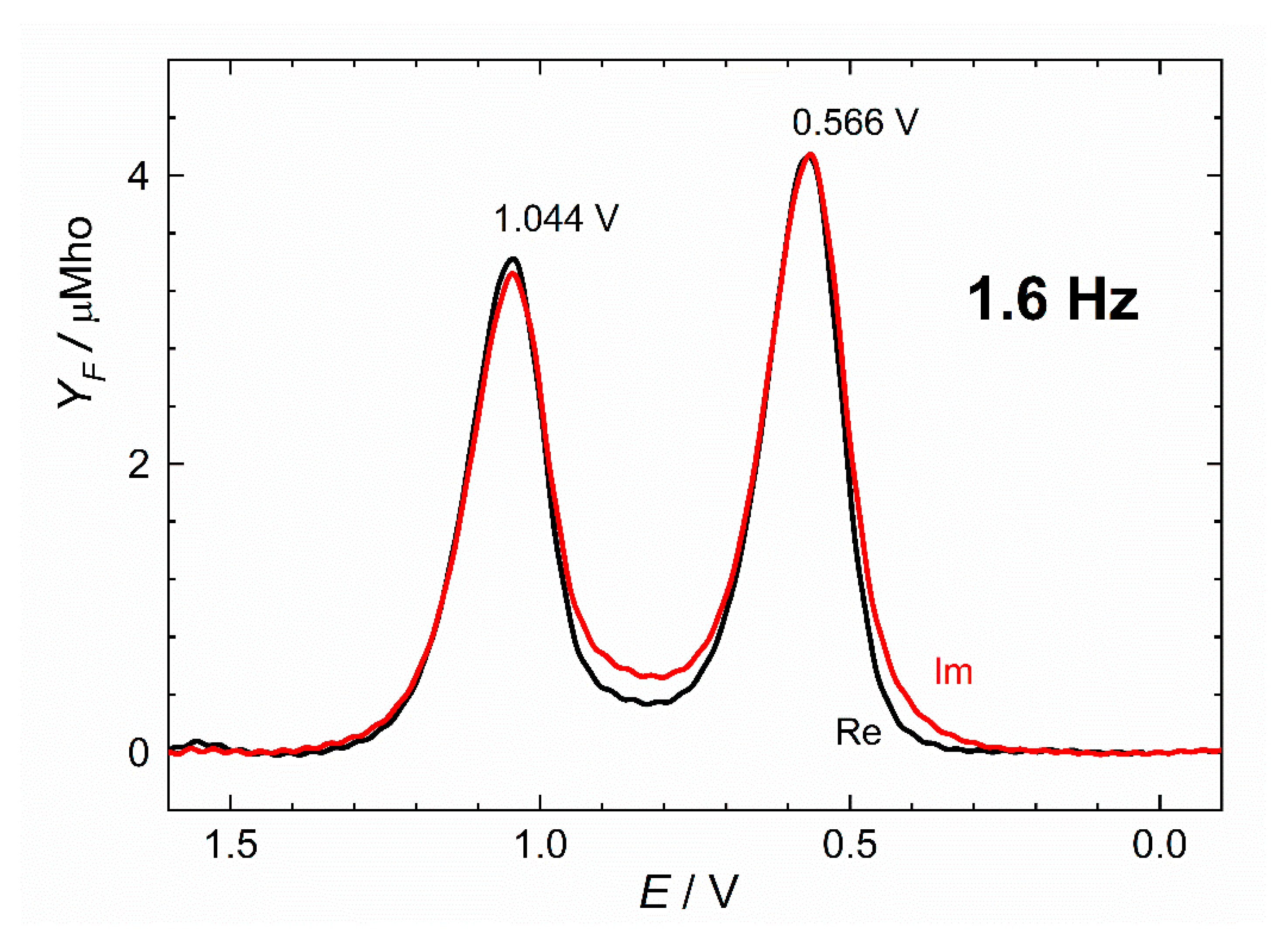
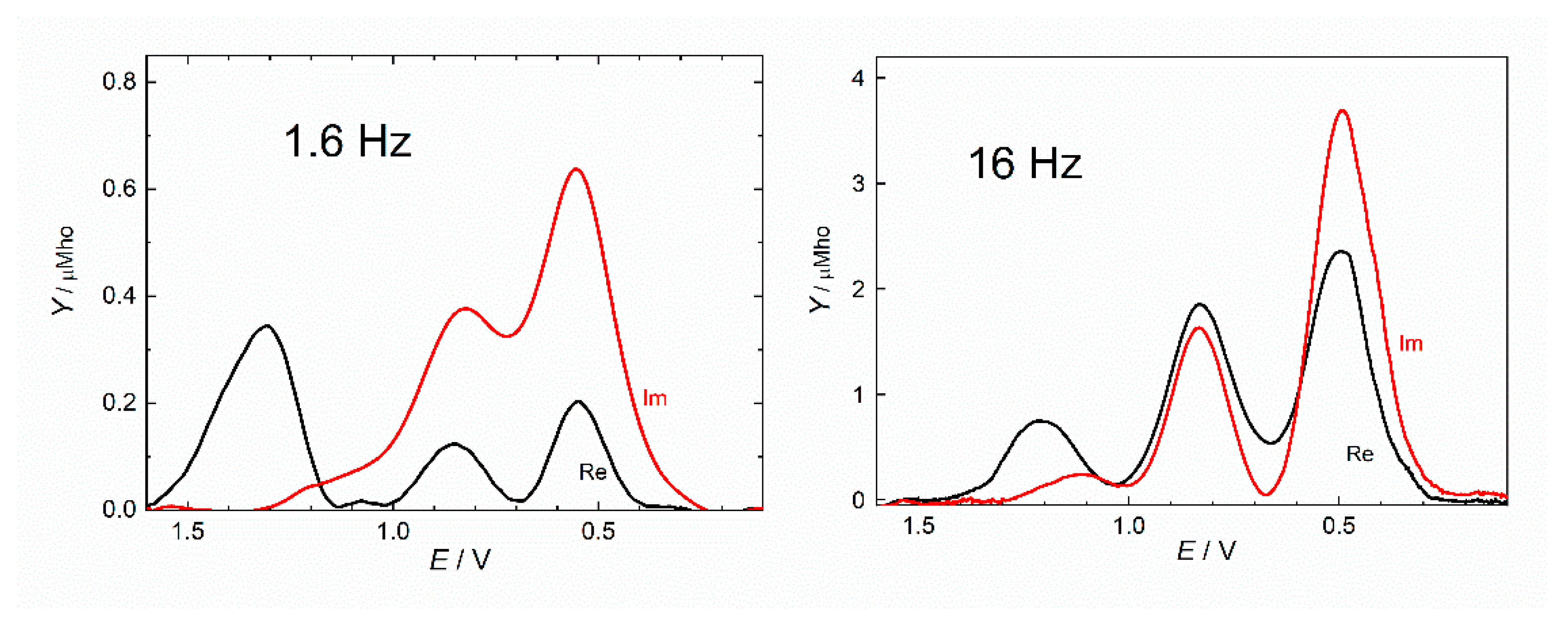
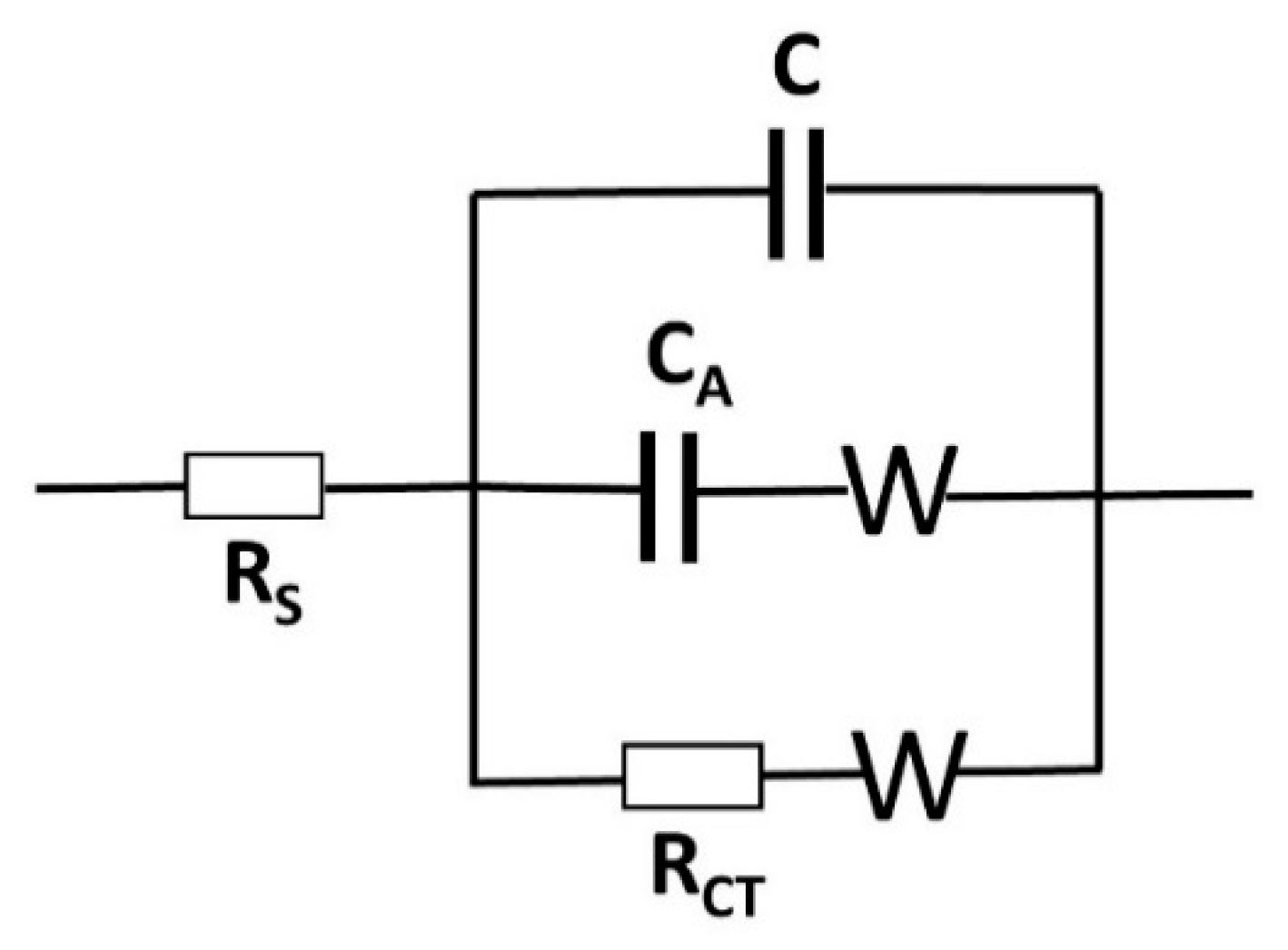
2.4. Electrochemistry
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Jeppesen, J.O.; Nielsen, M.B.; Becher, J. Tetrathiafulvalene Cyclophanes and Cage Molecules. Chem. Rev. 2004, 104, 5115–5132. [Google Scholar] [CrossRef] [PubMed]
- Segura, J.L.; Martín, N. New Concepts in Tetrathiafulvalene Chemistry. Angew. Chem. Int. Ed. 2001, 40, 1372–1709. [Google Scholar] [CrossRef]
- Schmid, G. (Ed.) Nanoparticles: From Theory to Application; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004. [Google Scholar]
- Saini, B.; Khamari, L.; Mukherjee, T.K. Kinetic and Mechanistic Insight into the Surfactant-Induced Aggregation of Gold Nanoparticles and Their Catalytic Efficacy: Importance of Surface Restructuring. J. Phys. Chem. B 2022, 126, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Kimpling, J.; Maier, M.; Okenve, B.; Kotaidis, V.; Ballot, H.; Plech, A. Turkevich Method for Gold Nanoparticle Synthesis Revisited. J. Phys. Chem. B 2006, 110, 15700–15707. [Google Scholar] [CrossRef]
- Parak, W.J.; Manna, L.; Simmel, F.C.; Gerion, D.; Alivisatos, P. Quantum Dots. In Nanoparticles; Schmid, G., Ed.; Wiley-VCH: Weinheim, Germany, 2004; pp. 4–49. [Google Scholar]
- Moores, A.; Goettmann, F. The plasmon band in noble metal nanoparticles: An introduction to theory and applications. New J. Chem. 2006, 30, 1121–1132. [Google Scholar] [CrossRef]
- Zabet-Khosousi, A.; Dhirani, A.A. Charge Transport in Nanoparticle Assemblies. Chem. Rev. 2008, 108, 4072–4124. [Google Scholar] [CrossRef]
- Weng, G.; Mahmoud, M.A.; El-Sayed, M.A. Nanocatalysts Can Change the Number of Electrons Involved in Oxidation–Reduction Reaction with the Nanocages Being the Most Efficient. J. Phys. Chem. C 2012, 116, 24171–24176. [Google Scholar] [CrossRef]
- Zhang, T.; Guérin, D.; Alibart, F.; Vuillaume, D.; Lmimouni, K.; Lenfant, S.; Yassin, A.; Oçafrain, M.; Blanchard, P.; Roncali, J. Negative Differential Resistance, Memory, and Reconfigurable Logic Functions Based on Monolayer Devices Derived from Gold Nanoparticles Functionalized with Electropolymerizable TEDOT Units. J. Phys. Chem. C 2017, 121, 10131–10139. [Google Scholar] [CrossRef] [Green Version]
- Wieckowska, A.; Jablonowska, E.; Dzwonek, M.; Jaskolowski, M.; Bilewicz, R. Tailored Lipid Monolayers Doped with Gold Nanoclusters: Surface Studies and Electrochemistry of Hybrid-film-covered Electrodes. ChemElectroChem 2022, 9, e202101367. [Google Scholar] [CrossRef]
- Wrzesińska, A.; Tomaszewska, E.; Ranoszek-Soliwoda, K.; Bobowska, I.; Grobelny, J.; Ulański, J.; Wypych-Puszkarz, A. Gold Nanoparticles as Effective ion Traps in Poly(dimethylsiloxane) Cross-Linked by Metal-Ligand Coordination. Molecules 2022, 27, 3579. [Google Scholar] [CrossRef] [PubMed]
- Oja, S.M.; Fan, Y.; Armstrong, C.M.; Defnet, P.; Zhang, B. Nanoscale Electrochemistry Revisited. Anal. Chem. 2016, 88, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cox, J.T.; Zhang, B. Electrochemical Responses and Electrocatalysis at Single Au Nanoparticles. J. Am. Chem. Soc. 2010, 132, 3047–3054. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, M.V.; Sun, T.; Yu, Y.; Zhou, M. Electrochemistry at One Nanoparticle. Acc. Chem. Res. 2016, 49, 2328–2335. [Google Scholar] [CrossRef]
- Nakai, H.; Yoshihara, M.; Fujihara, H. New Electroactive Tetrathiafulvalene-Derivatized Gold Nanoparticles and Their Re-markably Stable Nanoparticle Films on Electrodes. Langmuir 1999, 15, 8574–8576. [Google Scholar] [CrossRef]
- Dai, J.; Guo, L.; Jiang, Y.; Zhu, Q.Y.; Gu, R.A.; Jia, D.X.; Guo, W.J. Preparation of Gold Nanoparticles Modified with Tetrathiafulvalene via Direct Sulfur Bridge. J. Nanosci. Nanotechnol. 2005, 5, 474–478. [Google Scholar] [CrossRef]
- Guo, W.J.; Dai, J.; Zhang, D.Q.; Zhu, Q.Y.; Bian, G.Q. Redox active gold nanoparticles modified with tetrathiafulvalene de-rivative via direct sulfur bridge. Inorg. Chem. Commun. 2005, 8, 994–997. [Google Scholar] [CrossRef]
- Tiwari, P.; Azad, U.P.; Gupta, S.; Prakash, R. Pd@TTF Tailored Nanostructured Platform: Voltammetric Estimation of Ceftazidime. ChemistrySelect 2017, 2, 7432–7438. [Google Scholar] [CrossRef]
- Scheele, M.; Hanifi, D.; Zherebetskyy, D.; Chourou, S.T.; Axnanda, S.; Rancatore, B.J.; Thorkelsson, K.; Xu, T.; Liu, Z.; Wang, L.W.; et al. PbS Nanoparticles Capped with Tetrathiafulvalenetetracarboxylate: Utilizing Energy Level Alignment for Efficient Carrier Transport. ACS Nano 2014, 8, 2532–2540. [Google Scholar] [CrossRef] [Green Version]
- Garín, J.; Orduna, J.; Uriel, S.; Moore, A.J.; Bryce, M.R.; Wegener, S.; Yufit, D.S.; Howard, J.A.K. Improved Syntheses of Carboxytetrathiafulvalene, Formyltetrathiafulvalene and (Hydroxymethyl)tetrathiafulvalene1: Versatile Building Blocks for New Functionalised Tetrathiafulvalene Derivatives. Synthesis 1994, 489–493. [Google Scholar] [CrossRef]
- Bernard, L.; Kamdzhilov, Y.; Calame, M.; van der Molen, S.J.; Liao, J.; Schönenberger, C. Spectroscopy of Molecular Junction Networks Obtained by Place Exchange in 2D Nanoparticle Arrays. J. Phys. Chem C 2007, 111, 18445–18450. [Google Scholar] [CrossRef]
- Winter, R.F. Half-Wave Potential Splittings ΔE1/2 as a Measure of Electronic Coupling in Mixed-Valent Systems: Triumphs and Defeats. Organometallics 2014, 33, 4517–4536. [Google Scholar] [CrossRef]
- Heinze, J. Cyclic Voltammetry—“Electrochemical Spectroscopy”. New Analytical Methods. Angew. Chem. Int. Ed. 1984, 23, 831–847. [Google Scholar] [CrossRef]
- LeVanda, C.; Cowan, D.O.; Leitch, C.; Bechgaard, K. Mixed-valance diferrocenylacetylene cation. J. Am. Chem. Soc. 1974, 96, 6788–6789. [Google Scholar] [CrossRef]
- Morrison, W.H., Jr.; Krogsrud, S.; Hendrickson, D.N. Polarographic and magnetic susceptibility study of various biferrocene compounds. Inorg. Chem. 1973, 12, 1998–2004. [Google Scholar] [CrossRef]
- Flanagan, J.B.; Margel, S.; Bard, A.J.; Anson, F.C. Electron Transfer to and from Molecules Containing Multiple, Noninteracting Redox Centers. Electrochemical Oxidation of Poly(vinylferrocene). J. Am. Chem. Soc. 1978, 100, 4248–4253. [Google Scholar] [CrossRef]
- Sutton, J.E.; Taube, H. Metal to Metal Interactions in Weakly Coupled Mixed-Valence Complexes Based on Ruthenium Am-mines. Inorg. Chem. 1981, 20, 3125–3134. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulker, L.R. Electrochemical Methods: Fundamentals and Applications; Wiley: New York, NY, USA, 2001; pp. 367–416. [Google Scholar]
- Delahay, P. Electrode Processes without a Priori Separation of Double-Layer Charging. J. Phys. Chem. 1966, 70, 2373–2379. [Google Scholar] [CrossRef]
- Moreira, H.; de Levie, R. On the theory of the faradaic admittance with reactant adsorption. J. Electroanal. Chem. 1972, 35, 103–117. [Google Scholar] [CrossRef]
- Pospíšil, L. Faradaic admittance with reactant adsorption: Adsorption detection limits for redox systems with finite charge transfer rate. J. Electroanal. Chem. 1976, 74, 369–376. [Google Scholar] [CrossRef]
- Rasband, W.S.; ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA. 1997–2018. Available online: https://imagej.nih.gov/ij/ (accessed on 14 April 2019).
- Hermes, J.P.; Sander, F.; Peterle, T.; Urbani, R.; Pfohl, T.; Thompson, D.; Mayor, M. Gold Nanoparticles Stabilized by Thioether Dendrimers. Chemistry 2011, 17, 13473–13481. [Google Scholar] [CrossRef] [PubMed]
- Lipnická, Š.; Bělohradský, M.; Kolivoška, V.; Pospíšil, L.; Hromadová, M.; Pohl, R.; Vacek Chocholoušová, J.; Vacek, J.; Fiedler, P.; Stará, I.G.; et al. Tetrathiafulvalene–Oligo(para-phenyleneethynylene) Conjugates: Formation of Multiple Mixed-Valence Complexes upon Electrochemical Oxidation. Chem. Eur. J. 2013, 19, 6108–6121. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janoušek, J.; Rybáček, J.; Buděšínský, M.; Pospíšil, L.; Stará, I.G.; Starý, I. Electrochemistry of Tetrathiafulvalene Ligands Assembled on the Surface of Gold Nanoparticles. Molecules 2022, 27, 7639. https://doi.org/10.3390/molecules27217639
Janoušek J, Rybáček J, Buděšínský M, Pospíšil L, Stará IG, Starý I. Electrochemistry of Tetrathiafulvalene Ligands Assembled on the Surface of Gold Nanoparticles. Molecules. 2022; 27(21):7639. https://doi.org/10.3390/molecules27217639
Chicago/Turabian StyleJanoušek, Jiří, Jiří Rybáček, Miloš Buděšínský, Lubomír Pospíšil, Irena G. Stará, and Ivo Starý. 2022. "Electrochemistry of Tetrathiafulvalene Ligands Assembled on the Surface of Gold Nanoparticles" Molecules 27, no. 21: 7639. https://doi.org/10.3390/molecules27217639
APA StyleJanoušek, J., Rybáček, J., Buděšínský, M., Pospíšil, L., Stará, I. G., & Starý, I. (2022). Electrochemistry of Tetrathiafulvalene Ligands Assembled on the Surface of Gold Nanoparticles. Molecules, 27(21), 7639. https://doi.org/10.3390/molecules27217639