
Rapid Purification and Formulation of Radiopharmaceuticals via Thin-Layer Chromatography

 ,
,
Abstract
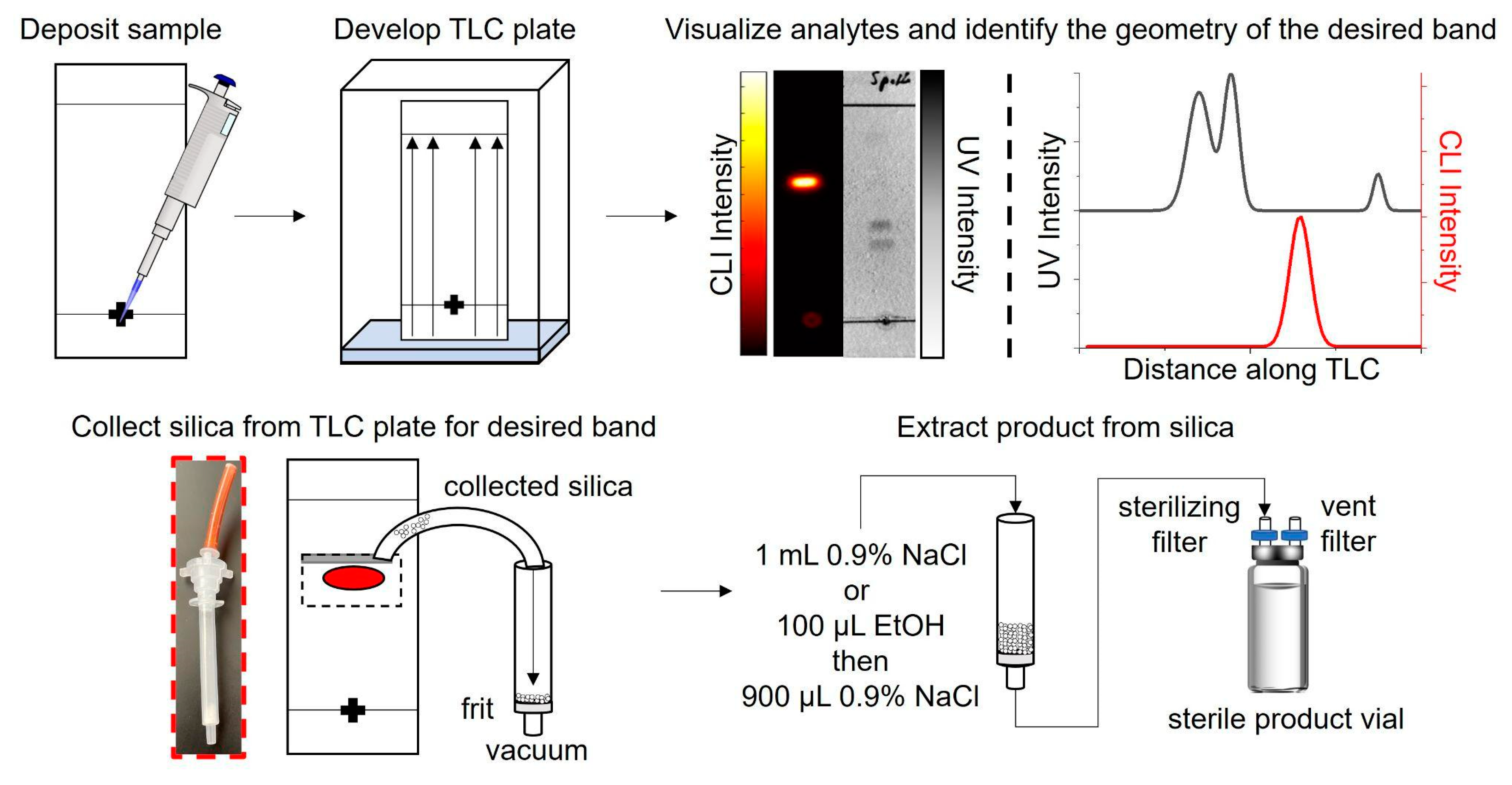
:1. Introduction
2. Results
2.1. Performance of TLC at the Scale of Crude Reaction Mixtures
2.2. Efficiency of Radiopharmaceutical Collection from the TLC Plate
2.3. Efficiency of Radiopharmaceutical Extraction from the Collected Sorbent
2.4. Scale-Up to Clinical Quantities
2.5. Quality Control Testing of Purified [18F]PBR-06
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Preparation of Radiopharmaceuticals and Reference Standards
4.3. Preparation of TLC Plates
4.4. Sample Spotting and Separation
4.5. Readout and Analysis of TLC Plates
4.6. TLC Purification of Radiopharmaceuticals
4.6.1. Collection of Sorbent from TLC
4.6.2. Extraction of the Radiopharmaceutical from Sorbent
4.7. HPLC Analyses
4.8. Quality Control Testing
4.9. ICP-MS Analysis for Silicon Content
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berti, V.; Osorio, R.S.; Mosconi, L.; Li, Y.; Santi, S.D.; de Leon, M.J. Early Detection of Alzheimer’s Disease with PET Imaging. NDD 2010, 7, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Dendl, K.; Koerber, S.A.; Kratochwil, C.; Cardinale, J.; Finck, R.; Dabir, M.; Novruzov, E.; Watabe, T.; Kramer, V.; Choyke, P.L.; et al. FAP and FAPI-PET/CT in Malignant and Non-Malignant Diseases: A Perfect Symbiosis? Cancers 2021, 13, 4946. [Google Scholar] [CrossRef]
- Aboagye, E.O.; Price, P.M.; Jones, T. In Vivo Pharmacokinetics and Pharmacodynamics in Drug Development Using Positron-Emission Tomography. Drug Discov. Today 2001, 6, 293–302. [Google Scholar] [CrossRef]
- Bhattacharyya, S. Application of Positron Emission Tomography in Drug Development. Biochem. Pharm. 2012, 1, 1000e128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckelman, W.C. The Use of Positron Emission Tomography in Drug Discovery and Development. In Positron Emission Tomography: Basic Sciences; Bailey, D.L., Townsend, D.W., Valk, P.E., Maisey, M.N., Eds.; Springer: London, UK, 2005; pp. 327–341. ISBN 978-1-84628-007-8. [Google Scholar]
- Avril, S.; Muzic, R.F.; Plecha, D.; Traughber, B.J.; Vinayak, S.; Avril, N. 18F-FDG PET/CT for Monitoring of Treatment Response in Breast Cancer. J. Nucl. Med. 2016, 57, 34S–39S. [Google Scholar] [CrossRef] [Green Version]
- Weber, W.A.; Figlin, R. Monitoring Cancer Treatment with PET/CT: Does It Make a Difference? J. Nucl. Med. 2007, 48, 36S–44S. [Google Scholar]
- Banister, S.; Roeda, D.; Dolle, F.; Kassiou, M. Fluorine-18 Chemistry for PET: A Concise Introduction. Curr. Radiopharm. 2010, 3, 68–80. [Google Scholar] [CrossRef]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjugate Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, A.F.; Makaravage, K.J.; Wright, J.; Sanford, M.S.; Scott, P.J.H. Fluorine-18 Radiochemistry. In Handbook of Radiopharmaceuticals; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020; pp. 251–289. ISBN 978-1-119-50057-5. [Google Scholar]
- Füchtner, F.; Angelberger, P.; Kvaternik, H.; Hammerschmidt, F.; Simovc, B.P.; Steinbach, J. Aspects of 6-[18F]Fluoro-L-DOPA Preparation: Precursor Synthesis, Preparative HPLC Purification and Determination of Radiochemical Purity. Nucl. Med. Biol. 2002, 29, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Boothe, T.E.; Emran, A.M. The Role of High Performance Liquid Chromatography in Radiochemical/Radiopharmaceutical Synthesis and Quality Assurance. In New Trends in Radiopharmaceutical Synthesis, Quality Assurance, and Regulatory Control; Emran, A.M., Ed.; Springer: Boston, MA, USA, 1991; pp. 409–422. ISBN 978-1-4899-0626-7. [Google Scholar]
- Wester, H.J.; Schottelius, M. Fluorine-18 Labeling of Peptides and Proteins. In Proceedings of the PET Chemistry; Schubiger, P.A., Lehmann, L., Friebe, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 79–111. [Google Scholar]
- Size Exclusion Chromatography: Principles and Methods; GE Healthcare: Chicago, IL, USA, 2014.
- Waldmann, C.M.; Gomez, A.; Marchis, P.; Bailey, S.T.; Momcilovic, M.; Jones, A.E.; Shackelford, D.B.; Sadeghi, S. An Automated Multidose Synthesis of the Potentiometric PET Probe 4-[18F]Fluorobenzyl-Triphenylphosphonium ([18F]FBnTP). Mol. Imaging Biol. 2018, 20, 205–212. [Google Scholar] [CrossRef]
- Simpson, N.J.K. Solid-Phase Extraction: Principles, Techniques, and Applications; CRC Press: Boca Raton, FL, USA, 2000; ISBN 978-1-4200-5624-2. [Google Scholar]
- Nandy, S.K.; Rajan, M.G.R. Simple, Column Purification Technique for the Fully Automated Radiosynthesis of [18F]Fluoroazomycinarabinoside ([18F]FAZA). Appl. Radiat. Isot. 2010, 68, 1944–1949. [Google Scholar] [CrossRef]
- Lee, S.J.; Hyun, J.S.; Oh, S.J.; Yu, K.H.; Ryu, J.S. Development of a New Precursor-Minimizing Base Control Method and Its Application for the Automated Synthesis and SPE Purification of [18F]Fluoromisonidazole ([18F]FMISO). J. Label. Compd. Radiopharm. 2013, 56, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Bogni, A.; Laera, L.; Cucchi, C.; Iwata, R.; Seregni, E.; Pascali, C. An Improved Automated One-Pot Synthesis of O-(2-[18F]Fluoroethyl)-L-Tyrosine ([18F]FET) Based on a Purification by Cartridges. Nucl. Med. Biol. 2019, 72–73, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.-Y.; Nickels, M.L.; McKinley, E.T.; Buck, J.R.; Manning, H.C. High-Yielding, Automated Production of 3′-Deoxy-3′-[18F]Fluorothymidine Using a Modified Bioscan Coincidence FDG Reaction Module. Appl. Radiat. Isot. 2015, 97, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Lazari, M.; Quinn, K.M.; Claggett, S.B.; Collins, J.; Shah, G.J.; Herman, H.E.; Maraglia, B.; Phelps, M.E.; Moore, M.D.; Dam, R.M. van ELIXYS—A Fully Automated, Three-Reactor High-Pressure Radiosynthesizer for Development and Routine Production of Diverse PET Tracers. EJNMMI Res. 2013, 3, 52. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, C.R. Formulation of Radiopharmaceuticals. In Radionuclide Imaging in Drug Research; Wilson, C.G., Hardy, J.G., Frier, M., Davis, S.S., Eds.; Springer: Dordrecht, The Netherlands, 1982; pp. 61–73. ISBN 978-94-011-9728-1. [Google Scholar]
- Lau, J.; Rousseau, E.; Kwon, D.; Lin, K.-S.; Bénard, F.; Chen, X. Insight into the Development of PET Radiopharmaceuticals for Oncology. Cancers 2020, 12, 1312. [Google Scholar] [CrossRef]
- Turiel, E.; Martin-Esteban, A. Molecularly Imprinted Polymers: Towards Highly Selective Stationary Phases in Liquid Chromatography and Capillary Electrophoresis. Anal. Bioanal. Chem. 2004, 378, 1876–1886. [Google Scholar] [CrossRef]
- Wang, J.; Chao, P.H.; Hanet, S.; van Dam, R.M. Performing Multi-Step Chemical Reactions in Microliter-Sized Droplets by Leveraging a Simple Passive Transport Mechanism. Lab Chip 2017, 17, 4342–4355. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chao, P.H.; van Dam, R.M. Ultra-Compact, Automated Microdroplet Radiosynthesizer. Lab Chip 2019, 19, 2415–2424. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.; Holloway, T.S.; Chao, P.H.; De Caro, C.; Okoro, C.C.; van Dam, R.M. Microliter-Scale Reaction Arrays for Economical High-Throughput Experimentation in Radiochemistry. Sci. Rep. 2022, 12, 10263. [Google Scholar] [CrossRef] [PubMed]
- Lisova, K.; Wang, J.; Chao, P.H.; van Dam, R.M. A Simple and Efficient Automated Microvolume Radiosynthesis of [18F]Florbetaben. EJNMMI Radiopharm. Chem. 2020, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Holloway, T.; Lisova, K.; van Dam, R.M. Green and Efficient Synthesis of the Radiopharmaceutical [18F]FDOPA Using a Microdroplet Reactor. React. Chem. Eng. 2020, 5, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; van Dam, R.M. High-Efficiency Production of Radiopharmaceuticals via Droplet Radiochemistry: A Review of Recent Progress. Mol. Imaging 2020, 19, 1–21. [Google Scholar] [CrossRef]
- Koag, M.C.; Kim, H.-K.; Kim, A.S. Efficient Microscale Synthesis of [18F]-2-Fluoro-2-Deoxy-d-Glucose. Chem. Eng. J. 2014, 258, 62–68. [Google Scholar] [CrossRef]
- Tarn, M.D.; Pascali, G.; De Leonardis, F.; Watts, P.; Salvadori, P.A.; Pamme, N. Purification of 2-[18F]Fluoro-2-Deoxy-d-Glucose by on-Chip Solid-Phase Extraction. J. Chromatogr. A 2013, 1280, 117–121. [Google Scholar] [CrossRef]
- Keng, P.Y.; Chen, S.; Ding, H.; Sadeghi, S.; Shah, G.J.; Dooraghi, A.; Phelps, M.E.; Satyamurthy, N.; Chatziioannou, A.F.; Kim, C.-J. “CJ”; et al. Micro-Chemical Synthesis of Molecular Probes on an Electronic Microfluidic Device. Proc. Natl. Acad. Sci. USA 2012, 109, 690–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koag, M.C.; Kim, H.-K.; Kim, A.S. Fast and Efficient Microscale Radiosynthesis of 3′-Deoxy-3′-[18F]Fluorothymidine. J. Fluor. Chem. 2014, 166, 104–109. [Google Scholar] [CrossRef]
- Javed, M.R.; Chen, S.; Kim, H.-K.; Wei, L.; Czernin, J.; Kim, C.-J. “CJ”; Dam, R.M. van; Keng, P.Y. Efficient Radiosynthesis of 3′-Deoxy-3′-18F-Fluorothymidine Using Electrowetting-on-Dielectric Digital Microfluidic Chip. J. Nucl. Med. 2014, 55, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liu, F.; Knapp, K.-A.; Nickels, M.L.; Manning, H.C.; Bellan, L.M. A Simple Microfluidic Platform for Rapid and Efficient Production of the Radiotracer [18F]Fallypride. Lab Chip 2018, 18, 1369–1377. [Google Scholar] [CrossRef]
- Lisova, K.; Wang, J.; Rios, A.; van Dam, R.M. Adaptation and Optimization of [F-18] Florbetaben ([F-18] FBB) Radiosynthesis to a Microdroplet Reactor. J. Label. Compd. Radiopharm. 2019, 62, S353–S354. [Google Scholar]
- Wang, J.; Chao, P.H.; Slavik, R.; van Dam, R.M. Multi-GBq Production of the Radiotracer [18F]Fallypride in a Droplet Microreactor. RSC Adv. 2020, 10, 7828–7838. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.L.; Steiner, R.R. Purification of Pharmaceutical Preparations Using Thin-Layer Chromatography to Obtain Mass Spectra with Direct Analysis in Real Time and Accurate Mass Spectrometry. Drug Test. Anal. 2011, 3, 345–351. [Google Scholar] [CrossRef]
- Wang, J.; Rios, A.; Lisova, K.; Slavik, R.; Chatziioannou, A.F.; van Dam, R.M. High-Throughput Radio-TLC Analysis. Nucl. Med. Biol. 2020, 82–83, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Nyiredy, S. Planar Chromatographic Method Development Using the PRISMA Optimization System and Flow Charts. J. Chromatogr. Sci. 2002, 40, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laferriere-Holloway, T.S.; Rios, A.; Lu, Y.; Okoro, C.C.; van Dam, R.M. A Rapid and Systematic Approach for the Optimization of Radio Thin-Layer Chromatography Resolution. J. Chromatogr. A 2022, 463656. [Google Scholar] [CrossRef]
- Wang, J.; van Dam, R.M. Economical Production of Radiopharmaceuticals for Preclinical Imaging Using Microdroplet Radiochemistry. In Biomedical Engineering Technologies: Volume 1; Ossandon, M.R., Baker, H., Rasooly, A., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2022; pp. 813–828. ISBN 978-1-07-161803-5. [Google Scholar]
- Holloway, T.; Rios, A.; Okoro, C.; van Dam, R.M. Replacing High-Performance Liquid Chromatography (HPLC) with High-Resolution Thin Layer Chromatography (TLC) for Rapid Radiopharmaceutical Analysis [ABSTRACT]. Nucl. Med. Biol. 2021, 96–97, S63. [Google Scholar] [CrossRef]
- Dooraghi, A.A.; Keng, P.Y.; Chen, S.; Javed, M.R.; Kim, C.-J. “CJ”; Chatziioannou, A.F.; van Dam, R.M. Optimization of Microfluidic PET Tracer Synthesis with Cerenkov Imaging. Analyst 2013, 138, 5654–5664. [Google Scholar] [CrossRef] [Green Version]
- King, E.J. The solubility of silica. Lancet 1938, 231, 1236–1238. [Google Scholar] [CrossRef]
- Lisova, K.; Wang, J.; Hajagos, T.J.; Lu, Y.; Hsiao, A.; Elizarov, A.; van Dam, R.M. Economical Droplet-Based Microfluidic Production of [18F]FET and [18F]Florbetaben Suitable for Human Use. Sci. Rep. 2021, 11, 20636. [Google Scholar] [CrossRef] [PubMed]
- Patt, M.; Schildan, A.; Barthel, H.; Becker, G.; Schultze-Mosgau, M.H.; Rohde, B.; Reininger, C.; Sabri, O. Metabolite Analysis of [18F]Florbetaben (BAY 94-9172) in Human Subjects: A Substudy within a Proof of Mechanism Clinical Trial. J. Radioanal. Nucl. Chem. 2010, 284, 557–562. [Google Scholar] [CrossRef]
- Rominger, A.; Brendel, M.; Burgold, S.; Keppler, K.; Baumann, K.; Xiong, G.; Mille, E.; Gildehaus, F.-J.; Carlsen, J.; Schlichtiger, J.; et al. Longitudinal Assessment of Cerebral β-Amyloid Deposition in Mice Overexpressing Swedish Mutant β-Amyloid Precursor Protein Using 18F-Florbetaben PET. J. Nucl. Med. 2013, 54, 1127–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CAMAG® Automatic TLC Sampler 4 (ATS 4). Available online: https://www.camag.com/product/camag-automatic-tlc-sampler-4-ats-4 (accessed on 11 November 2022).
- Arup, U.; Ekman, S.; Lindblom, L.; Mattsson, J.-E. High Performance Thin Layer Chromatography (HPTLC), an Improved Technique for Screening Lichen Substances. Lichenologist 1993, 25, 61–71. [Google Scholar] [CrossRef]
- Tuzimski, T. Basic Principles of Planar Chromatography and Its Potential for Hyphenated Techniques. In High-Performance Thin-Layer Chromatography (HPTLC); Srivastava, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 247–310. ISBN 978-3-642-14025-9. [Google Scholar]
- CAMAG® Horizontal Developing Chamber. Available online: https://www.camag.com/product/camag-horizontal-developing-chamber (accessed on 12 November 2022).
- Hałka-Grysińska, A.; Dzido, T.H.; Sitarczyk, E.; Klimek-Turek, A.; Chomicki, A. A New Semiautomatic Device with Horizontal Developing Chamber for Gradient Thin-Layer Chromatography. J. Liq. Chromatogr. Relat. Technol. 2016, 39, 257–263. [Google Scholar] [CrossRef]
- CAMAG® TLC-MS Interface 2. Available online: https://www.camag.com/product/camag-tlc-ms-interface-2 (accessed on 12 June 2020).
- Plate Express Automated TLC Plate Reader-Advion X Interchim. Available online: https://www.advion.com/products/plate-express/ (accessed on 11 November 2022).
- Läufer, K.; Lehmann, J.; Petry, S.; Scheuring, M.; Schmidt-Schuchardt, M. Simple, Inexpensive System for Using Thin-Layer Chromatography for Micro-Preparative Purposes. J. Chromatogr. A 1994, 684, 370–373. [Google Scholar] [CrossRef]
- Pasilis, S.P.; Van Berkel, G.J. Atmospheric Pressure Surface Sampling/Ionization Techniques for Direct Coupling of Planar Separations with Mass Spectrometry. J. Chromatogr. A 2010, 1217, 3955–3965. [Google Scholar] [CrossRef]
- Gerhardt, R.F.; Peretzki, A.J.; Piendl, S.K.; Belder, D. Seamless Combination of High-Pressure Chip-HPLC and Droplet Microfluidics on an Integrated Microfluidic Glass Chip. Anal. Chem. 2017, 89, 13030–13037. [Google Scholar] [CrossRef]
- Kagan, I.A.; Flythe, M.D. Thin-Layer Chromatographic (TLC) Separations and Bioassays of Plant Extracts to Identify Antimicrobial Compounds. J. Vis. Exp. 2014, 51411. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Gao, M.; Miller, K.D.; Zheng, Q.-H. Synthesis of [11C]PBR06 and [18F]PBR06 as Agents for Positron Emission Tomographic (PET) Imaging of the Translocator Protein (TSPO). Steroids 2011, 76, 1331–1340. [Google Scholar] [CrossRef]
- Lartey, F.M.; Ahn, G.-O.; Shen, B.; Cord, K.-T.; Smith, T.; Chua, J.Y.; Rosenblum, S.; Liu, H.; James, M.L.; Chernikova, S.; et al. PET Imaging of Stroke-Induced Neuroinflammation in Mice Using [18F]PBR06. Mol. Imaging Biol. 2014, 16, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Kuntzsch, M.; Lamparter, D.; Bruggener, N.; Muller, M.; Kienzle, G.J.; Reischl, G. Development and Successful Validation of Simple and Fast TLC Spot Tests for Determination of Kryptofix® 2.2.2 and Tetrabutylammonium in 18F-Labeled Radiopharmaceuticals. Pharmaceuticals 2014, 7, 621–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanzey, S.S.; Mossine, A.V.; Sowa, A.R.; Torres, J.; Brooks, A.F.; Sanford, M.S.; Scott, P.J.H. A Spot Test for Determination of Residual TBA Levels in 18F-Radiotracers for Human Use Using Dragendorff Reagent. Anal. Methods 2020, 12, 5004–5009. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Volume (μL) | Deposition Method | TLC Plate | Resolution |
|---|---|---|---|
| 6 | Spot | Analytical | 2.2 |
| 60 | Spot | Analytical | 1.0 |
| 60 | Streak | Analytical | 2.0 |
| 60 | Spot | HPTLC (with concentrating zone) | 1.7 |
| Radiotracer | Activity Level (MBq) | Crude RCY of Droplet Synthesis (%) (n = 8) | Silica Collection Efficiency (%) (n = 8) | Extraction Efficiency (%) | Overall Collection and Extraction Efficiency (%) | Overall RCY (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| Method 1 (n = 4) | Method 2 (n = 4) | Method 1 (n = 4) | Method 2 (n = 4) | Method 1 (n = 4) | Method 2 (n = 4) | ||||
| [18F]PBR-06 | 11 | 94.4 ± 1.2 | 98.7 ± 1.3 | 96.4 ± 3.4 | 97.9 ± 1.6 | 95.4 ± 4.6 | 96.3 ± 1.7 | 89.6 ± 3.9 | 91.3 ± 1.9 |
| 1110–1480 | 91.9 ± 1.8 | 98.1 ± 1.1 | 95.6 ± 2.9 | 98.2 ± 0.3 | 94.2 ± 2.6 | 95.9 ± 0.9 | 86.7 ± 3.7 | 87.9 ± 1.8 | |
| [18F]Fallypride | 7.5 | 96.5 ± 1.6 | 97.5 ± 1.6 | 95.4 ± 1.1 | 98.4 ± 0.3 | 92.6 ± 2.6 | 96.2 ± 1.3 | 89.4 ± 3.7 | 92.9 ± 2.6 |
| 740–1480 | 93.2 ± 2.5 | 97.5 ± 1.2 | 97.1 ± 1.0 | 97.8 ± 1.4 | 94.5 ± 1.9 | 95.6 ± 2.8 | 88.1 ± 3.8 | 89.2 ± 4.7 | |
| Test | Criteria | Batch 1 | Batch 2 | Batch 3 |
|---|---|---|---|---|
| Radioactivity | - | 821 MBq [22.2 mCi] | 744 MBq [20.1 mCi] | 829 MBq [22.4 mCi] |
| Molar Activity | - | 342 GBq/µmol | 315 GBq/μmol | 327 GBq/μmol |
| Appearance | Clear, colorless, and particulate-free | ✓ | ✓ | ✓ |
| Radiochemical Identity | Retention time ratio of radio peak vs. reference standard (0.90–1.10) | 1.01 | 1.01 | 1.01 |
| Residual TBAHCO3 | <104 mg/L | <45 mg/L | <45 mg/L | <45 mg/L |
| Residual Solvents | MeCN < 410 ppm | <1 | <1 | <1 |
| MeOH < 3000 ppm | 24 | 21 | 24 | |
| Hexanes < 290 ppm | 6 | 2 | 5 | |
| CHCl3 < 60 ppm | <1 | <1 | <1 | |
| Et2O < 5000 ppm | 104 | 47 | 102 | |
| EtOAc < 5000 ppm | 21 | 10 | 20 | |
| AcOH < 5000 ppm | 7 | 5 | 7 | |
| Thexyl alcohol < 5000 ppm | <1 | <1 | <1 | |
| Radiochemical Purity | >95% | >99% | >99% | >99% |
| Radionuclide Identity (half-life) | 105–115 min | 110.4 | 111.7 | 113.8 |
| pH | 4.5–7.5 | 5.5 | 5.5 | 5.5 |
| Shelf life | Pass appearance, pH, and radiochemical purity after 120 min | ✓ | ✓ | ✓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laferriere-Holloway, T.S.; Rios, A.; Carlucci, G.; van Dam, R.M. Rapid Purification and Formulation of Radiopharmaceuticals via Thin-Layer Chromatography. Molecules 2022, 27, 8178. https://doi.org/10.3390/molecules27238178
Laferriere-Holloway TS, Rios A, Carlucci G, van Dam RM. Rapid Purification and Formulation of Radiopharmaceuticals via Thin-Layer Chromatography. Molecules. 2022; 27(23):8178. https://doi.org/10.3390/molecules27238178
Chicago/Turabian StyleLaferriere-Holloway, Travis S., Alejandra Rios, Giuseppe Carlucci, and R. Michael van Dam. 2022. "Rapid Purification and Formulation of Radiopharmaceuticals via Thin-Layer Chromatography" Molecules 27, no. 23: 8178. https://doi.org/10.3390/molecules27238178
APA StyleLaferriere-Holloway, T. S., Rios, A., Carlucci, G., & van Dam, R. M. (2022). Rapid Purification and Formulation of Radiopharmaceuticals via Thin-Layer Chromatography. Molecules, 27(23), 8178. https://doi.org/10.3390/molecules27238178