3. Experimental
A. General. All reactions were performed under protection of N
2 in flame-dried glassware unless water was applied as solvent. Chromatographic purification was performed as flash chromatography with Silicycle SiliaFlash P60 silica gel (40–63 µm) or preparative thin-layer chromatography (prep-TLC) using silica gel F254 (1000 µm) plates and solvents indicated as eluent with 0.1–0.5 bar pressure. For quantitative flash chromatography, technical grades solvents were utilized. Analytical thin-layer chromatography (TLC) was performed on Silicycle SiliaPlate TLC silica gel F254 (250 µm) TLC glass plates. Visualization was accomplished with UV light. Infrared (IR) spectra were obtained via thin film IR on a salt plate using a Nicolet 6700 Fourier-transform infrared spectrophotometer. The IR bands are characterized as broad (br), weak (w), medium (m), and strong (s). Proton and carbon nuclear magnetic resonance spectra (
Supplementary Materials:
1H NMR,
13C NMR and
19F NMR) were recorded on a Bruker 400 MHz spectrometer or on a Bruker 500 MHz spectrometer or on a Bruker 700 MHz spectrometer with solvent resonances as the internal standard (
1H NMR: CDCl
3 at 7.26 ppm;
13C NMR: CDCl
3 at 77.0 ppm).
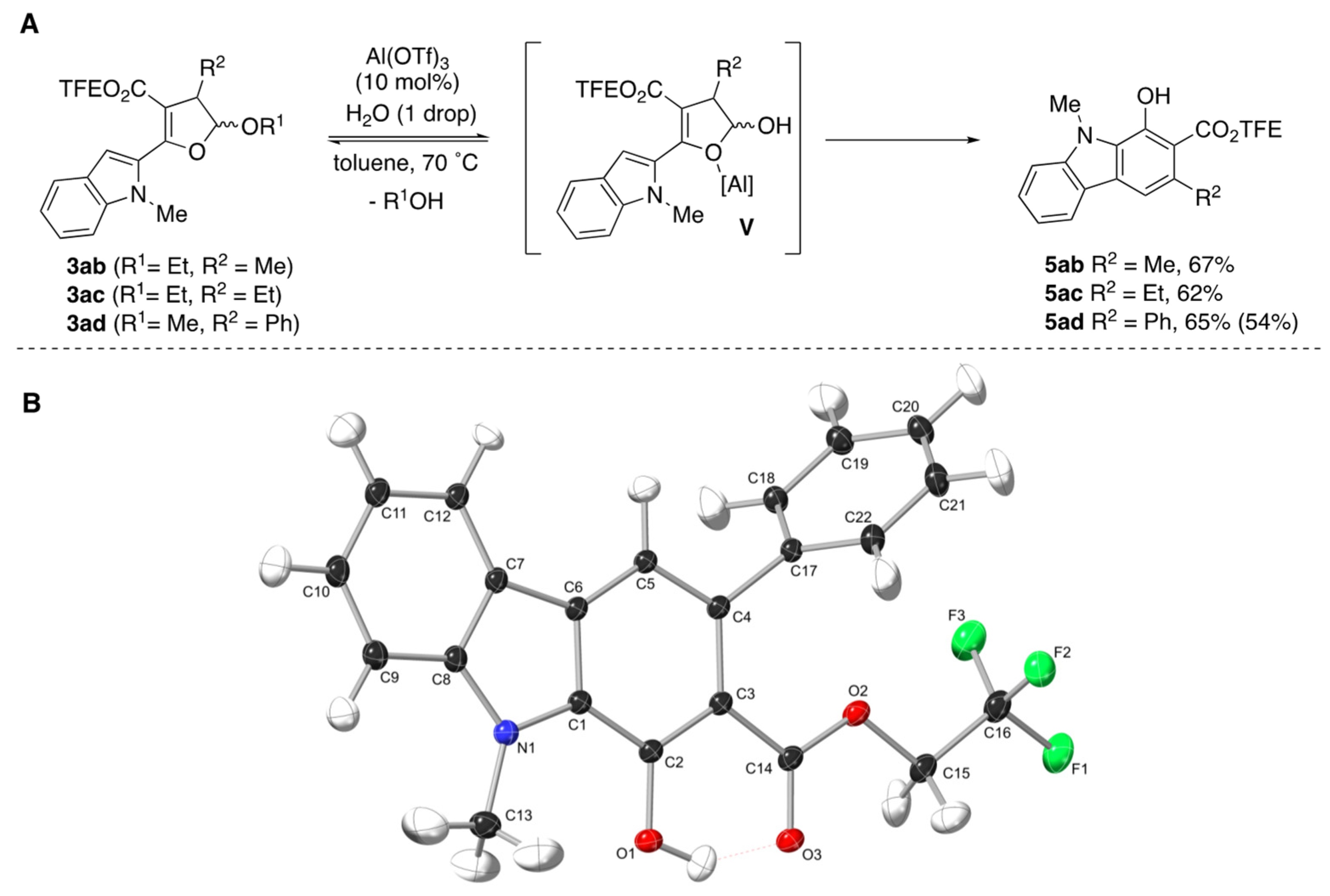
1H NMR data are reported as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, dd = doublet of doublets, dt = doublet of triplets, ddd = doublet of doublet of doublets, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad), coupling constants (Hz), and integration. Mass spectra were obtained through EI on a Micromass AutoSpec machine or through ESI on a Thermo Orbitrap XL. The accurate mass analyses run in EI mode were at a mass resolution of 10,000 and were calibrated using PFK (perfluorokerosene) as an internal standard. The accurate mass analyses run in EI mode were at a mass resolution of 30,000 using the calibration mixture supplied by Thermo. Crystal structures for carbazoles
5ad (Deposition Number 2213553) and
5ai (Deposition Number 2213554) were deposited in the Cambridge Structural Database.
B. Synthesis of Indolyl-α-diazo-β-ketoesters 1.
General Procedure: Using the following modified literature procedure [
55]: To a dry flask charged with a stir bar and the corresponding indole carboxylic acid (1.0 equiv.), dry CH
2Cl
2 was added to make a 0.5 M solution, followed by addition of catalytic DMF (a few drops). The solution was then cooled to 0 °C and oxalyl chloride (1.2 equiv.) was slowly added over 1 min. After 15 min, the reaction was allowed to warm to room temperature with continued stirring. After 3 h at room temperature, the reaction was concentrated under reduced pressure and the acid chloride residue was dissolved in dry THF to make a 1 M solution (keeping it under inert atmosphere). The solution was added slowly to the prepared enolate at −78 °C. The enolate was prepared by first adding LHMDS (1 M in THF, 3.0 equiv.) to a dry flask charged with a stir bar under nitrogen and cooling to −78 °C. The corresponding acetate (1.05 equiv.) was added to the solution of LHMDS in one shot and stirred for 45 min at −78 °C. After 30 min from the addition of the acid chloride to the enolate solution, the reaction was quenched with 0.5 M HCl at −78 °C, extracted with EtOAc three times, dried using Na
2SO
4, and filtered through celite. The combined organic layers were concentrated under reduced pressure and purified by flash chromatography on silica gel using 15–30% EtOAc/Hexanes as the mobile phase to produce pure product II. To an ice-cold solution of II (1.0 equiv.) and 4-acetamidobenzenesulfonyl azide (
p-ABSA, 1.05 equiv.) in MeCN (0.2 M), NEt
3 (1.1 equiv.) was slowly added. The reaction was left to warm gradually to room temperature overnight. Upon completion, the reaction mixture was vacuum-filtered through a celite plug to remove the formed solids. The filtrate was concentrated under vacuum and the obtained residue was purified on silica gel (15–30% EtOAC/Hexanes) to afford pure diazo compound
1.
2,2,2-Trifluoroethyl 2-diazo-3-(1-methyl-1H-indol-2-yl)-3-oxopropanoate (1a): Prepared by general diazo synthesis route, and started with N-methylindole-2-carboxylic acid (5.00 g, 28.5 mmol). It afforded 7.76 g (23.9 mmol, 84% over 3 steps) final product 1a as a yellow solid. 1H NMR (700 MHz, CDCl3) δ 7.69 (dt, J = 8.1, 1.0 Hz, 1H), 7.42–7.36 (m, 2H), 7.20 (s, 1H), 7.17 (ddd, J = 7.9, 5.0, 2.7 Hz, 1H), 4.65 (q, J = 8.2 Hz, 2H), 3.97 (s, 3H). 13C NMR (176 MHz, CDCl3) δ 175.5, 159.6, 140.1, 132.3, 126.1, 125.6, 123.1, 122.6 (q, J = 277.7 Hz), 120.9, 111.9, 110.2, 60.5 (q, J = 37.4 Hz), 31.8. IR 2950.79 (w), 2142.71 (s), 1741.70 (s), 1728.65 (s), 1613.26 (m), 1315.51 (s), 1274.18 (s), 1170. 92 (s), 1108.74 (m). HMRS (ESI) m/z: [M + H]+ calc. for. C14H10F3N3O3, 326.0747; Found 326.0748.
2,2,2-Trifluoroethyl 3-(1-benzyl-1H-indol-2-yl)-2-diazo-3-oxopropanoate (1b): Prepared by general diazo synthesis route, and started with
N-benzylindole-2-carboxylic acid (2.00 g, 7.96 mmol) [
55]. It afforded 2.46 g (6.14 mmol, 77% over 3 steps) final product
1b as a yellow gel.
1H NMR (500 MHz, CDCl
3) δ 7.74 (d,
J = 8.1 Hz, 1H), 7.34 (d,
J = 3.4 Hz, 2H), 7.30 (s, 1H), 7.28–7.17 (m, 4H), 7.06–7.02 (m, 2H), 5.71 (s, 2H), 4.63 (q,
J = 8.2 Hz, 2H).
13C NMR (126 MHz, CDCl
3) δ 175.6, 159.5, 139.8, 137.9, 132.2, 128.6, 127.2, 126.3, 126.2, 125.8, 123.1, 122.6 (q, J = 277.5 Hz), 121.2, 112.7, 110.8, 75.3, 60.5 (q, J = 37.1 Hz), 48.0.
IR 3031.90 (m), 2139.42 (s), 1764.60 (s), 1717.14 (m), 1665.47 (s), 1283.34 (m), 1167.42 (s), 1141.86 (m).
HMRS (ESI)
m/
z: [M + H]
+ calc. C
20H
14F
3N
3O
3, 402.1060; Found 402.1060.
2,2,2-Trifluoroethyl 2-diazo-3-(1-methyl-1H-indol-3-yl)-3-oxopropanoate (1c): Prepared by general diazo synthesis route, and started with N-methylindole-3-carboxylic acid (5.00 g, 28.5 mmol). It afforded 7.32 g (22.5 mmol, 79.0% over 3 steps) final product 1c as a yellow solid. 1H NMR (700 MHz, CDCl3) δ 8.38–8.34 (m, 1H), 8.19 (s, 1H), 7.39–7.28 (m, 3H), 4.63 (q, J = 8.3 Hz, 2H), 3.85 (s, 3H). 13C NMR (176 MHz, CDCl3) δ 176.2, 160.3, 137.6, 136.8, 127.6, 123.5, 122.9, 122.8 (q, J = 277.7 Hz), 122.5, 113.1, 109.6, 60.3 (q, J = 37.1 Hz), 33.7. IR 3148. 66 (m), 3012.10 (w), 2916.12 (w), 2148.29 (s), 1740.71 (s), 1731.42 (s), 1583.12 (m), 1307.18 (s), 1279.46 (s), 1167.31 (s), 1120.22 (m). HMRS (ESI) m/z: [M + H]+ calc. for. C14H10F3N3O3, 326.0747; Found 326.0731.
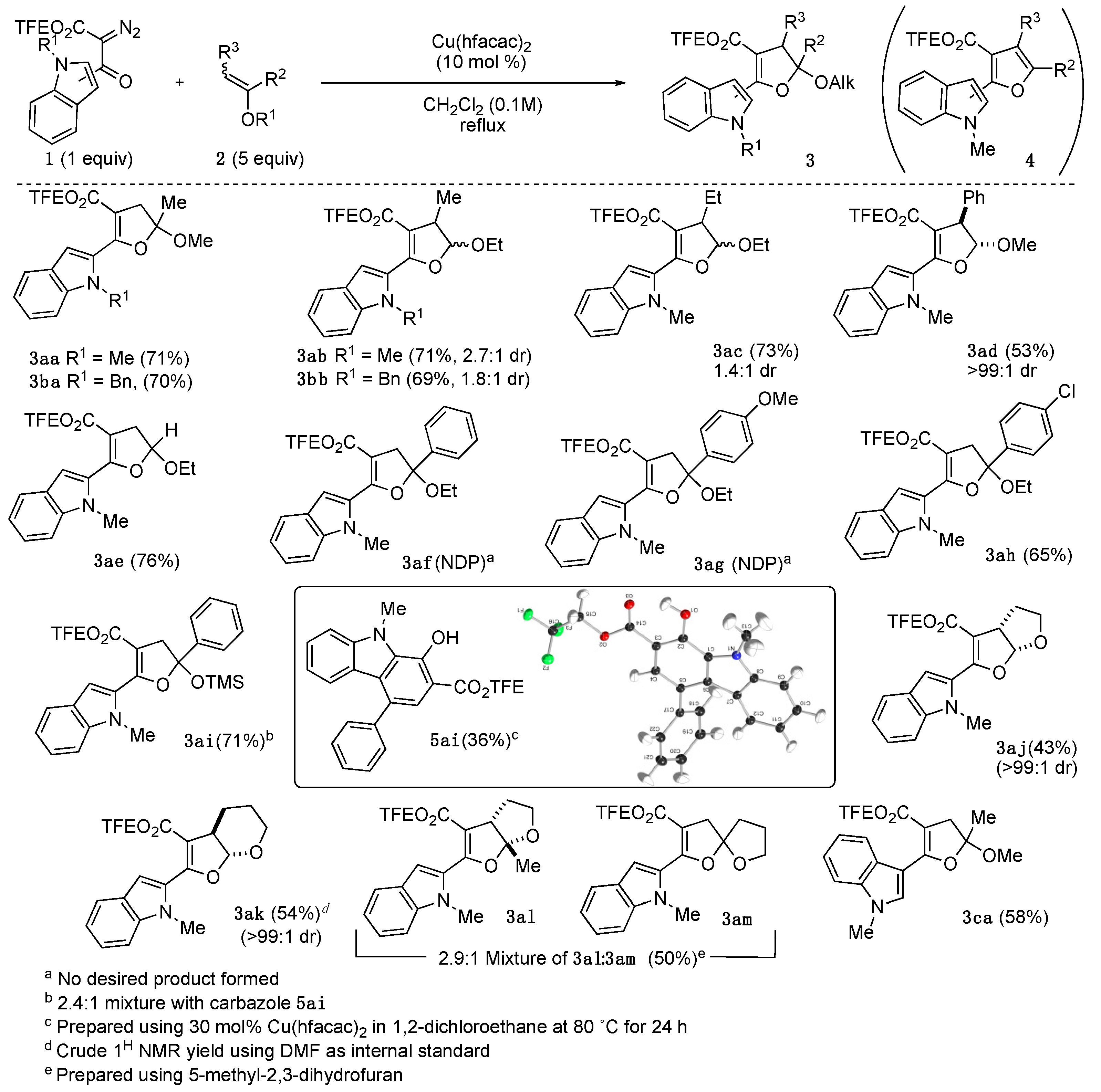
C. Synthesis of 2,3-dihydrofuran acetals 3.General Procedure: To a dry flask charged with a stir bar and a solution of Cu(hfacac)2 (29 mg, 61.5 μmol) and 2 (3.07 mmol) in anhydrous CH2Cl2 (6 mL) was added diazo 1 (615 μmol). The reaction was stirred vigorously under reflux condition for overnight or monitored by TLC for completion. After consuming all starting diazo, the mixture was diluted with Et2O (30 mL) and washed with saturated thiourea (20 mL). The organic layer was dried over Na2SO4, concentrated under reduced pressure, and purified by column chromatography. Since the 19F signals of all DHF acetal products have similar chemical shifts (−73.3 ppm to −73.8 ppm) and coupling constant (t, J = 8.5 Hz), only the 19F NMR of 3aa is reported and attached.
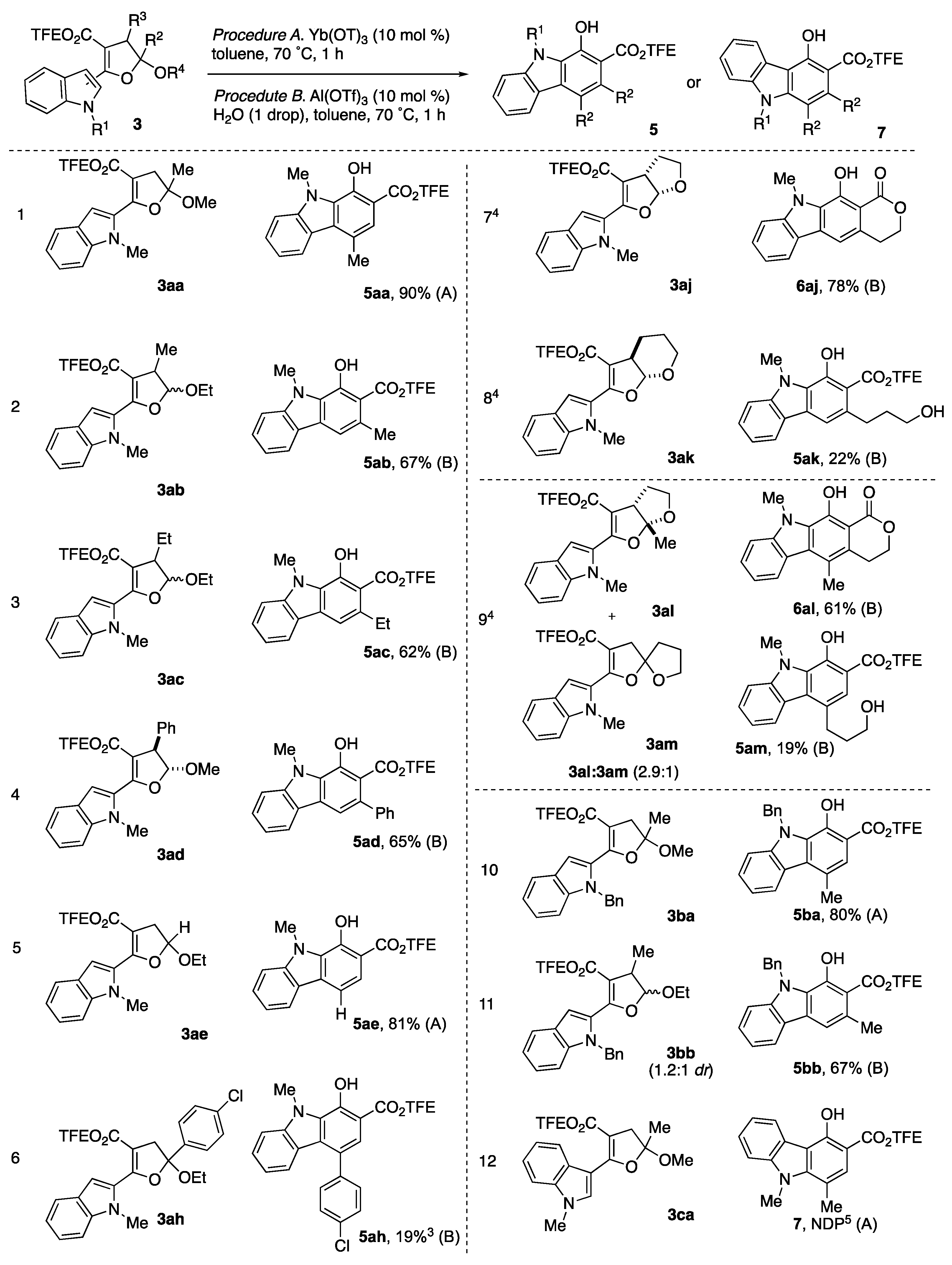
2,2,2-Trifluoroethyl 5-methoxy-5-methyl-2-(1-methyl-1H-indol-2-yl)-4,5-dihydrofuran-3-carboxylate (3aa): Prepared following general procedure using 2-methoxypropene 2a (221 mg, 3.07 mmol) and diazo 1a (200 mg, 615 μmol). Purification via silica gel column chromatography (5% EtOAC/Hexanes, Rf = 0.39 in 20% EtOAC/Hexanes) afforded 3aa as pale-yellow solid (181 mg, 79%). 1H NMR (700 MHz, CDCl3) δ 7.65 (dt, J = 8.0, 1.0 Hz, 1H), 7.34 (dq, J = 8.4, 1.0 Hz, 1H), 7.30 (ddd, J = 8.3, 6.9, 1.2 Hz, 1H), 7.17 (d, J = 1.0 Hz, 1H), 7.13 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 4.47 (qq, J = 8.5, 4.2 Hz, 2H), 3.81 (s, 3H), 3.43 (s, 3H), 3.21 (d, J = 16.6 Hz, 1H), 3.09 (d, J = 16.6 Hz, 1H), 1.73 (s, 3H). 13C NMR (176 MHz, CDCl3) δ 162.3, 158.3, 138.7, 127.7, 126.8, 123.9, 123.15 (q, J = 277.0 Hz), 121.9, 120.1, 111.6, 109.7, 108.9, 102.7, 59.82 (q, J = 36.4 Hz), 50.4, 40.5, 31.8, 24.5. 19F NMR (471 MHz, CDCl3) δ −73.43 (t, J = 8.5 Hz, 3F). IR 3050.91(m), 2945.23, 2836.57(m), 1720.57(s), 1711.31(s), 1168.64(s), 1106.59(s), 1047.59(s). HMRS (ESI) m/z: [M + H]+ calc. for C18H18F3NO4, 370.1260; Found 370.1262.
2,2,2-Trifluoroethyl 2-(1-benzyl-1H-indol-2-yl)-5-methoxy-5-methyl-4,5-dihydrofuran-3-carboxylate (3ba): Prepared following general procedure using 2-methoxypropene 2a (221 mg, 3.07 mmol) and diazo 1b (247 mg, 615 μmol). Purification via silica gel column chromatography (5% EtOAC/Hexanes, Rf = 0.52 in 20% EtOAC/Hexanes) afforded 3ba as pale-yellow solid (192 mg, 70%). 1H NMR (500 MHz, CDCl3) δ 7.69 (d, J = 7.9 Hz, 1H), 7.29 (s, 1H), 7.28–7.16 (m, 5H), 7.14 (ddd, J = 8.0, 6.3, 1.6 Hz, 1H), 7.00 (d, J = 6.9 Hz, 2H), 5.49 (s, 2H), 4.47 (qd, J = 8.5, 2.1 Hz, 2H), 3.17 (s, 3H), 3.13 (d, J = 16.7 Hz, 1H), 2.99 (d, J = 16.6 Hz, 1H), 1.57 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.3, 158.0, 138.3, 138.0, 128.5, 127.6, 127.1, 127.1, 126.0, 124.0, 123.1 (d, J = 277.4 Hz), 121.9, 120.4, 111.6, 110.3, 109.7, 102.9, 59.8 (q, J = 36.3 Hz), 50.2, 48.5, 40.6, 24.1. IR 3061.65(m), 3032.41(m), 2930.32(m), 2851.80(m), 1720.26(s), 1708.31(s), 1167.72(s), 1160.70(s), 1048.76(s). HMRS (ESI) m/z: [M + H]+ calc. for C24H22F3NO4, 446.1573; Found 446.1576.
2,2,2-Trifluoroethyl 5-ethoxy-4-methyl-2-(1-methyl-1H-indol-2-yl)-4,5-dihydrofuran-3-carboxylate (3ab): Prepared following general procedure using 1-ethoxypropene (mixture of cis:trans = 2.7:1) 2b (265 mg, 3.07 mmol) and diazo 1a (200 mg, 615 μmol). Purification via silica gel column chromatography (5% EtOAC/Hexanes, Rf = 0.45 in 20% EtOAC/Hexanes) afforded 3ab (167 mg, 70%) as a mixture of diastereomers (dr = 3.3:1). 1H NMR (500 MHz, CDCl3) for trans isomer: δ 7.64 (d, J = 7.7 Hz, 1H), 7.35–7.27 (m, 2H), 7.13 (d, J = 6.6 Hz, 1H), 7.03 (s, 1H), 5.74 (d, J = 7.5 Hz, 1H), 4.57–4.48 (m, 1H), 4.44–4.35 (m, 1H), 3.78 (s, 3H), 3.74–3.66 (m, 1H), 3.51 (p, J = 7.1 Hz, 1H), 1.34 (d, J = 7.0 Hz, 3H), 1.29 (t, J = 7.1 Hz, 3H); For cis isomer: δ 7.64 (d, J = 7.7 Hz, 1H), 7.35–7.27 (m, 2H), 7.16–7.09 (m, 2H), 5.27 (d, J = 1.8 Hz, 1H), 4.57–4.48 (m, 1H), 4.44–4.35 (m, 1H), 3.80 (s, 3H), 3.74–3.66 (m, 1H), 3.27 (qd, J = 7.1, 1.9 Hz, 1H),1.34 (d, J = 7.0 Hz, 3H), 1.29 (t, J = 7.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) for trans isomer (major isomer): δ 162.6, 158.1, 138.5, 128.2, 126.8, 123.51, 121.7, 120.0, 111.5, 109.6, 107.9, 107.8, 65.8, 59.6 (q, J = 36.3 Hz), 40.8, 31.5, 15.0, 11.8. The 13C signal that is directly coupled by fluorine is not reported due to the low intensity. IR 3058.43 (m), 2978.89 (s), 2933.93 (s), 1720.49 (s), 1711.45 (s), 1626.02 (m), 1281.91 (m), 1167.26 (s), 1084.81(s). HMRS (ESI) m/z: [M + H]+ calc. for C19H20F3NO4, 384.1417; Found 384.1417.
2,2,2-Trifluoroethyl 2-(1-benzyl-1H-indol-2-yl)-5-ethoxy-4-methyl-4,5-dihydrofuran-3-carboxylate (3bb): Prepared following general procedure but set up as 1 g (2.5 mmol) scale: using 1-ethoxypropene (mixture of cis:trans = 1.2:1) 2b (1.07 g, 12.46 mmol), diazo 1b (1.00 g, 2.49 mmol) and Cu(hfacac)2 (119 mg, 0.249 mmol) in anhydrous CH2Cl2 (24 mL). Purification via silica gel column chromatography (5% EtOAC/Hexanes, Rf = 0.48 in 20% EtOAC/Hexanes) afforded 3bb as the mixture of diastereomers (dr = 1.81: 1) (800 mg, 70%). 1H NMR (500 MHz, CDCl3) for trans isomer: δ 7.74–7.67 (m, 1H), 7.30–7.19 (m, 5H), 7.18–7.11 (m, 2H), 7.07 (d, J = 6.8 Hz, 2H), 5.60 (d, J = 7.5 Hz, 1H), 5.46 (d, J = 6.5 Hz, 1H), 5.44 (d, J = 6.5 Hz, 1H), 4.59–4.47 (m, 1H), 4.48–4.36 (m, 1H), 3.68–3.55 (m, 1H), 3.52–3.46 (m, 1H), 3.42 (p, J = 7.1 Hz, 1H), 1.28 (d, J = 7.1 Hz, 3H), 1.16 (d, J = 7.1 Hz, 3H); For cis isomer: δ 7.74–7.67 (m, 1H), 7.30–7.19 (m, 5H), 7.18–7.11 (m, 2H), 7.07 (d, J = 6.8 Hz, 2H), 5.49 (d, J = 6.5 Hz, 1H), 5.46 (d, J = 4.7 Hz, 1H), 5.17 (d, J = 1.9 Hz, 1H), 4.59–4.47 (m, 1H), 4.48–4.36 (m, 1H), 3.68–3.55 (m, 1H), 3.52–3.46 (m, 1H), 3.23 (qd, J = 7.1, 2.0 Hz, 1H), 1.28 (d, J = 7.1 Hz, 3H), 1.15 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) for both diastereomers: δ 162.4, 162.2, 157.80, 157.78, 138.2, 138.1, 138.0, 137.9, 128.42, 128.38, 127.2, 127.1, 126.2, 126.0, 123.9, 123.7, 121.9, 121.8, 120.3, 120.2, 111.5, 110.3, 110.2, 109.5, 109.4, 109.1, 109.0, 107.7, 65.5, 64.6, 59.7 (q, J = 36.3 Hz) 59.6 (q, J = 36.3 Hz), 48.49, 48.46, 44.0, 40.7, 17.4, 14.9, 14.8, 11.6. IR 3063.84 (w), 2977.64 (m), 2930.38 (m), 1711.27 (s), 1708.59 (s), 1281.37 (s), 1167.60 (s). HMRS (ESI) m/z: [M + H]+ calc. for C25H24F3NO4, 460.1730; Found 460.1731.
2,2,2-Trifluoroethyl 5-ethoxy-4-ethyl-2-(1-methyl-1H-indol-2-yl)-4,5-dihydrofuran-3-carboxylate (3ac): Prepared following general procedure using 1-ethoxybutene (mixture of cis:trans = 1.5:1) 2c (308 mg, 3.07 mmol) and diazo 1a (200 mg, 615 μmol). Purification via silica gel column chromatography (5% EtOAC/Hexanes, Rf = 0.48 in 20% EtOAC/Hexanes) afforded 3ac as the mixture of diastereomers (dr = 1.4:1) (179 mg, 73%). 1H NMR (400 MHz, CDCl3) for trans isomer: δ 7.65 (d, J = 7.0 Hz, 1H), 7.39–7.25 (m, 2H), 7.19–7.09 (m,1H), 7.02 (d, J = 0.9 Hz, 1H), 5.78 (d, J = 7.4 Hz, 1H), 4.58–4.33 (m, 2H), 3.98 (dqd, J = 9.6, 7.1, 3.9 Hz, 1H), 3.78 (s, 3H), 3.78–3.66 (m, 1H), 3.38 (ddd, J = 9.3, 7.3, 3.5 Hz, 1H), 1.96–1.79 (m, 2H), 1.30 (t, J = 6.9 Hz, 3H), 1.06 (t, J = 7.4 Hz, 3H); For cis isomer: δ 7.66 (d, J = 7.0 Hz, 1H), 7.39–7.25 (m, 2H), 7.19–7.09 (m, 2H), 5.37 (d, J = 1.9 Hz, 1H), 4.58–4.33 (m, 2H), 3.98 (dqd, J = 9.6, 7.1, 3.9 Hz, 1H), 3.78 (s, 3H), 3.78–3.66 (m, 1H), 3.21 (ddd, J = 8.6, 3.9, 1.9 Hz, 1H), 1.73–1.55 (m, 2H),1.32 (t, J = 6.9 Hz, 3H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) for both diastereomers: δ 162.6, 162.3, 158.3, 138.6, 138.4, 128.4, 128.1, 126.9, 126.8, 123.7, 123.5, 121.8, 121.6, 120.03, 119.95, 109.7, 109.6, 108.6, 107.8, 107.68, 107.65, 107.2, 65.9, 64.7, 59.7 (q, J = 36.3 Hz), 59.6 (q, J = 36.3 Hz), 50.4, 47.2, 31.5, 31.4, 24.3, 19.6, 15.1, 15.0, 12.3, 10.4. The 13C signal that is directly coupled by fluorine is not reported due to the low intensity. IR 2968.85 (m), 2936.33 (w), 2877.54 (w), 1720.23 (s), 1711.33 (m), 1277.33 (m), 1168.55 (s), 1084.69 (s). HMRS (ESI) m/z: [M + H]+ calc. for C20H22F3NO4, 398.1574; Found 398.1576.
2,2,2-Trifluoroethyl 5-methoxy-2-(1-methyl-1H-indol-2-yl)-4-phenyl-4,5-dihydrofuran-3-carboxylate (3ad): Prepared following general procedure using beta-methoxystyrene (pure cis isomer) 2d (413 mg, 3.07 mmol) and diazo 1a (200 mg, 615 μmol). Purification via silica gel column chromatography (5% EtOAC/Hexanes, Rf = 0.33 in 20% EtOAC/Hexanes) afforded 3ad as yellow gel (140 mg, 53%). 1H NMR (500 MHz, CDCl3) δ 7.69 (dt, J = 7.9, 1.0 Hz, 1H), 7.40–7.29 (m, 7H), 7.25 (d, J = 0.9 Hz, 1H), 7.16 (ddd, J = 7.9, 6.8, 1.1 Hz, 1H), 5.86 (d, J = 7.8 Hz, 1H), 4.66 (d, J = 7.8 Hz, 1H), 3.89 (s, 3H), 3.50 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.0, 159.0, 138.8, 135.7, 129.2, 128.0, 127.6, 127.2, 126.8, 123.9, 122.8 (q, J = 277.5 Hz), 121.9, 120.2, 109.7, 108.9, 108.6, 107.9, 59.6 (q, J = 36.5 Hz), 57.7, 52.5, 31.8. IR 3061.65 (w), 3030.38 (w), 2937.40 (m), 2845.72 (w), 1720.41 (s), 1711.54 (m), 1277.50 (m), 1168.60 (s), 1092.46 (s). HMRS (ESI) m/z: [M + H]+ calc. for C23H20F3NO4, 432.1417; Found 432.1418.
2,2,2-Trifluoroethyl 5-ethoxy-2-(1-methyl-1H-indol-2-yl)-4,5-dihydrofuran-3-carboxylate (3ae): Prepared following general procedure using ethoxyethene 2e (413 mg, 3.07 mmol) and diazo 1a (200 mg, 615 μmol). Purification via silica gel column chromatography (5% EtOAC/Hexanes, Rf = 0.39 in 20% EtOAC/Hexanes) afforded 3ae as yellow solid (171 mg, 75%). 1H NMR (500 MHz, CDCl3) δ 7.66 (dt, J = 8.0, 1.0 Hz, 1H), 7.37–7.28 (m, 2H), 7.16 (d, J = 0.8 Hz, 1H), 7.14 (ddd, J = 7.9, 6.8, 1.1 Hz, 1H), 5.75 (dd, J = 7.4, 2.8 Hz, 1H), 4.55–4.41 (m, 2H), 3.97 (dq, J = 9.5, 7.1 Hz, 1H), 3.81 (s, 3H), 3.71 (dq, J = 9.5, 7.1 Hz, 1H), 3.34 (dd, J = 16.6, 7.4 Hz, 1H), 3.05 (dd, J = 16.6, 2.8 Hz, 1H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 162.3, 158.1, 138.5, 127.8, 126.8, 123.7, 123.1 (q, J = 277.3 Hz), 121.8, 120.0, 109.7, 108.7, 105.2, 102.8, 64.8, 59.7 (q, J = 36.4 Hz), 37.2, 31.7, 15.1. IR 2977.71 (m), 2938.65 (w), 1720.30 (s), 1711.16 (m), 1629.92 (m), 1283.07 (s), 1247.21 (s), 1169.49 (s), 1080.27 (s). HMRS (ESI) m/z: [M + H]+ calc. for C18H18F3NO4, 370.1261; Found 370.1262.
2,2,2-Trifluoroethyl 5-(4-chlorophenyl)-5-ethoxy-2-(1-methyl-1H-indol-2-yl)-4,5-dihydrofuran-3-carboxylate (3ah): Prepared following general procedure using 1-chloro-4-(1-ethoxyvinyl)benzene
2h (562 mg, 3.07 mmol, which was prepared according to previous reported literature [
46]) and diazo
1a (200 mg, 615 μmol). Purification via silica gel column chromatography (5% EtOAC/Hexanes, R
f = 0.52 in 20% EtOAC/Hexanes) afforded
3ah as yellow solid (192 mg, 65%).
1H NMR (500 MHz, CDCl
3) δ 7.71 (dt,
J = 8.0, 1.0 Hz, 1H), 7.50–7.47 (m, 2H), 7.44–7.39 (m, 3H), 7.35 (ddd,
J = 8.3, 6.9, 1.2 Hz, 1H), 7.28 (d,
J = 0.8 Hz, 1H), 7.18 (ddd,
J = 8.0, 6.8, 1.1 Hz, 1H), 4.50 (qd,
J = 8.5, 2.6 Hz, 2H), 3.90 (s, 3H), 3.71 (dq,
J = 9.5, 7.1 Hz, 1H), 3.57 (d,
J = 16.6 Hz, 1H), 3.41 (dq,
J = 9.4, 7.1 Hz, 1H), 3.31 (d,
J = 16.6 Hz, 1H), 1.24 (t,
J = 7.0 Hz, 3H).
13C NMR (126 MHz, CDCl
3) δ 162.1, 157.7, 138.7, 138.6, 134.6, 128.9, 127.7, 127.1, 126.8, 123.9, 123.1 (q,
J = 277.5 Hz), 121.8, 120.2, 111.4, 109.7, 108.9, 103.5, 59.9 (d,
J = 36.4 Hz), 59.7, 44.8, 31.9, 15.3.
IR 3058.41 (w), 2978.18 (m), 2936.26 (w), 1724.57 (s), 1711.38 (m), 1283.63 (m), 1234.06 (m), 1171.08 (s), 1093.74 (s).
HMRS (ESI) m/
z: [M + H]
+ calc. for C
24H
21ClF
3NO
4, 480.1184; Found 480.1186.
Reaction of Diazo1aand Trimethyl((1-phenylvinyl)oxy)silane2i. Following the general procedure using trimethyl((1-phenylvinyl)oxy)silane 2i (591 mg, 3.07 mmol) and diazo 1a (200 mg, 615 μmol), an inseparable mixture of dihydrofuran 3ai, carbazole 5ai, and acetophenone (hydrolysis product of enol silane 2i) was isolated following column chromatography (1–5% EtOAC/Hexanes). According to quantitative 1H-NMR (with DMF as the internal standard), the yields of 3ai and 5ai were 50% and 21%, respectively.
2,2,2-Trifluoroethyl 2-(1-methyl-1H-indol-2-yl)-3a,4,5,6a-tetrahydrofuro[2,3-b]furan-3-carboxylate (3aj): Prepared following general procedure using dihydrofuran 2j (215 mg, 3.07 mmol), diazo 1a (200 mg, 615 μmol) and 20 mol% Cu(hfacac)2 (59 mg, 123 μmol). This reaction was kept at reflux for 2 d. Purification via silica gel column chromatography (5% EtOAC/Hexanes, Rf = 0.3 in 20% EtOAC/Hexanes) afforded 3aj as yellow gel (112 mg, 50%). 1H NMR (500 MHz, CDCl3) δ 7.68 (dt, J = 7.9, 1.0 Hz, 1H), 7.37–7.30 (m, 2H), 7.25 (s, 1H), 7.15 (ddd, J = 7.9, 6.6, 1.4 Hz, 1H), 6.34 (d, J = 6.3 Hz, 1H), 4.63 (dq, J = 12.7, 8.5 Hz, 1H), 4.45 (dq, J = 12.7, 8.5 Hz, 1H), 4.19 (ddd, J = 8.7, 5.6, 2.4 Hz, 1H), 4.07–4.02 (m, 1H), 3.90–3.84 (m, 1H), 3.82 (s, 3H), 2.25 (dt, J = 8.4, 4.0 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 162.0, 160.6, 138.7, 127.0, 126.6, 124.0, 123.1 (q, J = 277.5 Hz), 121.8, 120.1, 109.9, 109.7, 109.3, 104.1, 67.1, 59.7 (q, J = 36.3 Hz), 47.6, 31.9, 31.8. IR 2958.56 (m), 2881.14 (m), 1721.91 (s), 1711.23 (m), 1619.47 (m), 1280.55 (s), 1233.10 (s), 1168.90 (s), 1077.28 (s). HMRS (ESI) m/z: [M + H]+ calc. for C18H16F3NO4, 368.1104; Found 368.1104.
2,2,2-Trifluoroethyl 2-(1-methyl-1H-indol-2-yl)-3a,5,6,7a-tetrahydro-4H-furo[2,3-b]pyran-3-carboxylate (3ak): Prepared following general procedure using 2,3-dihydropyran 2k (259 mg, 3.07 mmol), diazo 1a (200 mg, 615 μmol) and 20 mol% Cu(hfacac)2 (59 mg, 123 μmol). The reaction was kept at reflux for 3 d for complete consumption of starting material. Purification via silica gel column chromatography (5% EtOAC/Hexanes, Rf = 0.36 in 20% EtOAC/Hexanes) afforded 3ak as yellow gel. DMF was applied as internal NMR standard to quantify the yield (54%), since there were some undefined impurities. 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 8.0 Hz, 1H), 7.36–7.28 (m, 2H), 7.21 (s, 1H), 7.12 (ddd, J = 7.9, 6.8, 1.2 Hz, 1H), 6.03 (d, J = 7.3 Hz, 1H), 4.55 (dq, J = 12.8, 8.5 Hz, 1H), 4.45 (dq, J = 12.7, 8.5 Hz, 1H), 3.94–3.86 (m, 2H), 3.82 (s, 3H), 3.21 (td, J = 7.3, 6.2 Hz, 1H), 2.23–2.13 (m, 1H), 1.79–1.70 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 162.5, 159.3, 138.9, 127.6, 126.8, 124.0, 123.2 (q, J = 277.7 Hz), 121.9, 120.2, 109.8, 109.4, 108.1, 104.8, 61.7, 59.7 (q, J = 36.4 Hz), 38.2, 31.9, 23.5, 20.3. IR 3057.59 (w), 2949.15 (m), 1720.38 (s), 1711.18 (m), 1618.58 (m), 1613.47 (m), 1279.06 (m), 1161.26 (s), 1093.65 (s). HMRS (ESI) m/z: [M + H]+ calc. for C19H18F3NO4, 382.1261; Found 382.1261.
Reaction of Diazo1aand 5-methyl 2,3-dihydrofuran (2l). Following the general procedure with commercial 5-methyl 2,3-dihydropyran
2l (259 mg, 3.07 mmol), diazo
1a (200 mg, 615 μmol) and 20 mol% Cu(hfacac)
2 (59 mg, 123 μmol), a 2.85:1 mixture (50% total yield according to
1H-NMR) of dihydrofurans
3al and
3am was isolated due to the in situ isomerization of
2l to 2-methylene tetrahydrofuran (
2m) in the reaction pot. Interestingly, if synthesized 5-methyl 2,3-dihydropyran
2l [
56] was used (see
Supporting Information for
1H-NMR) instead of the commercial material, only
3am was isolated as the major product when the reaction was run at 65 °C. Carbazole
5ai could be prepared directly in 36% yield by the reaction of diazo
1a and
2i using Cu(hfacac)
2 (30 mol%) with 1,2-dichloroethane as the solvent at 80 °C for 24 h.
2,2,2-Trifluoroethyl 2-(1-methyl-1H-indol-2-yl)-1,6-dioxaspiro[4.4]non-2-ene-3-carboxylate (3am): Prepared according to what is described previously. Purification via silica gel column chromatography (5% EtOAc/Hexanes, Rf = 0.3 in 20% EtOAc/Hexanes) afforded 3am as yellow gel (132 mg, 56%). 1H NMR (500 MHz, CDCl3) δ 7.63 (dd, J = 7.9, 1.0 Hz, 1H), 7.34–7.26 (m, 2H), 7.14–7.09 (m, 2H), 4.53–4.40 (m, 2H), 4.19–4.09 (m, 2H), 3.78 (s, 3H), 3.31 (d, J = 16.6 Hz, 1H), 3.27 (d, J = 16.6 Hz, 1H), 2.47–2.42 (m, 1H), 2.26–2.17 (m, 1H), 2.16–2.07 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 162.3, 157.8, 138.6, 127.9, 126.8, 123.7, 123.2 (q, J = 277.3 Hz), 121.8, 120.0, 117.7, 109.6, 108.6, 102.6, 69.1, 59.7 (q, J = 36.4 Hz), 38.5, 36.7, 31.6, 23.9. IR 3058.34 (w), 2922.83 (s), 2853.16 (m), 1720.57 (s), 1614.31 (s), 1463.21 (m), 1284.80 (s), 1171.14 (s). HMRS (ESI) m/z: [M + H]+ calc. for 382.1261; Found 382.1261.
2,2,2-Trifluoroethyl 5-methoxy-5-methyl-2-(1-methyl-1H-indol-3-yl)-4,5-dihydrofuran-3-carboxylate (3ca): Prepared following general procedure using 2-methoxypropene 2a (221 mg, 3.07 mmol) and diazo 1c (200 mg, 615 μmol). The reaction reached completion within 1 h under reflux. Purification via silica gel column chromatography (5% EtOAC/Hexanes, Rf = 0.5 in 20% EtOAC/Hexanes) afforded 3ca as yellow solid (132 mg, 58%). 1H NMR (500 MHz, CDCl3) δ 8.88 (s, 1H), 8.19 (d, J = 8.0 Hz, 1H), 7.38 (d, J = 8.2 Hz, 1H), 7.32 (t, 1H), 7.27 (d, J = 1.1 Hz, 1H), 4.63–4.50 (m, 2H), 3.89 (s, 3H), 3.44 (s, 3H), 3.24–3.04 (m, 2H), 1.78 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 164.2, 163.5, 136.8, 136.6, 126.9, 123.5 (d, J = 277.6 Hz), 122.7, 122.7, 121.5, 110.5, 109.7, 104.4, 94.5, 59.5 (q, J = 35.8 Hz), 50.2, 39.7, 33.5, 25.1. IR 3113.53 (w), 2938.52 (w), 1763.42 (s), 1759.62 (s), 1653.54 (s), 1530.37 (m), 1282.47 (m), 1169.54 (s), 1087.58 (m). HMRS (ESI) m/z: [M + H]+ calc. for C18H18F3NO4, 370.1260; Found 370.1262.
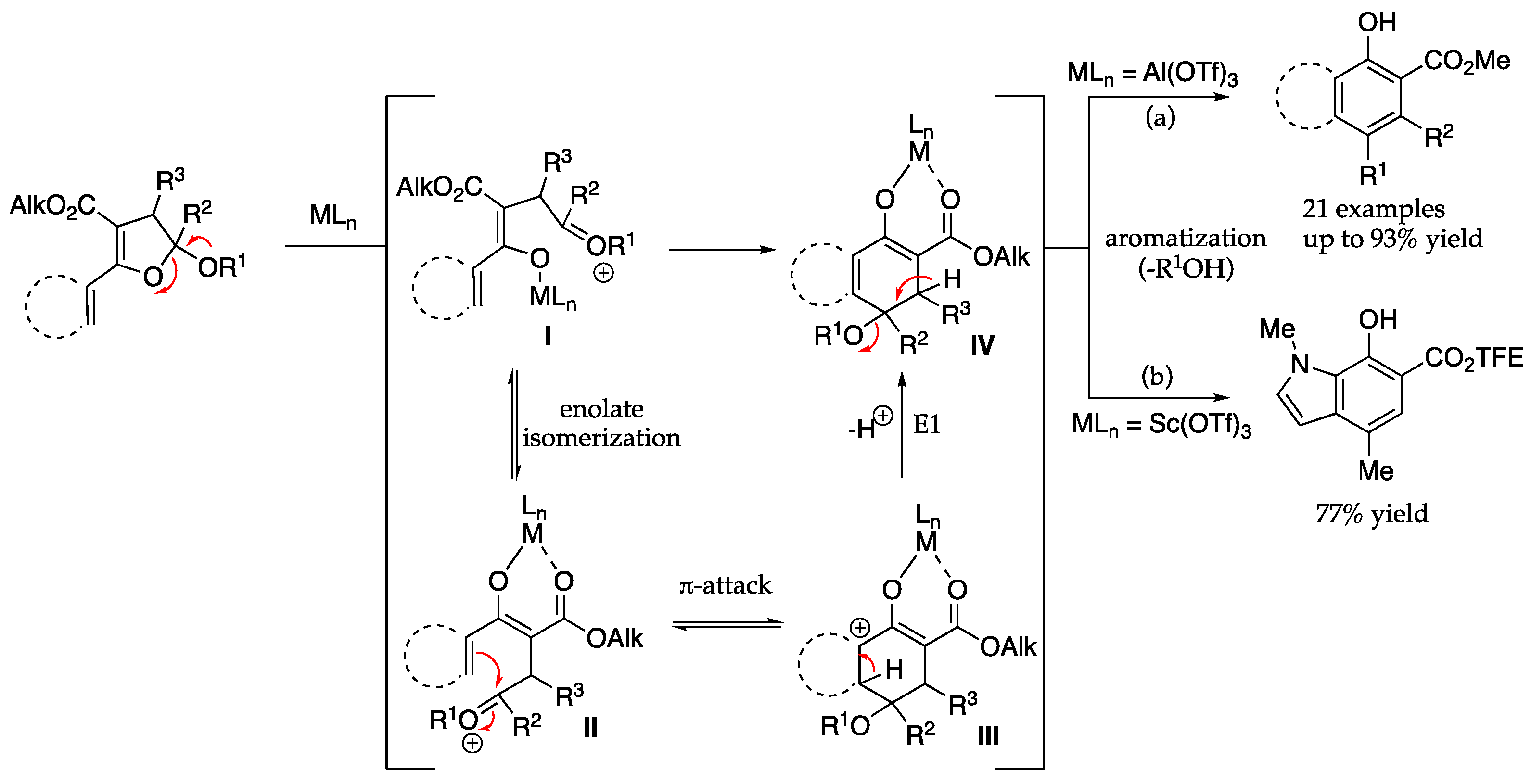
D. Ring-opening benzannulation of DHF toward 1-hydroxycarbazoles 5 and 6.
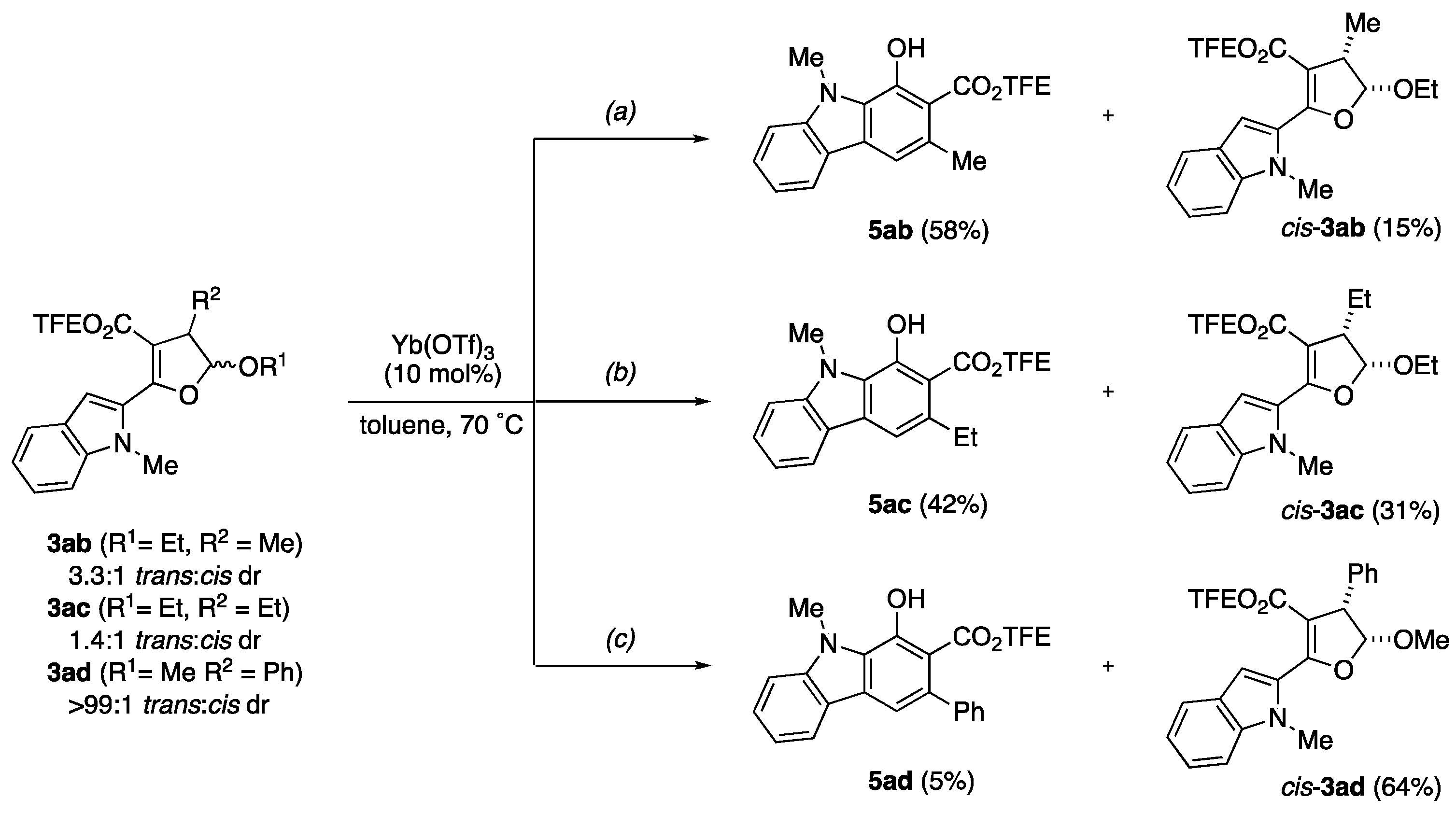
General procedure A: To a vial or pressured tube charged with a stir bar and a solution of DHF 3 in toluene (0.1 M) was added Yb(OTf)3 (10 mol%). Then, the suspension was set under sonication for 1 min. The reaction was stirred under 70 °C for 1 h or monitored by TLC for completion. After consuming all DHF, the mixture was concentrated under reduced pressure, and purified by column chromatography.
General procedure B: To a vial or pressured tube charged with a stir bar and a solution of DHF 3 in toluene (0.1 M) was added Al(OTf)3 (10 mol%). Then, the suspension was set under sonication for 1 min, and 1 small drop of water (~4 mg) was added to the mixture. After the mixture was sonicated for another 5 min, the reaction was stirred vigorously under 70 °C for 1 h. After consuming all DHF, the mixture was concentrated under reduced pressure, and purified by column chromatography.
Note: Since the 19F signals of all carbazole products have similar chemical shifts (−73.3 ppm to −73.8 ppm) with same coupling constant (t, J = 8.5 Hz), only the 19F NMR of 5aa is reported and attached.
2,2,2-Trifluoroethyl 1-hydroxy-4,9-dimethyl-9H-carbazole-2-carboxylate (5aa): Prepared following general procedure A using DHF 3aa (89 mg, 0.24 mmol) and Yb(OTf)3 (15 mg, 24 μmol) in toluene (2.4 mL). Purification via silica gel column chromatography (1–10% EtOAC/Hexanes, Rf = 0.59 in 20% EtOAC/Hexanes) afforded 5aa as yellow crystal (73 mg, 90%). 1H NMR (700 MHz, CDCl3) δ 10.99 (s, 1H), 8.21 (dt, J = 8.0, 1.0 Hz, 1H), 7.56 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.46 (d, J = 8.3 Hz, 1H), 7.40 (s, 1H), 7.28 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 4.75 (q, J = 8.4 Hz, 2H), 4.25 (s, 3H), 2.80 (s, 3H). 13C NMR (176 MHz, CDCl3) δ 169.7, 149.6, 142.5, 128.4, 128.1, 126.8, 124.1, 123.3, 123.0 (q, J = 277.7 Hz), 122.8, 119.6, 119.5, 109.1, 105.7, 60.6 (q, J = 36.7 Hz), 32.1, 20.4. 19F NMR (471 MHz, CDCl3) δ −73.44 (t, J = 8.5 Hz, 3F). IR 3057.01 (w), 2963.78 (w), 1670.40 (s), 1278.20 (s), 1234.57 (s), 1157.37 (s), 1127.22 (s). HMRS (ESI) m/z: [M + H]+ calc. for C17H14F3NO3, 338.0999; Found 338.0998.
2,2,2-Trifluoroethyl 1-hydroxy-3,9-dimethyl-9H-carbazole-2-carboxylate (5ab): Prepared following general procedure B using DHF 3ab (110 mg, 0.287 mmol) and Al(OTf)3 (14 mg, 29 μmol) in toluene (2.8 mL). Purification via silica gel column chromatography (1–10% EtOAC/Hexanes, Rf = 0.59 in 20% EtOAC/Hexanes) afforded 5ab as yellow crystal (65 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 11.83 (s, 1H), 7.52 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.41–7.37 (m, 2H), 7.23 (ddd, J = 7.9, 7.1, 1.0 Hz, 1H), 4.72 (q, J = 8.4 Hz, 2H), 4.19 (s, 3H), 2.67 (d, J = 0.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.3, 152.7, 142.8, 130.4, 128.0, 127.3, 123.1 (q, J = 277.8 Hz) 121.7, 121.0, 119.2, 114.3, 109.2, 106.3, 60.9 (q, J = 36.8 Hz), 32.0, 24.5. IR 3030.13 (w), 2961.61 (m), 2937.58 (m), 1649.38 (s), 1637.42 (s), 1316.84 (s), 1272.05 (s), 1158.44 (s), 1036.63 (s). HMRS (ESI) m/z: [M + H]+ calc. for C17H14F3NO3, 338.0999; Found 338.0998.
2,2,2-Trifluoroethyl 3-ethyl-1-hydroxy-9-methyl-9H-carbazole-2-carboxylate (5ac): Prepared following general procedure B using DHF 3ac (108 mg, 0.272 mmol) and Al(OTf)3 (13 mg, 27 μmol) in toluene (2.7 mL). Purification via silica gel column chromatography (1–10% EtOAC/Hexanes, Rf = 0.59 in 20% EtOAC/Hexanes) afforded 5ac as yellow crystal (59 mg, 62%). 1H NMR (500 MHz, CDCl3) δ 11.80 (s, 1H), 8.03 (dt, J = 7.8, 1.0 Hz, 1H), 7.53 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.44 (s, 1H), 7.40 (dt, J = 8.4, 0.9 Hz, 1H), 7.24 (ddd, J = 7.9, 7.1, 0.9 Hz, 1H), 4.74 (q, J = 8.4 Hz, 2H), 4.19 (s, 3H), 3.07 (q, J = 7.4 Hz, 2H), 1.29 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 171.1, 152.6, 142.8, 136.9, 128.1, 127.3 (d, J = 2.3 Hz), 123.0 (q, J = 277.2 Hz), 121.8, 121.0, 119.2, 113.1, 109.2, 105.7, 61.0 (q, J = 36.8 Hz), 32.0, 30.1, 16.8. IR 3026.24 (w), 2954.17 (m), 2870.75 (w), 1649.94 (s), 1315.64 (s), 1259.43 (s), 1158.68 (s), 1123.55 (s), 1036.82 (m). HMRS (ESI) m/z: [M + H]+ calc. for C18H16F3NO3, 352.1155; Found 352.1153.
2,2,2-Trifluoroethyl 1-hydroxy-9-methyl-3-phenyl-9H-carbazole-2-carboxylate (5ad): Prepared following general procedure B using DHF 3ad (100 mg, 0.232 mmol) and Al(OTf)3 (11 mg, 23 μmol) in toluene (2.3 mL). Purification via silica gel column chromatography (1–10% EtOAC/Hexanes, Rf = 0.59 in 20% EtOAC/Hexanes) afforded 5ad as yellow crystal (60 mg, 65%). 1H NMR (500 MHz, CDCl3) δ 11.38 (s, 1H), 8.02 (dt, J = 7.9, 1.0 Hz, 1H), 7.56 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.51 (s, 1H), 7.46–7.33 (m, 6H), 7.26 (ddd, J = 7.9, 6.6, 0.9 Hz, 1H), 4.38 (q, J = 8.4 Hz, 2H), 4.26 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 170.7, 151.2, 143.5, 142.8, 135.0, 128.4, 127.7, 127.4, 126.4, 122.2 (q, J = 277.5 Hz), 122.0, 121.1, 119.6, 114.9, 109.3, 106.1, 60.6 (q, J = 37.2 Hz), 32.1. IR 3022.72 (w), 2916.53 (w), 1655.22 (s), 1321.40 (s), 1281.60 (m), 1159.52 (s), 1030.67 (m). HMRS (ESI) m/z: [M + H]+ calc. for C22H16F3NO3, 400.1155; Found 400.1154.
2,2,2-Trifluoroethyl 1-hydroxy-9-methyl-9H-carbazole-2-carboxylate (5ae): Prepared following general procedure A using DHF 3ae (150 mg, 0.406 mmol) and Yb(OTf)3 (25 mg, 41 μmol) in toluene (4 mL). Purification via silica gel column chromatography (1–10% EtOAC/Hexanes, Rf = 0.59 in 20% EtOAC/Hexanes) afforded 5ae as yellow crystal (106 mg, 81%). 1H NMR (500 MHz, CDCl3) δ 11.15 (s, 1H), 8.07 (dt, J = 7.8, 1.0 Hz, 1H), 7.65 (d, J = 8.4 Hz, 1H), 7.60–7.51 (m, 2H), 7.43 (dt, J = 8.4, 0.9 Hz, 1H), 7.26 (ddd, J = 7.9, 7.1, 0.9 Hz, 1H), 4.75 (q, J = 8.3 Hz, 2H), 4.23 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 169.7, 151.2, 142.4, 129.4, 128.3, 127.4, 123.0 (q, J = 277.3 Hz), 122.0, 121.1, 119.5, 119.5, 111.5, 109.2, 106.2, 60.6 (q, J = 36.9 Hz), 32.1. IR 3061.58 (w), 2964.60 (m), 2933.49 (w), 1668.50 (s), 1630.09 (m), 1464.87 (s), 1250.61 (s), 1144.95 (s), 1040.08 (m). HMRS (ESI) m/z: [M + H]+ calc. for C16H12F3NO3, 324.0842; Found 324.0840.
Reaction of dihydrofuran3ahwith Al(OTf)3: Following the general procedure B with dihydrofuran 3ah, Al(OTf)3, and no added, an inseparable mixture of carbazole 5ah and furan 4ah was formed. Since product 5ah and 4ah run at similar Rf value, q-NMR was applied to quantify the yield of each one. According to quantitative 1H-NMR (with DMF as internal standard), the respective yield of 5ah and 4ah in the mixture were 19% and 68%.
2,2,2-Trifluoroethyl 1-hydroxy-9-methyl-4-phenyl-9H-carbazole-2-carboxylate (5ai): To a dry flask charged with a stir bar and the mixture of 3ai and 5ai prepared in previous section, dry 1,2-DCE (6 mL) was added, followed by addition of Cu(hfacac)2 (59 mg, 123 μmol, 20 mol%.). The solution was then heated to 80 °C for 24 h, diluted with Et2O (30 mL) and washed with saturated thiourea (20 mL). The organic layer was dried over Na2SO4, concentrated under reduced pressure, and purified by column chromatography (silica gel, 0–5% EtOAC/Hexanes), which afforded 5ai as pale-yellow crystal (88 mg, 36% over two steps). 1H NMR (500 MHz, CDCl3) δ 11.18 (s, 1H), 7.57–7.46 (m, 7H), 7.43 (dt, J = 8.4, 0.9 Hz, 1H), 7.35 (dt, J = 8.1, 1.0 Hz, 1H), 6.99 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 4.75 (q, J = 8.4 Hz, 2H), 4.30 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 169.7, 150.5, 142.7, 140.3, 129.4, 129.1, 128.6, 128.5, 127.6, 127.1, 127.0, 123.1, 122.9 (q, J = 277.4 Hz), 121.8, 120.2, 119.2, 109.1, 105.9, 60.6 (q, J = 37.2 Hz), 32.2. IR 3056.30 (w), 2946.60 (w), 1658.71 (s), 1465.88 (m), 1236.68 (s), 1234.71 (s) 1155.21 (s), 1128.94 (m). HMRS (ESI) m/z: [M-H]− calc. for C22H16F3NO3, 398.1010; Found 398.1004.
11-Hydroxy-10-methyl-4,10-dihydropyrano[3,4-b]carbazol-1(3H)-one (6aj): Prepared following general procedure B using DHF 3aj (88 mg, 0.24 mmol) and Al(OTf)3 (11 mg, 24 μmol) in toluene (2.4 mL) without addition of water. Purification via silica gel column chromatography (1–10% EtOAC/Hexanes, Rf = 0.38 in 20% EtOAC/Hexanes) afforded 5ae as yellow crystal (50 mg, 78%). 1H NMR (500 MHz, CDCl3) δ 12.01 (s, 1H), 8.02 (dt, J = 7.8, 1.0 Hz, 1H), 7.54 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.42 (dt, J = 8.4, 0.9 Hz, 1H), 7.36 (t, J = 1.1 Hz, 1H), 7.24 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 4.62 (dd, J = 6.4, 5.6 Hz, 2H), 4.21 (s, 3H), 3.19 (ddd, J = 6.6, 5.5, 1.1 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 171.3, 151.5, 142.6, 128.7, 128.4, 127.5, 121.8, 121.0, 119.4, 109.3, 109.0, 104.0, 68.8, 31.9, 28.1. IR 3056.02 (w), 2914.82 (m), 1648.69 (s), 1468.19 (m), 1302.74 (s), 1253.14 (s), 1124.21 (s). HMRS (ESI) m/z: [M + H]+ calc. for C16H13NO3, 268.0968; Found 268.0968.
2,2,2-Trifluoroethyl 1-hydroxy-3-(3-hydroxypropyl)-9-methyl-9H-carbazole-2-carboxylate (5ak): Prepared following general procedure B using DHF 3ak (75 mg, 0.24 mmol) and Al(OTf)3 (9 mg, 20 μmol) in toluene (2 mL) without addition of water. Purification via silica gel column chromatography (10–30% EtOAC/Hexanes, Rf = 0.14 in 20% EtOAC/Hexanes) afforded 5ak as yellow crystal (25 mg, 33%). 1H NMR (500 MHz, CDCl3) δ 11.81 (s, 1H), 8.03 (dt, J = 7.8, 1.0 Hz, 1H), 7.53 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.46 (s, 1H), 7.41 (dt, J = 8.4, 0.9 Hz, 1H), 7.24 (ddd, J = 7.9, 7.0, 0.9 Hz, 1H), 4.78 (q, J = 8.4 Hz, 2H), 4.21 (s, 3H), 3.72 (t, J = 6.4 Hz, 2H), 3.16–3.12 (m, 2H), 1.95–1.88 (m, 2H), 1.49 (s, br, 1H). 13C NMR (126 MHz, CDCl3) δ 171.0, 152.9, 142.8, 134.1, 128.1, 127.5, 127.4, 123.1 (q, J = 277.3 Hz), 121.7, 121.1, 119.3, 114.1, 109.2, 105.7, 62.4, 61.0 (q, J = 36.8 Hz), 35.2, 33.3, 32.1. IR 3324.04 (w, br), 2958.85 (m), 2925.22 (m), 1647.07 (s), 1434.70 (m), 1313.12 (s), 1256.98 (s), 1157.55 (s), 1035.10 (s). HMRS (ESI) m/z: [M + H]+ calc. for C19H18F3NO4, 382.1261; Found 382.1259.
11-Hydroxy-5,10-dimethyl-4,10-dihydropyrano[3,4-b]carbazol-1(3H)-one (6al) and 2,2,2-Trifluoroethyl 1-hydroxy-4-(3-hydroxypropyl)-9-methyl-9H-carbazole-2-carboxylate (5am): To a vial charged with a stir bar and a 2.85:1 mixture of 3al and 3am (100 mg, 0.262 mmol) in toluene (2.5 mL) was added Al(OTf)3 (12 mg, 26.2 μmol). The suspension was set under sonication for 1 min, followed by stirred and heated under 70 °C for 1 h. Then, another portion of Al(OTf)3 (12 mg, 26.2 μmol) was added to the reaction followed by increasing temperature to 85 °C for additional 1 h. After consuming all DHF, the mixture was concentrated under reduced pressure, and purified by column chromatography.
6al: Purification on a silica gel column chromatography (1–10% EtOAC/Hexanes, Rf = 0.38 in 20% EtOAC/Hexanes) afforded 5al as yellow crystal (45 mg, 61%). 1H NMR (400 MHz, CDCl3) δ 12.06 (s, 1H), 8.24 (d, J = 8.1 Hz, 1H), 7.55 (ddd, J = 8.3, 7.0, 1.1 Hz, 2H), 7.44 (dt, J = 8.3, 0.9 Hz, 2H), 7.25 (ddd, J = 8.1, 7.1, 1.1 Hz, 1H), 4.60 (dd, J = 6.5, 5.6 Hz, 3H), 4.22 (s, 5H), 3.15 (t, J = 6.1 Hz, 3H), 2.69 (s, 5H). 13C NMR (101 MHz, CDCl3) δ 171.8, 150.0, 142.7, 127.9, 127.4, 126.8, 125.4, 123.5, 122.7, 119.6, 119.3, 109.1, 103.9, 68.3, 32.0, 24.9, 15.6. IR 2994.64 (w), 2922.00 (m), 2851.13 (w), 1654.70 (s), 1626.95 (m), 1317.00 (s), 1224. 95 (s), 1158. 46 (m), 1134.11 (m). HMRS (ESI) m/z: [M + H]+ calc. for C17H15NO3, 282.1125; Found 282.1118.
5am: Purification via silica gel column chromatography (10–30% EtOAC/Hexanes, Rf = 0.14 in 20% EtOAC/Hexanes) afforded 5am as pale-yellow crystal (19 mg, 19%). 1H NMR (500 MHz, CDCl3) δ 11.03 (s, 1H), 8.20 (dt, J = 8.1, 0.9 Hz, 1H), 7.56 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.47 (dt, J = 8.3, 0.9 Hz, 1H), 7.43 (s, 1H), 7.28 (ddd, J = 8.1, 7.1, 1.1 Hz, 1H), 4.76 (q, J = 8.3 Hz, 2H), 4.26 (s, 3H), 3.84 (t, J = 6.3 Hz, 2H), 3.30–3.21 (m, 2H), 2.13–2.05 (m, 2H). 1.41 (s, br, 1H). 13C NMR (126 MHz, CDCl3) δ 169.6, 149.8, 142.6, 128.8, 128.1, 127.4, 126.8, 123.3, 123.0 (q, J = 277.5 Hz), 121.9, 119.7, 118.8, 109.2, 105.8, 62.6, 60.6 (q, J = 37.0 Hz), 32.5, 32.1, 30.0. IR 3325.97 (s, br), 2931.32 (m), 2891.86 (w), 1662.28 (s), 1332.21 (s), 1278.97 (s), 1239.36 (s), 1153.52(s), 1040.43 (s). HMRS (ESI) m/z: [M + H]+ calc. for C19H18F3NO4, 382.1261; Found 382.1258.
2,2,2-Trifluoroethyl 9-benzyl-1-hydroxy-4-methyl-9H-carbazole-2-carboxylate (5ba): Prepared following general procedure A using DHF 3ba (175 mg, 0.393 mmol) and Yb(OTf)3 (24 mg, 39 μmol) in toluene (3.9 mL). Purification via silica gel column chromatography (1–10% EtOAC/Hexanes, Rf = 0.59 in 20% EtOAC/Hexanes) afforded 5ba as yellow crystal (133 mg, 82%). 1H NMR (500 MHz, CDCl3) δ 11.36 (s, 1H), 8.58 (dt, J = 8.0, 0.9 Hz, 1H), 7.84 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.80–7.74 (m, 2H), 7.67–7.52 (m, 4H), 7.53–7.47 (m, 2H), 6.30 (s, 2H), 5.07 (q, J = 8.3 Hz, 2H), 3.16 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 169.5, 149.20, 142.0, 138.6, 128.5, 128.3, 127.9, 127.1, 126.9, 126.3, 124.0, 123.3, 123.04, 122.97 (q, J = 277.1 Hz), 119.9, 119.8, 109.8, 105.9, 60.6 (q, J = 36.8 Hz), 48.5, 20.4. IR 2969.19 (w), 2918.79 (w), 1664.19 (s), 1459.67 (s), 1302.85 (m), 1156.55 (s), 1122.03 (s). HMRS (ESI) m/z: [M + H]+ calc. for C23H18F3NO3, 414.1311; Found 414.1313.
2,2,2-Trifluoroethyl 9-benzyl-1-hydroxy-3-methyl-9H-carbazole-2-carboxylate (5bb): Prepared following general procedure B using DHF 3bb (624 mg, 1.36 mmol) and Al(OTf)3 (64 mg, 136 μmol) in toluene (14 mL) with couple drops of water. Purification on a silica gel column chromatography (1–10% EtOAC/Hexanes, Rf = 0.59 in 20% EtOAC/Hexanes) afforded 5bb as yellow crystal (376 mg, 67%). 1H NMR (500 MHz, CDCl3) δ 11.88 (s, 1H), 8.06 (d, J = 7.8 Hz, 1H), 7.49–7.45 (m, 2H), 7.38 (d, J = 8.3 Hz, 1H), 7.26–7.18 (m, 4H), 7.17–7.13 (m, 2H), 5.93 (s, 2H), 4.74 (q, J = 8.3 Hz, 2H), 2.71 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.3, 152.5, 142.4, 138.8, 130.9, 128.5, 128.4, 127.5, 127.1, 126.9, 126.4, 123.0 (q, J = 277.4 Hz), 122.0, 121.1, 119.6, 114.4, 109.9, 106.7, 60.9 (q, J = 37.0 Hz), 48.5, 24.6. IR 3030.93 (w), 2974.09 (w), 2936.23 (m), 1645.01 (s), 1435.07 (s), 1314.23 (s), 1251.83 (s), 1150.59 (s), 1038.06 (s). HMRS (ESI) m/z: [M + H]+ calc. for C23H18F3NO3, 414.1311; Found 414.1313.
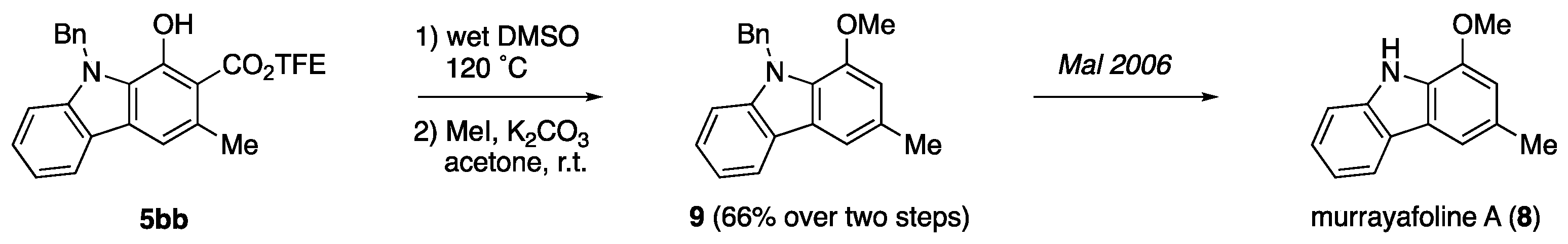
E. Synthesis of benzyl-protected Murrayafoline A (9) from Carbazole 5bb: The conversion from
5bb to murrayafoline A was inspired by a reported literature procedure [
57]. Wet DMSO (10 mL) was added to a vial charged with a stir bar and
5bb (310 mg, 0.75 mmol). The mixture was heating at 120 °C for overnight (~18 h), poured into 40 mL brine, extracted with 50 mL EtOAc. The organic layer was washed with brine (2 × 40 mL) and the combined aqueous layer was extracted with another 50 mL EtOAc. The combined organic layer was dried over Na
2SO
4, concentrated under reduced pressure, and purified by column chromatography to afford the decarboxylated carbazole product as a yellow solid (151 mg, 70%, with 18% recovery of
5bb). Next, anhydrous acetone (20 mL) was added to a flask charged with a stir bar, decarboxylated product (151 mg, 0.525 mmol), and K
2CO
3 (363 mg, 2.63 mmol, 5 eq.). The mixture was stirred at 0 °C for 20 min, then MeI (373 mg, 2.63 mmol, 5 eq.) was slowly added to cold suspension. The reaction mixture was removed from the ice-bath stirred at room temperature overnight (~18 h). The reaction was concentrated under reduced pressure, re-dissolved in Et
2O (60 mL), and washed with brine (30 mL). The organic layer was dried over Na
2SO
4, concentrated under reduced pressure, and purified by column chromatography to afford
N-benzyl murrayafoline A (
9) as a yellow gel (150 mg, 94%). Characterizations were consistent with previously reported literature [
48] and
1H-NMR spectrum is attached.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
