
Acetylsalicylic Acid–Primus Inter Pares in Pharmacology

 , , ,
, , , 
Abstract
:1. Introduction
2. History of ASA
3. ASA Structure
3.1. The Basic Physical, Chemical and Biological Properties of ASA
3.2. Mechanism of Action for ASA
4. The Potential Repurposing of ASA
4.1. ASA for Preventing Cardiovascular and Cerebrovascular Diseases
4.2. ASA Use for Cancer Therapy
4.3. ASA as Promising Candidate for Repositioning in the Treatment of Severe Bacterial Infections of MDR Bacteria, Extensively Resistant (XDR) and Pan-Resistant (PDR) to Drugs
4.4. ASA Potential for Neuropsychiatric Disorders
4.5. ASA Use for COVID-19
5. ASA Dose and Formulation
6. ASA Derivatives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anatomical Therapeutic Chemical Classification System. Available online: https://www.whocc.no/atc_ddd_index/?code=N02BA01 (accessed on 10 October 2022).
- DrugBank. Available online: https://go.drugbank.com/drugs/DB00945 (accessed on 10 October 2022).
- Drug Index A to Z. Available online: https://www.drugs.com/search.php?searchterm=aspirin&sources%5B%5D= (accessed on 10 October 2022).
- Balescu, A.; Nedelcu, L. The aspirin-the first drug obtained by sinthesys-frequently used currently. Bull. Transilv. Univ. Brasov. Med. Sci. Ser. VI 2009, 6, 145. [Google Scholar]
- Hybiak, J.; Broniarek, I.; Kiryczyński, G.; Los, L.D.; Rosik, J.; Machaj, F.; Sławiński, H.; Jankowska, K.; Urasińska, E. Aspirin and its pleiotropic application. Eur. J. Pharmacol. 2020, 866, 172762. [Google Scholar] [CrossRef]
- Marko, V. From Aspirin to Viagra: Stories of the Drugs That Changed the World; Springer Nature: Berlin, Germany, 2020. [Google Scholar]
- Warner, T.D.; Mitchell, J.A. Cyclooxygenase-3 (COX-3): Filling in the gaps toward a COX continuum? Proc. Natl. Acad. Sci. USA 2002, 99, 13371–13373. [Google Scholar] [CrossRef] [Green Version]
- Zdrojewicz, Z.; Jagodziński, A.; Kowalik-Jagodzińska, M.; Zielińska, E. Stare i nowe oblicze aspiryny. Pediatr. Med. Rodz. 2018, 14, 369–375. [Google Scholar] [CrossRef]
- Wood, J.N. From plant extract to molecular panacea: A commentary on Stone (1763) ‘An account of the success of the bark of the willow in the cure of the agues’. Philos. Transac. R. Soc. B Biol. Sci. 2015, 370, 20140317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, D.B. One hundred years of aspirin. Lancet 1997, 350, 437–439. [Google Scholar] [CrossRef]
- Rocke, A.J. Hermann Kolbe, Encyclopedia Britannica. Available online: https://www.britannica.com/biography/Hermann-Kolbe (accessed on 10 October 2022).
- Rocke, A.J. The Quiet Revolution: Hermann Kolbe and the Science of Organic Chemistry; University of California Press: Berkeley, CA, USA, 1993. [Google Scholar]
- Alegbeleye, B.J.; Akpoveso, O.O.P.; Mohammed, R.K.; Asare, B.Y.A. Pharmacology, Pharmaceutics and Clinical Use of Aspirin: A Narrative Review. J. Drug Deliv. Ther. 2020, 10, 236–253. [Google Scholar] [CrossRef]
- Gulyás, A.; Heszberger, Z.; Biró, J. Amazing Scientific Discoveries: Aspirin, Cattle, Business Communication and Others. In Paths; Springer: Berlin, Germany, 2021; pp. 67–71. [Google Scholar]
- Montinari, M.R.; Minelli, S.; De Caterina, R. The first 3500 years of aspirin history from its roots—A concise summary. Vasc. Pharmacol. 2019, 113, 1–8. [Google Scholar] [CrossRef]
- Leggat, P.A. A brief history of the discovery and applications of aspirin. Ann. ACTM 2012, 13, 5–9. [Google Scholar]
- Desborough, M.J.; Keeling, D.M. The aspirin story–from willow to wonder drug. Br. J. Haematol. 2017, 177, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Wick, J. Aspirin: A history, a love story. Consult. Pharm. 2012, 27, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Nowaczyk, A. 120 urodziny aspiryny. Wiad. Akad. 2019, 76, 21–25. [Google Scholar]
- Nowaczyk, A. Fenomen aspiryny. Głos Uczelni 2019, XI–XII, 39–44. [Google Scholar]
- The U.S. Food and Drug Administration (FDA). Clinical Trials Register. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=aspirin&cntry=&state=&city=&dist= (accessed on 1 October 2022).
- The European Union. Clinical Trials Register. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=aspirin (accessed on 1 October 2022).
- The Australian New Zealand Clinical Trials Registry. Available online: https://www.australianclinicaltrials.gov.au/anzctr_feed/form (accessed on 1 October 2022).
- Saleh, I. The Pattern of Aspirin Use in Patients with Heart Failure at Mohammad Hoesin Hospital Palembang from 1st June 2013–30th July 2014. Int. J. Health Sci. Res. 2016, 13–24. [Google Scholar]
- The Cambridge Structural Database (CSD). Available online: https://www.ccdc.cam.ac.uk/structures/? (accessed on 10 October 2022).
- Wheatley, P. 1163. The crystal and molecular structure of aspirin. J. Chem. Soc. (Resumed) 1964, 6036–6048. [Google Scholar] [CrossRef]
- Kim, Y.; Machida, K.; Taga, T.; Osaki, K. Structure redetermination and packing analysis of aspirin crystal. Chem. Pharm. Bull. 1985, 33, 2641–2647. [Google Scholar] [CrossRef] [Green Version]
- Ouvrard, C.; Price, S.L. Toward crystal structure prediction for conformationally flexible molecules: The headaches illustrated by aspirin. Cryst. Growth Des. 2004, 4, 1119–1127. [Google Scholar] [CrossRef]
- Payne, R.; Rowe, R.C.; Roberts, R.; Charlton, M.; Docherty, R. Potential polymorphs of aspirin. J. Comput. Chem. 1999, 20, 262–273. [Google Scholar] [CrossRef]
- Bond, A.D.; Solanko, K.A.; Parsons, S.; Redder, S.; Boese, R. Single crystals of aspirin form II: Crystallisation and stability. Cryst. Eng. Comm. 2011, 13, 399–401. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals 2e; International Union of Crystal: Chester UK; Oxford University Press: Oxford, UK, 2020; Volume 30. [Google Scholar]
- FDA Guidance for Industry: Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q6a-specifications-test-procedures-and-acceptance-criteria-new-drug-substances-and-new-drug-products (accessed on 1 October 2022).
- Tsuri, Y.; Maruyama, M.; Fujimoto, R.; Okada, S.; Adachi, H.; Yoshikawa, H.Y.; Takano, K.; Murakami, S.; Matsumura, H.; Inoue, T. Crystallization of aspirin form II by femtosecond laser irradiation. Appl. Phys. Express 2018, 12, 015507. [Google Scholar] [CrossRef]
- Yadav, A.R.; Mohite, S.K. Different techniques and characterization of polymorphism with their evaluation: A Review. Asian J. Pharm. Technol. 2020, 10, 213–216. [Google Scholar] [CrossRef]
- Vishweshwar, P.; McMahon, J.A.; Oliveira, M.; Peterson, M.L.; Zaworotko, M.J. The predictably elusive form II of aspirin. J. Am. Chem. Soc. 2005, 127, 16802–16803. [Google Scholar] [CrossRef] [PubMed]
- Crowell, E.L.; Dreger, Z.A.; Gupta, Y.M. High-pressure polymorphism of acetylsalicylic acid (aspirin): Raman spectroscopy. J. Mol. Struct. 2015, 1082, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Shtukenberg, A.G.; Hu, C.T.; Zhu, Q.; Schmidt, M.U.; Xu, W.; Tan, M.; Kahr, B. The third ambient aspirin polymorph. Cryst. Growth Des. 2017, 17, 3562–3566. [Google Scholar] [CrossRef]
- Choudhary, A.; Kamer, K.J.; Raines, R.T. An n→ π* interaction in aspirin: Implications for structure and reactivity. J. Organ. Chem. 2011, 76, 7933–7937. [Google Scholar] [CrossRef]
- Lee, E.H. A practical guide to pharmaceutical polymorph screening & selection. Asian J. Pharm. Sci. 2014, 9, 163–175. [Google Scholar]
- Dymczuk, K. Study of Phase Equilibria in Binary System: Pharmaceuticals + Solvent; Diss. Zakład Chemii Fizycznej, Politechnika Warszwska: Warsaw, Poland, 2012. [Google Scholar]
- Poźniak, B.; Świtała, M. Salicylany w farmakoterapii weterynaryjnej. Część I. Współczesny stan wiedzy o właściwościach farmakologicznych. Życie Weter 2012, 87, 942–947. [Google Scholar]
- Perlovich, G.L.; Bauer-Brandl, A. Thermodynamics of solutions I: Benzoic acid and acetylsalicylic acid as models for drug substances and the prediction of solubility. Pharm. Res. 2003, 20, 471–478. [Google Scholar] [CrossRef]
- Gaohua, L.; Miao, X.; Dou, L. Crosstalk of physiological pH and chemical pKa under the umbrella of physiologically based pharmacokinetic modeling of drug absorption, distribution, metabolism, excretion, and toxicity. Expert Opin. Drug Met. 2021, 17, 1103–1124. [Google Scholar] [CrossRef]
- Nordström, F.L.; Rasmuson, Å.C. Solubility and melting properties of salicylic acid. J. Chem. Eng. Data 2006, 51, 1668–1671. [Google Scholar] [CrossRef]
- Chen, C.-C.; Song, Y. Solubility modeling with a nonrandom two-liquid segment activity coefficient model. Ind. Eng. Chem. Res. 2004, 43, 8354–8362. [Google Scholar] [CrossRef]
- Vane, J.; Bakhle, Y.; Botting, R. Cyclooxygenase 1 and 2. Annu. Rev. Pharmacol. 1998, 38, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Cryer, B.; Feldman, M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res. 2013, 191, 39–65. [Google Scholar] [PubMed]
- Nokhodchi, A.; Ghafourian, T.; Nashed, N.; Asare-Addo, K.; Behboudi, E.; Sefid-Sefidehkhan, Y.; Zarghampour, A.; Rahimpour, E.; Jouyban, A. Solubility Study of Acetylsalicylic Acid in Ethanol+ Water Mixtures: Measurement, Mathematical Modeling, and Stability Discussion. AAPS Pharm. Sci. Tech. 2022, 23, 1–12. [Google Scholar] [CrossRef]
- Shalmashi, A.; Eliassi, A. Solubility of salicylic acid in water, ethanol, carbon tetrachloride, ethyl acetate, and xylene. J. Chem. Eng. Data 2008, 53, 199–200. [Google Scholar] [CrossRef]
- Belhachemi, B.; Makhlouf, H.; Belhachemi, M.H. Determination and Correlation of Solubilities of Benzoic Acid, Salicylic Acid, Resorcinol and Hydroquinone in Water and in 1-Octanol at Temperatures from 297.25 K to 334.45 K. Int. J. Thermophys. 2021, 42, 1–16. [Google Scholar] [CrossRef]
- Stevens, H.; Voelker, M.; Gow, L.; MacDougall, F.; Bieri, G. In-vivo disintegration and absorption of two fast acting aspirin tablet formulations compared to ibuprofen tablets using pharmacoscintigraphy. J. Drug Deliv. mSci. Technol. 2019, 51, 535–541. [Google Scholar] [CrossRef]
- Jirmář, R.; Widimský, P. Enteric-coated aspirin in cardiac patients: Is it less effective than plain aspirin? Cor et Vasa 2018, 60, e165–e168. [Google Scholar] [CrossRef] [Green Version]
- Kanani, K.; Gatoulis, S.C.; Voelker, M. Influence of differing analgesic formulations of aspirin on pharmacokinetic parameters. Pharmaceutics 2015, 7, 188–198. [Google Scholar] [CrossRef]
- Brenda The Compensive Enzyme Information Sysytem. Available online: https://www.brenda-enzymes.org/enzyme.php?ecno=1.14.99.1 (accessed on 10 October 2022).
- Sankaranarayanan, R.; Kumar, D.R.; Altinoz, M.A.; Bhat, G.J. Mechanisms of colorectal cancer prevention by aspirin—a literature review and perspective on the role of COX-dependent and-independent pathways. Int. J. Mol. Sci. 2020, 21, 9018. [Google Scholar] [CrossRef] [PubMed]
- Zacharias-Millward, N.; Menter, D.G.; Davis, J.S.; Lichtenberger, L.; Hawke, D.; Hawk, E.; Vilar, E.; Bhattacharya, P.; Millward, S. Beyond COX-1: The effects of aspirin on platelet biology and potential mechanisms of chemoprevention. Cancer Metast. Rev. 2017, 36, 289–303. [Google Scholar]
- Fuster, V.; Sweeny, J.M. Aspirin: A historical and contemporary therapeutic overview. Circulation 2011, 123, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.; Selinsky, B.S.; Kaub, C.J.; Katz, A.K.; Loll, P.J. The 2.0 Å resolution crystal structure of prostaglandin H2 synthase-1: Structural insights into an unusual peroxidase. J. Mol. Biol. 2004, 335, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Garavito, R.M.; Malkowski, M.G.; DeWitt, D.L. The structures of prostaglandin endoperoxide H synthases-1 and-2. Prostaglandins Other Lipid Mediat. 2002, 68, 129–152. [Google Scholar] [CrossRef]
- Miciaccia, M.; Belviso, B.D.; Iaselli, M.; Cingolani, G.; Ferorelli, S.; Cappellari, M.; Loguercio Polosa, P.; Perrone, M.G.; Caliandro, R.; Scilimati, A. Three-dimensional structure of human cyclooxygenase (hCOX)-1. Sci. Rep. 2021, 11, 4312. [Google Scholar] [CrossRef]
- Orlando, B.J.; Malkowski, M.G. Substrate-selective inhibition of cyclooxygeanse-2 by fenamic acid derivatives is dependent on peroxide tone. J. Biol. Chem. 2016, 291, 15069–15081. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The CLASS study: A randomized controlled trial. JAMA 2000, 284, 1247–1255. [Google Scholar] [CrossRef]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Schnitzer, T.J. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Dequeker, J.; Hawkey, C.; Kahan, A.O.; Steinbrück, K.; Alegre, C.; Baumelou, E.; Begaud, B.; Isomäki, H.; Littlejohn, G.; Mau, J. Improvement in gastrointestinal tolerability of the selective cyclooxygenase (COX)-2 inhibitor, meloxicam, compared with piroxicam: Results of the Safety and Efficacy Large-scale Evaluation of COX-inhibiting Therapies (SELECT) trial in osteoarthritis. Br. J. Rheumatol. 1998, 37, 946–951. [Google Scholar] [CrossRef] [Green Version]
- Nowaczyk, A.; Szwedowski, D.; Dallo, I.; Nowaczyk, J. Overview of First-Line and Second-Line Pharmacotherapies for Osteoarthritis with Special Focus on Intra-Articular Treatment. IJMS 2022, 23, 1566. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.; Botting, R. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K. Aspirin and Other Cyclooxygenase Inhibitors: New Therapeutic Insights; Seminars in vascular medicine; Thieme Medical Publishers, Inc.: New York, NY, USA, 2003; pp. 107–112. [Google Scholar]
- Bianconi, V.; Violi, F.; Fallarino, F.; Pignatelli, P.; Sahebkar, A.; Pirro, M. Is acetylsalicylic acid a safe and potentially useful choice for adult patients with COVID-19? Drugs 2020, 80, 1383–1396. [Google Scholar] [CrossRef] [PubMed]
- Stolarek, W.; Kasprzak, M.; Sikora, J.; Siemińska, E.; Grześk, G. High on-treatment platelet reactivity to aspirin in patients after myocardial infarction. Biomed. Pharmacother. 2022, 147, 112618. [Google Scholar] [CrossRef] [PubMed]
- Original Aspirin. The Summary of Product Characteristic. pp. 129–152. Available online: https://www.medicines.org.uk/emc/product/13691/pil (accessed on 1 October 2022).
- Zarghi, A.; Arfaei, S. Selective COX-2 inhibitors: A review of their structure-activity relationships. IJPR 2011, 10, 655. [Google Scholar] [PubMed]
- Altman, R.; Luciardi, H.L.; Muntaner, J.; Herrera, R.N. The antithrombotic profile of aspirin. Aspirin resistance, or simply failure? Thromb. J. 2004, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Patrono, C. The multifaceted clinical readouts of platelet inhibition by low-dose aspirin. J. Am. Coll. Cardiol. 2015, 66, 74–85. [Google Scholar] [CrossRef]
- Meade, E.A.; Smith, W.L.; Dewitt, D.L. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1993, 268, 6610–6614. [Google Scholar] [CrossRef]
- Costello, P.B.; Green, F.A. Identification and partial purification of the major aspirin hydrolyzing enzyme in human blood. Arthritis Rheum. 1983, 26, 541–547. [Google Scholar] [CrossRef]
- Patrono, C.; Patrignani, P.; Rodríguez, L.A.G. Cyclooxygenase-selective inhibition of prostanoid formation: Transducing biochemical selectivity into clinical read-outs. J. Clin. Investig. 2001, 108, 7–13. [Google Scholar] [CrossRef]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Roy, A.; Kundu, M.; Jana, M.; Luan, C.-H.; Gonzalez, F.J.; Pahan, K. Aspirin binds to PPARα to stimulate hippocampal plasticity and protect memory. Proc. Natl. Acad. Sci. USA 2018, 115, E7408–E7417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clissold, S.P. Aspirin and related derivatives of salicylic acid. Drugs 1986, 32, 8–26. [Google Scholar] [CrossRef]
- Shishodia, S.; Aggarwal, B.B. Nuclear factor-κB activation: A question of life or death. BMB Rep. 2002, 35, 28–40. [Google Scholar] [CrossRef]
- Kutuk, O.; Basaga, H. Aspirin inhibits TNFα-and IL-1-induced NF-κB activation and sensitizes HeLa cells to apoptosis. Cytokine 2004, 25, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.H.; Lee, B.-J.; Tran, T.T. Current studies of aspirin as an anticancer agent and strategies to strengthen its therapeutic application in cancer. Curr. Pharm. Des. 2021, 27, 2209–2220. [Google Scholar] [CrossRef]
- Kim, J.; Lee, K.-S.; Kim, J.-H.; Lee, D.-K.; Park, M.; Choi, S.; Park, W.; Kim, S.; Choi, Y.K.; Hwang, J.Y. Aspirin prevents TNF-α-induced endothelial cell dysfunction by regulating the NF-κB-dependent miR-155/eNOS pathway: Role of a miR-155/eNOS axis in preeclampsia. Free Radic. Biol. Med. 2017, 104, 185–198. [Google Scholar] [CrossRef]
- Li, X.; Wu, S.; Yu, Y. Aspirin Use and the Incidence of Hepatocellular Carcinoma in Patients with Hepatitis B Virus or Hepatitis C Virus Infection: A Meta-Analysis of Cohort Studies. Front. Med. 2021, 7, 968. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, J.; Jiao, Y.; Chen, Q.; Li, M.; Wang, Z.; Yu, Z.; Huang, X.; Yao, A.; Gao, Q. Aspirin inhibits natural killer/T-cell lymphoma by modulation of VEGF expression and mitochondrial function. Front. Oncol. 2019, 8, 679. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Murphy, E.; Olaniyi, P.; Bryant, R. The multiple effects of aspirin in prostate cancer patients. Cancer Treat. Res. Commun. 2021, 26, 100267. [Google Scholar] [CrossRef]
- Patrono, C.; Baigent, C. Role of aspirin in primary prevention of cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 675–686. [Google Scholar] [CrossRef] [PubMed]
- RCSB Database. Available online: https://www.rcsb.org/ligand/AIN (accessed on 1 October 2022).
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Revi. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.; Mestres, J. Drug repurposing: Far beyond new targets for old drugs. AAPS J. 2012, 14, 759–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langedijk, J.; Mantel-Teeuwisse, A.K.; Slijkerman, D.S.; Schutjens, M.-H.D. Drug repositioning and repurposing: Terminology and definitions in literature. Drug Discov. Today 2015, 20, 1027–1034. [Google Scholar] [CrossRef]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018, 14, 1232. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Li, M.; Yang, M.; Wu, F.-X.; Li, Y.; Wang, J. Biomedical data and computational models for drug repositioning: A comprehensive review. Brief. Bioinform. 2021, 22, 1604–1619. [Google Scholar] [CrossRef]
- Sudeep, P.; Francesco, I.; Patrick, A.E.; Jane, E.K.; Shirley, H.; Andrew, W.; Andrew, D.; Tim, G.; Joanna, L.; Christine, M.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar]
- TRS-615-P; World Health Organization Technical Report Series 615. World Health Organization: Geneva, Switzerland, 1977; pp. 1–36.
- The WHO Model Lists of Essential Medicines and Essential Medicines for Children. Available online: https://list.essentialmeds.org/medicines/13 (accessed on 10 October 2022).
- Koellner, C.M.; Mensink, K.A.; Highsmith, W.E., Jr. Chapter 5-Basic concepts in human molecular genetics. In Essential Concepts in Molecular Pathology, 2nd ed.; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Jourdan, J.-P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Du, L.; Wang, S.; Kong, L.; Zhang, S.; Li, S.; Zhang, W.; Du, G. Differences in the prevention and control of cardiovascular and cerebrovascular diseases. Pharmacol. Res. 2021, 170, 105737. [Google Scholar] [CrossRef]
- Craven, L.L. Experiences with aspirin (Acetylsalicylic acid) in the nonspecific prophylaxis of coronary thrombosis. Miss Valley Med. J. 1953, 75, 38–44. [Google Scholar]
- Hennekens, C.H. Aspirin in the treatment and prevention of cardiovascular disease. Annu. Rev. Public Health 1997, 18, 37–49. [Google Scholar] [CrossRef]
- Stolarek, W.; Kasprzak, M.; Obońska, K.; Ostrowska, M.; Wiciński, M.; Kubica, A.; Kubica, J.; Grześk, G. Acetylsalicylic acid resistance risk factors in patients with myocardial infarction. Pharmacol. Rep. 2015, 67, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Aspirin for heart patients. FDA Drug Bull. 1985, 15, 34–36.
- Peto, R.; Gray, R.; Collins, R.; Wheatley, K.; Hennekens, C.; Jamrozik, K.; Warlow, C.; Hafner, B.; Thompson, E.; Norton, S. Randomised trial of prophylactic daily aspirin in British male doctors. Br. Med. J. (Clin. Res. Ed.) 1988, 296, 313–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steering Committee of the Physicians’ Health Study Research Group. Final report on the aspirin component of the ongoing Physicians’ Health Study. N. Engl. J. Med. 1989, 321, 129–135. [Google Scholar] [CrossRef]
- The Medical Research Council’s General Practice Research Framework Thrombosis prevention trial: Randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998, 351, 233–241.
- De Gaetano, G. Low-dose aspirin and vitamin E in people at cardiovascular risk: A randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 2001, 357, 89–95. [Google Scholar]
- Ridker, P.M.; Cook, N.R.; Lee, I.-M.; Gordon, D.; Gaziano, J.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N. Engl. J. Med. 2005, 352, 1293–1304. [Google Scholar] [CrossRef]
- Belch, J.; MacCuish, A.; Campbell, I.; Cobbe, S.; Taylor, R.; Prescott, R.; Lee, R.; Bancroft, J.; MacEwan, S.; Shepherd, J. The prevention of progression of arterial disease and diabetes (POPADAD) trial: Factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008, 337, a1840. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, H.; Nakayama, M.; Morimoto, T.; Uemura, S.; Kanauchi, M.; Doi, N.; Jinnouchi, H.; Sugiyama, S.; Saito, Y.; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: A randomized controlled trial. JAMA 2008, 300, 2134–2141. [Google Scholar] [CrossRef] [Green Version]
- Fowkes, F.G.R.; Price, J.F.; Stewart, M.C.; Butcher, I.; Leng, G.C.; Pell, A.C.; Sandercock, P.A.; Fox, K.A.; Lowe, G.D.; Murray, G.D. Aspirin for prevention of cardiovascular events in a general population screened for a low ankle brachial index: A randomized controlled trial. JAMA 2010, 303, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Shimada, K.; Teramoto, T.; Uchiyama, S.; Yamazaki, T.; Oikawa, S.; Sugawara, M.; Ando, K.; Murata, M.; Yokoyama, K. Low-dose aspirin for primary prevention of cardiovascular events in Japanese patients 60 years or older with atherosclerotic risk factors: A randomized clinical trial. JAMA 2014, 312, 2510–2520. [Google Scholar] [CrossRef] [PubMed]
- Raber, I.; McCarthy, C.P.; Vaduganathan, M.; Bhatt, D.L.; Wood, D.A.; Cleland, J.G.; Blumenthal, R.S.; McEvoy, J.W. The rise and fall of aspirin in the primary prevention of cardiovascular disease. Lancet 2019, 393, 2155–2167. [Google Scholar] [CrossRef] [PubMed]
- Eikelboom, J.W.; Connolly, S.J.; Bosch, J.; Dagenais, G.R.; Hart, R.G.; Shestakovska, O.; Diaz, R.; Alings, M.; Lonn, E.M.; Anand, S.S. Rivaroxaban with or without aspirin in stable cardiovascular disease. N. Engl. J. Med. 2017, 377, 1319–1330. [Google Scholar] [CrossRef]
- Zannad, F.; Anker, S.D.; Byra, W.M.; Cleland, J.G.; Fu, M.; Gheorghiade, M.; Lam, C.S.; Mehra, M.R.; Neaton, J.D.; Nessel, C.C. Rivaroxaban in patients with heart failure, sinus rhythm, and coronary disease. N. Engl. J. Med. 2018, 379, 1332–1342. [Google Scholar] [CrossRef]
- Bonaca, M.P.; Bauersachs, R.M.; Anand, S.S.; Debus, E.S.; Nehler, M.R.; Patel, M.R.; Fanelli, F.; Capell, W.H.; Diao, L.; Jaeger, N. Rivaroxaban in peripheral artery disease after revascularization. N. Engl. J. Med. 2020, 382, 1994–2004. [Google Scholar] [CrossRef]
- Krantz, M.J.; Debus, S.E.; Hsia, J.; Patel, M.R.; Anand, S.S.; Nehler, M.R.; Hess, C.N.; Capell, W.H.; Bracken, T.; Szarek, M. Low-dose rivaroxaban plus aspirin in older patients with peripheral artery disease undergoing acute limb revascularization: Insights from the VOYAGER PAD trial. Eur. Heart J. 2021, 42, 4040–4048. [Google Scholar] [CrossRef]
- Collaboration, A.T. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002, 324, 71–86. [Google Scholar] [CrossRef] [Green Version]
- Ten Cate, H.; Guzik, T.J.; Eikelboom, J.; Spronk, H.M. Pleiotropic actions of factor Xa inhibition in cardiovascular prevention: Mechanistic insights and implications for anti-thrombotic treatment. Cardiovasc. Res. 2021, 117, 2030–2044. [Google Scholar] [CrossRef]
- Hansson, L.; Zanchetti, A.; Carruthers, S.G.; Dahlöf, B.; Elmfeldt, D.; Julius, S.; Ménard, J.; Rahn, K.H.; Wedel, H.; Westerling, S. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: Principal results of the Hypertension Optimal Treatment (HOT) randomised trial. Lancet 1998, 351, 1755–1762. [Google Scholar] [CrossRef]
- Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J. Effects of aspirin for primary prevention in persons with diabetes mellitus: The ASCEND Study Collaborative Group. J. Vasc. Surg. 2019, 69, 305. [Google Scholar] [CrossRef] [Green Version]
- Kernan, W.N.; Ovbiagele, B.; Black, H.R.; Bravata, D.M.; Chimowitz, M.I.; Ezekowitz, M.D.; Fang, M.C.; Fisher, M.; Furie, K.L.; Heck, D.V. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014, 45, 2160–2236. [Google Scholar] [CrossRef] [PubMed]
- Abdelaziz, H.K.; Saad, M.; Pothineni, N.V.K.; Megaly, M.; Potluri, R.; Saleh, M.; Kon, D.L.C.; Roberts, D.H.; Bhatt, D.L.; Aronow, H.D. Aspirin for primary prevention of cardiovascular events. J. Am. Coll. Cardiol. 2019, 73, 2915–2929. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Okada, S.; Ogawa, H.; Soejima, H.; Sakuma, M.; Nakayama, M.; Doi, N.; Jinnouchi, H.; Waki, M.; Masuda, I. Low-dose aspirin for primary prevention of cardiovascular events in patients with type 2 diabetes mellitus: 10-year follow-up of a randomized controlled trial. Circulation 2017, 135, 659–670. [Google Scholar] [CrossRef]
- Gaziano, J.M.; Brotons, C.; Coppolecchia, R.; Cricelli, C.; Darius, H.; Gorelick, P.B.; Howard, G.; Pearson, T.A.; Rothwell, P.M.; Ruilope, L.M. Use of aspirin to reduce risk of initial vascular events in patients at moderate risk of cardiovascular disease (ARRIVE): A randomised, double-blind, placebo-controlled trial. Lancet 2018, 392, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- McNeil, J.J.; Nelson, M.R.; Woods, R.L.; Lockery, J.E.; Wolfe, R.; Reid, C.M.; Kirpach, B.; Shah, R.C.; Ives, D.G.; Storey, E. Effect of aspirin on all-cause mortality in the healthy elderly. N. Engl. J. Med. 2018, 379, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- McNeil, J.J.; Wolfe, R.; Woods, R.L.; Tonkin, A.M.; Donnan, G.A.; Nelson, M.R.; Reid, C.M.; Lockery, J.E.; Kirpach, B.; Storey, E. Effect of aspirin on cardiovascular events and bleeding in the healthy elderly. N. Engl. J. Med. 2018, 379, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- PROSPERO 2018 CRD42018115612. Available online: https://www.crd.york.ac.uk/PROSPERO/display_record.php?RecordID=115612 (accessed on 10 October 2022).
- Singal, A.K.; Karthikeyan, G. Aspirin for primary prevention: Is this the end of the road? Indian Heart J. 2019, 71, 113–117. [Google Scholar] [CrossRef]
- Members, W.C.; Otto, C.M.; Nishimura, R.A.; Bonow, R.O.; Carabello, B.A.; Erwin III, J.P.; Gentile, F.; Jneid, H.; Krieger, E.V.; Mack, M. 2020 ACC/AHA guideline for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2021, 77, e25–e197. [Google Scholar]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 74, e177–e232. [Google Scholar] [CrossRef]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B. 2018 guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2018, 49, e46–e99. [Google Scholar] [CrossRef]
- Cappelaere, P.; Adenis, L.; Driessens, J. Value of injectable aspirin in cancerology. Lille Med. 1971, 17, 566–571. [Google Scholar]
- Thorat, M.A.; Cuzick, J. Role of aspirin in cancer prevention. Curr. Oncol. Rep. 2013, 15, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Patrono, C. Aspirin, platelet inhibition and cancer prevention. Platelets 2018, 29, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhong, X.; Gao, P.; Zhou, C.; Shi, J.; Wu, Z.; Guo, Z.; Wang, Z. Aspirin and its potential preventive role in cancer: An umbrella review. Front. Endocrinol. 2020, 11, 3. [Google Scholar] [CrossRef]
- Patrignani, P.; Patrono, C. Aspirin and cancer. J. Am. Coll. Cardiol. 2016, 68, 967–976. [Google Scholar] [CrossRef]
- Joharatnam-Hogan, N.; Cafferty, F.; Hubner, R.; Swinson, D.; Sothi, S.; Gupta, K.; Falk, S.; Patel, K.; Warner, N.; Kunene, V. Aspirin as an adjuvant treatment for cancer: Feasibility results from the Add-Aspirin randomised trial. Lancet Gastroenterol. Hepatol. 2019, 4, 854–862. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.; Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y. Complex roles of the old drug aspirin in cancer chemoprevention and therapy. Med. Res. Rev. 2019, 39, 114–145. [Google Scholar] [CrossRef] [Green Version]
- Qorri, B.; Harless, W.; Szewczuk, M.R. Novel Molecular Mechanism of Aspirin and Celecoxib Targeting Mammalian Neuraminidase-1 Impedes Epidermal Growth Factor Receptor Signaling Axis and Induces Apoptosis in Pancreatic Cancer Cells. Drug Des. Dev. Ther. 2020, 14, 4149. [Google Scholar] [CrossRef]
- Kolawole, O.R.; Kashfi, K. NSAIDs and Cancer Resolution: New Paradigms beyond Cyclooxygenase. Int. J. Mol. Sci. 2022, 23, 1432. [Google Scholar] [CrossRef]
- Baron, J.A.; Cole, B.F.; Sandler, R.S.; Haile, R.W.; Ahnen, D.; Bresalier, R.; McKeown-Eyssen, G.; Summers, R.W.; Rothstein, R.; Burke, C.A. A randomized trial of aspirin to prevent colorectal adenomas. N. Engl. J. Med. 2003, 348, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.; Halton, D.E.; Hague, A.; Paraskeva, C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: Independence from COX-2 protein expression. Clin. Cancer Res. 1997, 3, 1679–1683. [Google Scholar] [PubMed]
- Baandrup, L.; Kjaer, S.; Olsen, J.; Dehlendorff, C.; Friis, S. Low-dose aspirin use and the risk of ovarian cancer in Denmark. Ann. Oncol. 2015, 26, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Shebl, F.; Sakoda, L.; Black, A.; Koshiol, J.; Andriole, G.; Grubb, R.; Church, T.; Chia, D.; Zhou, C.; Chu, L. Aspirin but not ibuprofen use is associated with reduced risk of prostate cancer: A PLCO study. Br. J. Cancer 2012, 107, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takiuchi, T.; Blake, E.A.; Matsuo, K.; Sood, A.K.; Brasky, T.M. Aspirin use and endometrial cancer risk and survival. Gynecol. Oncol. 2018, 148, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabuddhe, V.V.; Gunja, M.Z.; Graubard, B.I.; Trabert, B.; Schwartz, L.M.; Park, Y.; Hollenbeck, A.R.; Freedman, N.D.; McGlynn, K.A. Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. JNCI 2012, 104, 1808–1814. [Google Scholar] [CrossRef] [Green Version]
- Clouser, M.C.; Roe, D.J.; Foote, J.A.; Harris, R.B. Effect of non-steroidal anti-inflammatory drugs on non-melanoma skin cancer incidence in the SKICAP-AK trial. Pharmacoepidemiol. Drug Saf. 2009, 18, 276–283. [Google Scholar] [CrossRef]
- Sun, L.; Yu, S. Meta-analysis: Non-steroidal anti-inflammatory drug use and the risk of esophageal squamous cell carcinoma. Dis. Esophagus 2011, 24, 544–549. [Google Scholar] [CrossRef]
- Cui, X.-J.; He, Q.; Zhang, J.-M.; Fan, H.-J.; Wen, Z.-F.; Qin, Y.-R. High-dose aspirin consumption contributes to decreased risk for pancreatic cancer in a systematic review and meta-analysis. Pancreas 2014, 43, 135–140. [Google Scholar] [CrossRef]
- de Pedro, M.; Baeza, S.; Escudero, M.-T.; Dierssen-Sotos, T.; Gómez-Acebo, I.; Pollán, M.; Llorca, J. Effect of COX-2 inhibitors and other non-steroidal inflammatory drugs on breast cancer risk: A meta-analysis. Breast Cancer Res. Treat. 2015, 149, 525–536. [Google Scholar] [CrossRef]
- Yiannakopoulou, E.C. Aspirin and NSAIDs for breast cancer chemoprevention. Eur. J. Cancer Prev. 2015, 24, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Holmes, M.D. Role of aspirin in breast cancer survival. Curr. Oncol. Rep. 2017, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, S.E.; Pfeiffer, R.M.; Sigurdson, A.J.; Hayes, R.B.; Leitzmann, M.; Schatzkin, A.; Hollenbeck, A.R.; Silverman, D.T. Nonsteroidal antiinflammatory drugs and bladder cancer: A pooled analysis. Am. J. Epidemiol. 2011, 173, 721–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, C.; Wilson, J.C.; Jick, S.S.; Meier, C.R. Non-steroidal anti-inflammatory drugs and the risk of head and neck cancer: A case-control analysis. Int. J. Cancer 2015, 137, 2424–2431. [Google Scholar] [CrossRef] [Green Version]
- Elwood, P.C.; Gallagher, A.M.; Duthie, G.G.; Mur, L.A.; Morgan, G. Aspirin, salicylates, and cancer. Lancet 2009, 373, 1301–1309. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.R.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Algra, A.M.; Rothwell, P.M. Effects of regular aspirin on long-term cancer incidence and metastasis: A systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012, 13, 518–527. [Google Scholar] [CrossRef]
- Elwood, P.C.; Pickering, J.E.; Morgan, G.; Galante, J.; Weightman, A.L.; Morris, D.; Longley, M.; Mason, M.; Adams, R.; Dolwani, S. Systematic review update of observational studies further supports aspirin role in cancer treatment: Time to share evidence and decision-making with patients? PLoS ONE 2018, 13, e0203957. [Google Scholar] [CrossRef] [Green Version]
- Stark, L.; Din, F.; Zwacka, R.; Dunlop, M. Aspirin-induced activation of the NF-κB signaling pathway: A novel mechanism for aspirin-mediated apoptosis in colon cancer cells. FASEB J. 2001, 15, 1273–1275. [Google Scholar] [CrossRef]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [Green Version]
- Park, A.; Lee, Y.; Kim, M.S.; Kang, Y.J.; Park, Y.-J.; Jung, H.; Kim, T.-D.; Lee, H.G.; Choi, I.; Yoon, S.R. Prostaglandin E2 secreted by thyroid cancer cells contributes to immune escape through the suppression of natural killer (NK) cell cytotoxicity and NK cell differentiation. Front. Immunol. 2018, 9, 1859. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gaynor, R.B. Role of the NF-kB pathway in the pathogenesis of human disease states. Curr. Mol. Med. 2001, 1, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Foletto, V.S.; da Rosa, T.F.; Serafin, M.B.; Bottega, A.; Hörner, R. Repositioning of non-antibiotic drugs as an alternative to microbial resistance: A systematic review. Int. J. Antimicrob. Agents 2021, 58, 106380. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.T.; Denkin, S.M.; Zhang, Y. Aspirin and ibuprofen enhance pyrazinamide treatment of murine tuberculosis. J. Antimicrob. Chemother. 2007, 59, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.R. Mitigating antimicrobial resistance with aspirin (acetylsalicylic acid) and paracetamol (acetaminophen): Conversion of doxycycline and minocycline resistant bacteria into sensitive in presence of aspirin and paracetamol. bioRxiv 2021. [CrossRef]
- Launer, L.J. Nonsteroidal anti-inflammatory drug use and the risk for Alzheimer’s disease. Drugs 2003, 63, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Westwell-Roper, C.; Stewart, S.E. Commentary: Neurobiology and therapeutic potential of cyclooxygenase-2 (COX-2) inhibitors for inflammation in neuropsychiatric disorders. Front. Psychiatry 2020, 11, 264. [Google Scholar] [CrossRef] [Green Version]
- Hirohata, M.; Ono, K.; Naiki, H.; Yamada, M. Non-steroidal anti-inflammatory drugs have anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. Neuropharmacology 2005, 49, 1088–1099. [Google Scholar] [CrossRef] [Green Version]
- Yagami, T.; Koma, H.; Yamamoto, Y. Pathophysiological roles of cyclooxygenases and prostaglandins in the central nervous system. Mol. Neurobiol. 2016, 53, 4754–4771. [Google Scholar] [CrossRef]
- Sethi, R.; Gómez-Coronado, N.; Walker, A.J.; Robertson, O.D.A.; Agustini, B.; Berk, M.; Dodd, S. Neurobiology and therapeutic potential of cyclooxygenase-2 (COX-2) inhibitors for inflammation in neuropsychiatric disorders. Front. Psychiatry 2019, 10, 605. [Google Scholar] [CrossRef] [Green Version]
- Ajmone-Cat, M.A.; Bernardo, A.; Greco, A.; Minghetti, L. Non-steroidal anti-inflammatory drugs and brain inflammation: Effects on microglial functions. Pharmaceuticals 2010, 3, 1949–1965. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, I.G.; Machado-Vieira, R.; Soares, J.C.; Teixeira, A.L. The immunology of bipolar disorder. Neuroimmunomodulation 2014, 21, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarada, T.N.; Rokne, J.G.; Alhajj, R. A review of computational drug repositioning: Strategies, approaches, opportunities, challenges, and directions. J. Cheminform. 2020, 12, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Rameshrad, M.; Ghafoori, M.; Mohammadpour, A.H.; Nayeri, M.J.D.; Hosseinzadeh, H. A comprehensive review on drug repositioning against coronavirus disease 2019 (COVID19). Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Prasher, P.; Sharma, M.; Gunupuru, R. Targeting cyclooxygenase enzyme for the adjuvant COVID-19 therapy. Drug Dev. Res. 2021, 82, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Wijaya, I.; Andhika, R.; Huang, I.; Purwiga, A.; Budiman, K.Y. The effects of aspirin on the outcome of COVID-19: A systematic review and meta-analysis. Clin. Epidemiol. Glob. Health 2021, 12, 100883. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Karl, N.; Ziebuhr, J.; Pleschka, S. L-lysine acetylsalicylate+ glycine impairs coronavirus replication. J. Antivir. Antiretrovir. 2016, 8, 142–150. [Google Scholar] [CrossRef]
- Chow, J.H.; Khanna, A.K.; Kethireddy, S.; Yamane, D.; Levine, A.; Jackson, A.M.; McCurdy, M.T.; Tabatabai, A.; Kumar, G.; Park, P. Aspirin use is associated with decreased mechanical ventilation, intensive care unit admission, and in-hospital mortality in hospitalized patients with coronavirus disease 2019. Anesth. Analg. 2021, 132, 930–941. [Google Scholar] [CrossRef]
- Sahai, A.; Bhandari, R.; Koupenova, M.; Freedman, J.E.; Godwin, M.; McIntyre, T.; Chung, M.K.; Iskandar, J.-P.; Kamran, H.; Hariri, E. SARS-CoV-2 receptors are expressed on human platelets and the effect of aspirin on clinical outcomes in COVID-19 patients. Res. Square 2020, preprint. [Google Scholar] [CrossRef]
- Mohamed-Hussein, A.A.; Aly, K.M.; Ibrahim, M.-E.A. Should aspirin be used for prophylaxis of COVID-19-induced coagulopathy? Med. Hypotheses 2020, 144, 109975. [Google Scholar] [CrossRef]
- Grześk, G.; Kozinski, M.; Tantry, U.S.; Wicinski, M.; Fabiszak, T.; Navarese, E.P.; Grzesk, E.; Jeong, Y.-H.; Gurbel, P.A.; Kubica, J. High-dose, but not low-dose, aspirin impairs anticontractile effect of ticagrelor following ADP stimulation in rat tail artery smooth muscle cells. BioMed Res. Int. 2013, 2013, 928271. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Brummel-Ziedins, K.E.; Mann, K.G. Antithrombotic properties of aspirin and resistance to aspirin: Beyond strictly antiplatelet actions. Blood 2007, 109, 2285–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Farre, A.; Caramelo, C.; Esteban, A.; Alberola, M.; Millas, I.; Monton, M.; Casado, S. Effects of aspirin on platelet-neutrophil interactions: Role of nitric oxide and endothelin-1. Circulation 1995, 91, 2080–2088. [Google Scholar] [CrossRef]
- Dovizio, M.; Tacconelli, S.; Sostres, C.; Ricciotti, E.; Patrignani, P. Mechanistic and pharmacological issues of aspirin as an anticancer agent. Pharmaceuticals 2012, 5, 1346–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, G.; Dachineni, R.; Kumar, D.R.; Marimuthu, S.; Alfonso, L.F.; Bhat, G.J. Aspirin acetylates wild type and mutant p53 in colon cancer cells: Identification of aspirin acetylated sites on recombinant p53. Tumor Biol. 2016, 37, 6007–6016. [Google Scholar] [CrossRef]
- Ai, G.; Dachineni, R.; Kumar, D.R.; Alfonso, L.F.; Marimuthu, S.; Bhat, G.J. Aspirin inhibits glucose-6-phosphate dehydrogenase activity in HCT 116 cells through acetylation: Identification of aspirin-acetylated sites. Mol. Med. Rep. 2016, 14, 1726–1732. [Google Scholar] [CrossRef] [Green Version]
- Ai, G.; Dachineni, R.; Muley, P.; Tummala, H.; Bhat, G.J. Aspirin and salicylic acid decrease c-Myc expression in cancer cells: A potential role in chemoprevention. Tumor Biol. 2016, 37, 1727–1738. [Google Scholar] [CrossRef]
- Bos, C.L.; Kodach, L.L.; van den Brink, G.R.; Diks, S.H.; van Santen, M.M.; Richel, D.J.; Peppelenbosch, M.P.; Hardwick, J.C. Effect of aspirin on the Wnt/β-catenin pathway is mediated via protein phosphatase 2A. Oncogene 2006, 25, 6447–6456. [Google Scholar] [CrossRef] [Green Version]
- Szczeklik, A.; Krzanowski, M.; Gora, P.; Radwan, J. Antiplatelet drugs and generation of thrombin in clotting blood. Blood 1992, 80, 2006–2011. [Google Scholar] [CrossRef] [Green Version]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515.e3. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.; Huang, Y.-W.; Oshima, K.; Yearsley, M.; Zhang, J.; Yu, J.; Arnold, M.; Wang, L.-S. Could aspirin and diets high in fiber act synergistically to reduce the risk of colon cancer in humans? Int. J. Mol. Sci. 2018, 19, 166. [Google Scholar] [CrossRef] [Green Version]
- Babbar, N.; Gerner, E.W.; Casero, R.A., Jr. Induction of spermidine/spermine N 1-acetyltransferase (SSAT) by aspirin in Caco-2 colon cancer cells. Biochem. J. 2006, 394, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, H.; Aggarwal, S. Salicylic acid (Aspirin). Available online: https://europepmc.org/article/NBK/nbk519032 (accessed on 1 October 2022).
- Alhamdany, H.F.; Alfahad, M.A. Transdermal patch of acetylsalicylic acid as an alternative dosage form: A review. Iraqi J. Pharm. 2022, 18, 64–72. [Google Scholar] [CrossRef]
- Watkinson, A.C.; Kearney, M.-C.; Quinn, H.L.; Courtenay, A.J.; Donnelly, R.F. Future of the transdermal drug delivery market–have we barely touched the surface? Expert Opin. Drug Deliv. 2016, 13, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gong, M.; Huang, Z.; Wang, F.; Wang, Y.; Hu, Z.; Zeng, Z.; Wang, Y. Alleviating Aspirin-Induced Gastric Injury by Binding Aspirin to β-Lactoglobulin. Drug Des. Dev. Ther. 2022, 16, 571. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Pollack, C.V., Jr. The Future of Aspirin Therapy in Cardiovascular Disease. Am. J. Cardiol. 2021, 144, S40–S47. [Google Scholar] [CrossRef]
- Bais, N.; Birthare, A.; Choudhary, G.; Darwhekar, G. Prophylaxis and Prevention of Heart Attack by Pulmonary Delivery of Aspirin by Dry Powder Inhaler. Int. J. Pharm. Life Sci. 2016, 7, 5047–5050. [Google Scholar]
- Chime, S.A.; Akpa, P.A.; Ugwuanyi, C.C.; Attama, A.A. Anti-inflammatory and gastroprotective properties of aspirin-entrapped solid lipid microparticles. Recent Pat. Inflamm. Allergy Drug Discov. 2020, 14, 78–88. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Bliden, K.P.; Chaudhary, R.; Tantry, U.S. First in-human experience with inhaled acetylsalicylic acid for immediate platelet inhibition: Comparison with chewed and swallowed acetylsalicylic acid. Circulation 2020, 142, 1305–1307. [Google Scholar] [CrossRef]
- Aspirin Suppositories 150 mg. The Summary of Product Characteristic. Available online: https://www.medicines.org.uk/emc/product/2404/smpc (accessed on 1 October 2022).
- Aspirin Suppositories 300 mg. The Summary of Product Characteristic. Available online: https://www.medicines.org.uk/emc/product/2405/smpc (accessed on 1 October 2022).
- Aspirin 300 mg Tablets. The Summary of Product Characteristic. Available online: https://www.medicines.org.uk/emc/product/11864/smpc (accessed on 1 October 2022).
- Ali, H.S.; Suliman, R.S.; Elhaj, B.M.; Suliman, R. The effect of modified release dosage forms on absorption of medications. Int. J. Pharm. Clin. Res. 2019, 11, 30–33. [Google Scholar]
- Kedir, H.M.; Sisay, E.A.; Abiye, A.A. Enteric-coated aspirin and the risk of gastrointestinal side effects: A systematic review. Int. J. Gen. Med. 2021, 14, 4757. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Prats, J.; Deliargyris, E.N.; Schneider, D.J.; Scheiman, J.; Kimmelstiel, C.; Steg, P.; Alberts, M.; Rosengart, T.; Mehran, R. Pharmacokinetic and Pharmacodynamic Profile of a Novel Phospholipid Aspirin Formulation. Clin. Pharmacokinet. 2022, 61, 465–479. [Google Scholar] [CrossRef] [PubMed]
- 209. Aspirin 75 mg Gastro-Resistant Tablets. The Summary of Product Characteristic. Available online: https://www.medicines.org.uk/emc/product/2614/smpc (accessed on 1 October 2022).
- Felton, L.A.; Porter, S.C. An update on pharmaceutical film coating for drug delivery. Expert Opin. Drug Deliv. 2013, 10, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Awtry, E.H.; Loscalzo, J. Aspirin. Circulation 2000, 101, 1206–1218. [Google Scholar] [CrossRef]
- Dasa, O.; Pepine, C.J.; Pearson, T.A. Aspirin in primary prevention: What changed? A critical appraisal of current evidence. Am. J. Cardiol. 2021, 141, 38–48. [Google Scholar] [CrossRef]
- Gupta, P.; Jain, M.; Sharma, S.; Vidhyadhari, A.; Singh, S.; Sharma, D. Medicated Chewing Gum–A Modernistic Drug Delivery System. Int. J. Pharm. Life Sci. 2019, 10, 6319–6330. [Google Scholar]
- John, A.; Kumar, K.K.; Dinesh Kumar, B. Medicated chewing gum: Modern drug delivery system. UJPSR 2019, 5, 29–39. [Google Scholar]
- ARE, L.W.S. Aspirin: 4000 years and still learning. Clevel. Clin. J. Med. 2019, 86, 523. [Google Scholar]
- Sequeira, I.R. Higher doses of ascorbic acid may have the potential to promote nutrient delivery via intestinal paracellular absorption. World J. Gastroenterol. 2021, 27, 6750. [Google Scholar] [CrossRef]
- ASPIRIN C. Patient Information Leaflet. Available online: https://www.aspirin.me/sites/g/files/vrxlpx16276/files/2021-02/Aspirin-C-en.pdf (accessed on 1 October 2022).
- Aspirin 300mg Dispersible tablet. The Summary of Product Characteristic. Available online: https://www.medicines.org.uk/emc/product/1910/pil (accessed on 1 October 2022).
- Schick, P.; Sager, M.; Voelker, M.; Weitschies, W.; Koziolek, M. Application of the GastroDuo to study the interplay of drug release and gastric emptying in case of immediate release Aspirin formulations. Eur. J. Pharm. Biopharm. 2020, 151, 9–17. [Google Scholar] [CrossRef]
- Kayesh, R.; Bhuiya, M.; Islam, M.; Chowdhury, J. Quality-by-Design Approach and Optimization of Risk Factors by Box-Behnken Design in Formulation Development of Aspirin and Glycine Orally Disintegrating Tablet. J. Sci. Res. 2021, 13, 935–950. [Google Scholar] [CrossRef]
- Cooper, S.; Voelker, M. Evaluation of onset of pain relief from micronized aspirin in a dental pain model. Inflammopharmacology 2012, 20, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kweekel-DeVries, W.; Spierdijk, J.; Mattie, H.; Hermans, J. A new soluble acetylsalicylic acid derivative in the treatment of postoperative pain. Br. J. Anaesth. 1974, 46, 133–135. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.; Cashman, J.; Foster, M.; Wedley, J.; Adams, A. Comparison of infusions of morphine and lysine acetyl salicylate for the relief of pain following thoracic surgery. BJA Br. J. Anaesth. 1985, 57, 259–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivas, D.; Martín, A.; Bernardo, E.; Ortega-Pozzi, M.A.; Tirado, G.; Fernández, C.; Vilacosta, I.; Núñez-Gil, I.; Macaya, C.; Fernández-Ortiz, A. Impact of Intravenous Lysine Acetylsalicylate Versus Oral Aspirin on Prasugrel-Inhibited Platelets: Results of a Prospective, Randomized, Crossover Study (the ECCLIPSE Trial). Results of a Prospective, Randomized, Crossover Study (the ECCLIPSE Trial). Circ. Cardiovasc. Interv. 2015, 8, e002281. [Google Scholar] [CrossRef] [Green Version]
- McAteer, E.; Dundee, J. Injectable aspirin as a postoperative analgesic. BJA Br. J. Anaesth. 1981, 53, 1069–1071. [Google Scholar] [CrossRef] [Green Version]
- Dinis-Oliveira, R.J.; Pontes, H.; Bastos, M.L.; Remiao, F.; Duarte, J.A.; Carvalho, F. An effective antidote for paraquat poisonings: The treatment with lysine acetylsalicylate. Toxicology 2009, 255, 187–193. [Google Scholar] [CrossRef]
- Bretagne, J.; Feuillu, A.; Gosselin, M.; Gastard, J. Aspirin and gastroduodenal toxicity. A double-blind endoscopic study of the effects of placebo, aspirin and lysine acetylsalicylate in healthy subjects. Gastroen. Clin. Biol. 1984, 8, 28–32. [Google Scholar]
- Majluf-Cruz, A.; Chavez-Ochoa, A.R.d.; Majluf-Cruz, K.; Coria-Ramirez, E.; Pineda Del Aguila, I.; Treviño-Perez, S.; Matías-Aguilar, L.; López-Armenta, J.C.; Corona de la Peña, N. Effect of combined administration of clopidogrel and lysine acetylsalicylate versus clopidogrel and aspirin on platelet aggregation and activated GPIIb/IIIa expression in healthy volunteers. Platelets 2006, 17, 105–107. [Google Scholar] [CrossRef]
- Weatherall, M.; Telzerow, A.; Cittadini, E.; Kaube, H.; Goadsby, P. Intravenous aspirin (lysine acetylsalicylate) in the inpatient management of headache. Neurology 2010, 75, 1098–1103. [Google Scholar] [CrossRef]
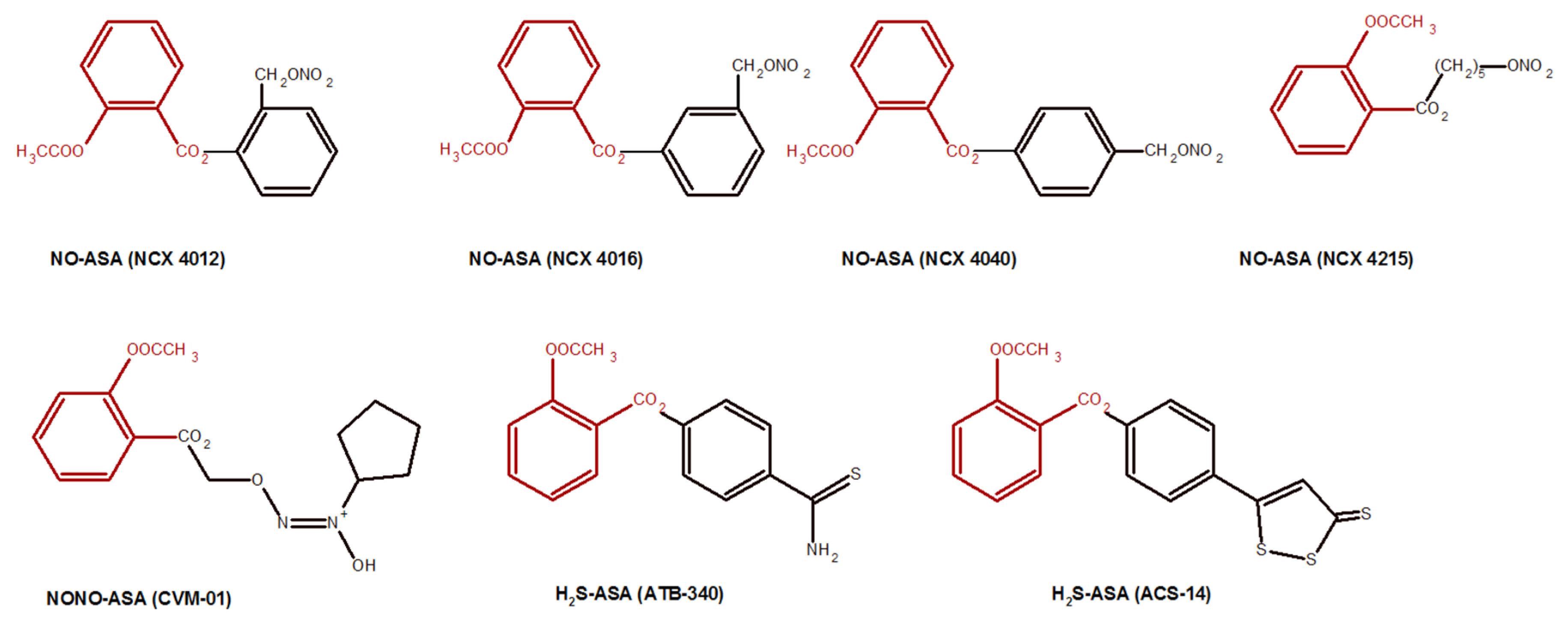
- Fiorucci, S.; Santucci, L.; Gresele, P.; Faccino, R.M.; Del Soldato, P.; Morelli, A. Gastrointestinal safety of NO-aspirin (NCX-4016) in healthy human volunteers: A proof of concept endoscopic study. Gastroenterology 2003, 124, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Velazquez, C.A.; Pruski, A.; Nia, K.V.; Abdelatif, K.; Keefer, L.K.; Kashfi, K. Comparison between NO-ASA and NONO-ASA as safe anti-inflammatory, analgesic, antipyretic, anti-oxidant, prodrugs. J. Pharmacol. Exp. Ther. 2010, 33, 171017. [Google Scholar] [CrossRef] [Green Version]
- Magli, E.; Perissutti, E.; Santagada, V.; Caliendo, G.; Corvino, A.; Esposito, G.; Esposito, G.; Fiorino, F.; Migliaccio, M.; Scognamiglio, A. H2S donors and their use in medicinal chemistry. Biomolecules 2021, 11, 1899. [Google Scholar] [CrossRef] [PubMed]
- Nowaczyk, A.; Kowalska, M.; Nowaczyk, J.; Grześk, G. Carbon monoxide and nitric oxide as the examples of the youngest class of transmitters. Int. J. Mol. Sci. 2021, 22, 6029. [Google Scholar] [CrossRef]
- Kashfi, K.; Rigas, B. The mechanism of action of nitric oxide-donating aspirin. Biochem. Bioph. Res. Commun. 2007, 358, 1096–1101. [Google Scholar] [CrossRef]
- Gao, J.; Liu, X.; Rigas, B. Nitric oxide-donating aspirin induces apoptosis in human colon cancer cells through induction of oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 17207–17212. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, K. Insight into the biological activity of organometallic acetylsalicylic acid (aspirin) derivatives. ChemPlusChem 2019, 84, 403–415. [Google Scholar] [CrossRef]
- Mandal, P.; Kundu, B.K.; Vyas, K.; Sabu, V.; Helen, A.; Dhankhar, S.S.; Nagaraja, C.; Bhattacherjee, D.; Bhabak, K.P.; Mukhopadhyay, S. Ruthenium (ii) arene NSAID complexes: Inhibition of cyclooxygenase and antiproliferative activity against cancer cell lines. Dalton Trans. 2018, 47, 517–527. [Google Scholar] [CrossRef]
- Ott, I.; Kircher, B.; Bagowski, C.P.; Vlecken, D.H.; Ott, E.B.; Will, J.; Bensdorf, K.; Sheldrick, W.S.; Gust, R. Modulation of the biological properties of aspirin by formation of a bioorganometallic derivative. Angew. Chem. Int. Ed. 2009, 48, 1160–1163. [Google Scholar] [CrossRef]
- Lu, S.; Obianom, O.N.; Ai, Y. Novel cinnamaldehyde-based aspirin derivatives for the treatment of colorectal cancer. Bioorg. Med. Chem. Lett. 2018, 28, 2869–2874. [Google Scholar] [CrossRef]
- Uzzaman, M.; Mahmud, T. Structural modification of aspirin to design a new potential cyclooxygenase (COX-2) inhibitors. In Silico Pharmacol. 2020, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ngaini, Z.; Mortadza, N.A. Synthesis of halogenated azo-aspirin analogues from natural product derivatives as the potential antibacterial agents. Nat. Prod. Res. 2019, 33, 3507–3514. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhang, Y.; Wang, Z.; Zhang, S.; Li, Y.; Fan, Y.; Li, D.; Li, S.; Bai, Y. Preparation of novel anthraquinone-based aspirin derivatives with anti-cancer activity. Eur. J. Pharmacol. 2021, 900, 174020. [Google Scholar] [CrossRef] [PubMed]
- Rosetti, M.; Tesei, A.; Ulivi, P.; Fabbri, F.; Vannini, I.; Brigliadori, G.; Amadori, D.; Bolla, M.; Zoli, W. Molecular characterization of cytotoxic and resistance mechanisms induced by NCX 4040, a novel NO-NSAID, in pancreatic cancer cell lines. Apoptosis 2006, 11, 1321–1330. [Google Scholar] [CrossRef]
- El-Ashmawy, I.M.; Ebeid, M.A.; Aljohani, M.S.; Alhumaydhi, F.A.; Aljohani, A.S. Assessment of the hepatorenal and hematological parameters of rats exposed to graded doses of lysine acetylsalicylate. J. Taibah Univ. Sci. 2022, 16, 214–223. [Google Scholar] [CrossRef]








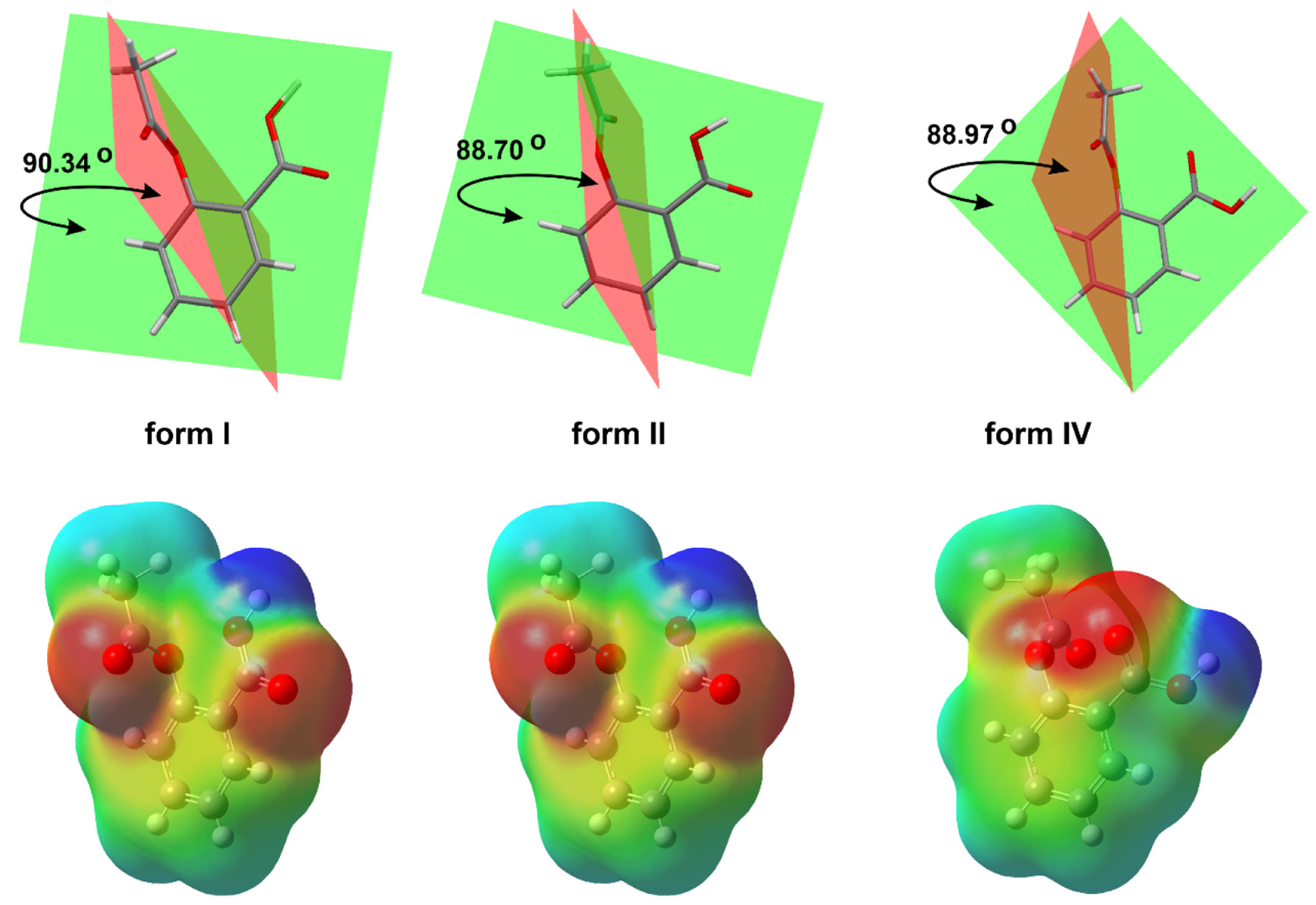{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Solubility of ASA at 298 K, g/100 g [a] | Solubility of SA at 298 K, g/100 g | pKa | IC50 * | ||
|---|---|---|---|---|---|---|
| COX-1 | COX-2 | |||||
| H2O | 0.46 [d,e] | 0.22 [f] | ASA | 3.5 | 1.67 [b] | 278 [b] |
| EtOH | 20 | 32.54 [f] | SA | 3 | >100.00 [c] | 14.08 [c] |
| n-C8H17OH | 3 | 1.25 [g] | paracetamol | 9.7 | 42.23 [c] | 10.69[c] |
| MeOH | 33 | 38.46 [h] | ibuprofen | 4.4 | 5.9 [c] | 9.9 [c] |
| CO(CH3)2 | 29 | 33.33 [h] | diclofenac | 3.9 | 0.26 [c] | 0.01 [c] |
| AcOH | 12 | 7.59 [h] | ketoprofen | 3.9 | 0.11 [c] | 0.88 [c] |
| CHCl3 | 6 | 2.22 [h] | piroxicam | 6.3 | 2.68 [c] | 2.11 [c] |
| O(Et)2 | 5 | 32.82 * [h] | celecoxib | 11.1 | 15 [b] | 0.04 [b] |
| Drug Targets Name | Action Type |
|---|---|
| Prostaglandin G/H synthase 1, Prostaglandin G/H synthase 2, Aldo-keto reductase family 1 member C1, Endothelin-1 receptor, Ribosomal protein S6 kinase alpha-3, NF-kappa-B inhibitor alpha, tumor necrosis factor-inducible gene 6 protein, Caspase-1, Caspase-3, Solute carrier family 22 member 6, Solute carrier family 22 member 8 | Inhibitor |
| 5′-AMP-activated protein kinase, | Activator |
| Cellular tumor antigen p53, Cytochrome P450 2C19, P-glycoprotein 1, | Inducer |
| Cytochrome P450 2C9, UDP-glucuronosyltransferase 1–6, Arylamine N-acetyltransferase 2, P-glycoprotein 1 | Substrate |
| 78 kDa glucose-regulated protein, | Binding |
| P-glycoprotein 1 | Modulator |
| Tumor necrosis factor-inducible gene 6 protein, Caspase-1, Caspase-3, G1/S-specific cyclin-D1, Myc proto-oncogene protein, Proliferating cell nuclear antigen, Cyclin A, | Downregulator |
| Dose/Concentration Ranging of ASA | Therapeutic Goal | Cellular Concentration of ASA Need for Therapeutic Effect | |
|---|---|---|---|
| COX Dependent | COX Independent | ||
| 70–150 mg/day [55,186,187,188,189] | (COX-1) platelet effect antiplatelet | NF-κB | 0.05–5 mM [54,77] |
| 325–600 mg/4–6 h [55,189] | (COX-1, COX-2) megakaryocyte analgesic | Acetylation of p53, Glucose-6-Phosphate Dehydrogenase and Other Proteins | ≥100 μM [190,191] |
| 1.2 g/4–6 h [55,189] | COX-2) endothelial/stromal anti-inflammatory | c-Myc, Cyclin A2, CDK2 and Wnt/–catenin pathway | 0.5–2.5 mM [192,193] |
| Single dose of 500 mg/day [194] | Fibrinolysis antiplatelet | mTOR | 5 mM [195] |
| ≤150 mg/day [196] | Activation of the gene encoding SSAT (spermidine/spermine N1-acetyltransferase) antitumor | ODC | 20–100 μM [197] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fijałkowski, Ł.; Skubiszewska, M.; Grześk, G.; Koech, F.K.; Nowaczyk, A. Acetylsalicylic Acid–Primus Inter Pares in Pharmacology. Molecules 2022, 27, 8412. https://doi.org/10.3390/molecules27238412
Fijałkowski Ł, Skubiszewska M, Grześk G, Koech FK, Nowaczyk A. Acetylsalicylic Acid–Primus Inter Pares in Pharmacology. Molecules. 2022; 27(23):8412. https://doi.org/10.3390/molecules27238412
Chicago/Turabian StyleFijałkowski, Łukasz, Magdalena Skubiszewska, Grzegorz Grześk, Frankline Kiptoo Koech, and Alicja Nowaczyk. 2022. "Acetylsalicylic Acid–Primus Inter Pares in Pharmacology" Molecules 27, no. 23: 8412. https://doi.org/10.3390/molecules27238412
APA StyleFijałkowski, Ł., Skubiszewska, M., Grześk, G., Koech, F. K., & Nowaczyk, A. (2022). Acetylsalicylic Acid–Primus Inter Pares in Pharmacology. Molecules, 27(23), 8412. https://doi.org/10.3390/molecules27238412