


Structural Investigation of DHICA Eumelanin Using Density Functional Theory and Classical Molecular Dynamics Simulations

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Geometry Optimization and Validation of the DHICA-Eumelanin Model
2.2. Classical MD Simulations
3. Results and Discussion
3.1. Radial Distribution Functions
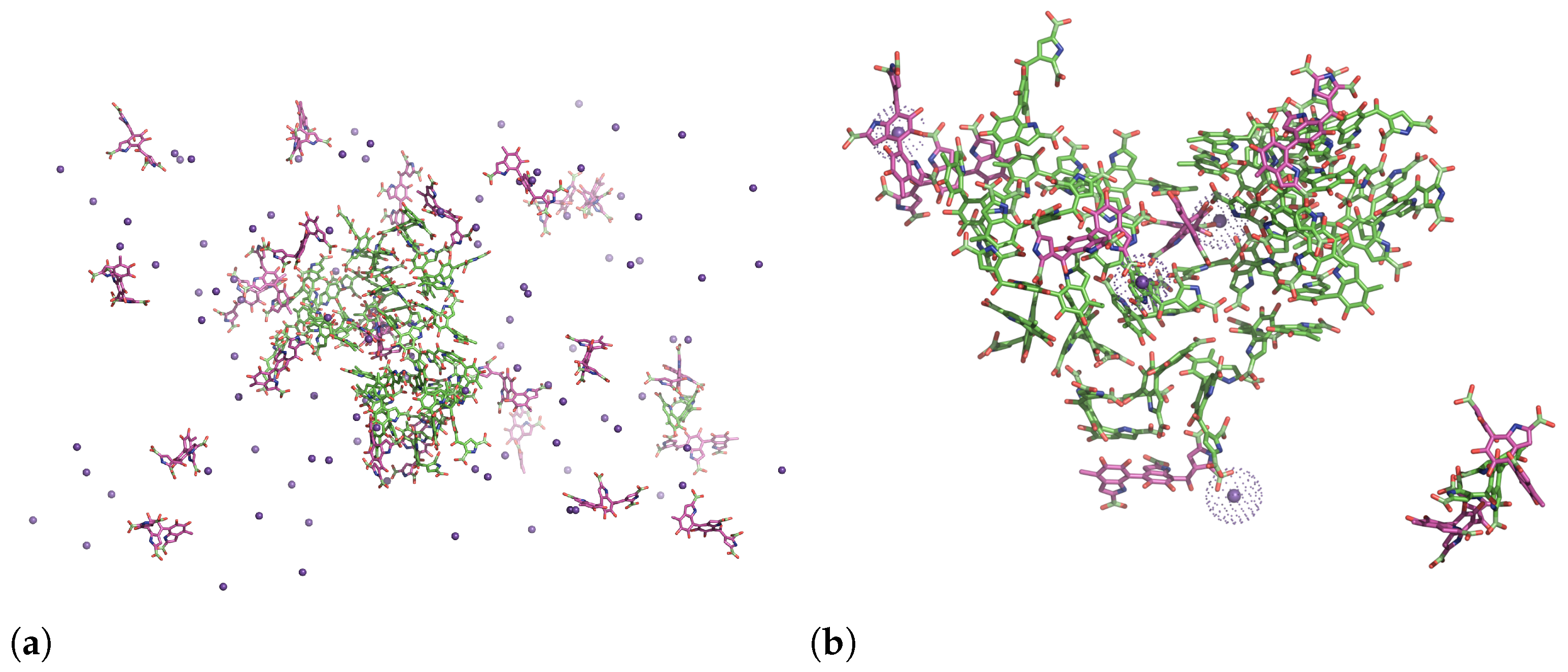
3.2. Aggregation
3.3. Dihedral Angles
3.4. Hydrogen Bonding
3.5. Dipole Moment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DHICA | 5,6-DiHydroxyIndole-2-Carboxylic Acid |
| DHI | 5,6-DiHydroxyIndole |
| PTCA | Pyrrole-2,3,5-TriCarboxylic Acid |
| DFT | Density Functional Theory |
| STM | scanning tunneling microscopy |
| UHR-SEM | ultra-high resolution scanning electron microscopy |
| MD | Molecular Dynamics |
| OPLS-AA | Optimized Potentials for Liquid Simulations for All Atoms |
| SPC | Simple Point Charge |
| PME | Particle-Mesh Ewald |
| P-LINCS | Parallel LINear Constraint Solver |
| NVT | constant particle Number, Volume, and Temperature |
| NPT | constant particle Number, Pressure, and Temperature |
| VMD | Visual Molecular Dynamics |
| RDF | Radial Distribution Functions |
| COM | Center of Mass |
| ESP | ElectroStatic Potential |
| B3LYP | Becke three-parameter Lee–Yang–Parr |
| MK | Merz–Singh–Kollman |
References
- d’Ischia, M.; Napolitano, A.; Pezzella, A.; Meredith, P.; Buehler, M. Melanin Biopolymers: Tailoring Chemical Complexity for Materials Design. Angew. Chem. Int. Ed. 2020, 59, 11196–11205. [Google Scholar] [CrossRef] [PubMed]
- d’Ischia, M.; Napolitano, A.; Pezzella, A.; Meredith, P.; Sarna, T. Chemical and Structural Diversity in Eumelanins: Unexplored Bio-Optoelectronic Materials. Angew. Chem. Int. Ed. 2009, 48, 3914–3921. [Google Scholar] [CrossRef] [PubMed]
- Panzella, L.; Gentile, G.; D’Errico, G.; Della Vecchia, N.F.; Errico, M.E.; Napolitano, A.; Carfagna, C.; d’Ischia, M. Atypical Structural and π-Electron Features of a Melanin Polymer That Lead to Superior Free-Radical-Scavenging Properties. Angew. Chem. Int. Ed. 2013, 52, 12684–12687. [Google Scholar] [CrossRef] [PubMed]
- Schraermeyer, U.; Heimann, K. Current understanding on the role of retinal pigment epithelium and its pigmentation. Pigment Cell Res. 1999, 12, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Ortonne, J.P. Photoprotective properties of skin melanin. Br. J. Dermatol. 2002, s61, 7–10. [Google Scholar] [CrossRef]
- Sarna, F. Properties and function of the ocular melanin–a photobiophysical view. J. Photochem. Photobiol. B 1992, 12, 215–258. [Google Scholar] [CrossRef]
- Ju, K.Y.; Kang, J.; Chang, J.H.; Lee, J.K. Clue to Understanding the Janus Behavior of Eumelanin: Investigating the Relationship between Hierarchical Assembly Structure of Eumelanin and Its Photophysical Properties. Biomacromolecules 2016, 17, 2860–2872. [Google Scholar] [CrossRef]
- Micillo, R.; Iacomino, M.; Perfetti, M.; Panzella, L.; Koike, K.; D’Errico, G.; d’Ischia, M.; Napolitano, A. Unexpected impact of esterification on the antioxidant activity and (photo)stability of a eumelanin from 5,6-dihydroxyindole- 2-carboxylic acid. Pigment Cell Melanoma Res. 2018, 31, 475–483. [Google Scholar] [CrossRef]
- Liberti, D.; Alfieri, M.L.; Monti, D.M.; Panzella, L.; Napolitano, A. A Melanin-Related Phenolic Polymer with Potent Photoprotective and Antioxidant Activities for Dermo-Cosmetic Applications. Antioxidants 2018, 9, 270. [Google Scholar] [CrossRef] [Green Version]
- Wünsche, J.; Deng, Y.; Kumar, P.; Mauro, E.D.; Josberger, E.; Sayago, J.; Pezzella, A.; Soavi, F.; Cicoira, F.; Rolandi, M.; et al. Protonic and Electronic Transport in Hydrated Thin Films of the Pigment Eumelanin. Chem. Mater. 2015, 27, 436–442. [Google Scholar] [CrossRef]
- Matta, M.; Pezzella, A.; Troisi, A. Relation between Local Structure, Electric Dipole, and Charge Carrier Dynamics in DHICA Melanin: A Model for Biocompatible Semiconductors. J. Phys. Chem. Lett. 2020, 11, 1045–1051. [Google Scholar] [CrossRef]
- Tran, M.L.; Powell, B.J.; Meredith, P. Chemical and Structural Disorder in Eumelanins: A Possible Explanation for Broadband Absorbance. Biophys. J. 2006, 90, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Abbas, M.; D’Amico, F.; Morresi, L.; Pinto, N.; Ficcadenti, M.; Natali, R.; Ottaviano, L.; Passacantando, M.; Cuccioloni, M.; Angeletti, M.; et al. Structural, electrical, electronic and optical properties of melanin films. Eur. Phys. J. E 2009, 28, 285–291. [Google Scholar] [CrossRef]
- d’Ischia, M.; Napolitano, A.; Ball, V.; Chen, C.T.; Buehler, M.J. Polydopamine and Eumelanin: From Structure–Property Relationships to a Unified Tailoring Strategy. Acc. Chem. Res. 2014, 47, 3541–3550. [Google Scholar] [CrossRef]
- Mostert, A.B. Melanin, the What, the Why and the How: An Introductory Review for Materials Scientists Interested in Flexible and Versatile Polymers. Polymers 2021, 13, 1670. [Google Scholar] [CrossRef]
- Cheng, J.; Moss, S.; Eisner, M.; Zschack, P. X-ray Characterization of Melanins—I. Pigment Cell Res. 1994, 7, 255–262. [Google Scholar] [CrossRef]
- Cheng, J.; Moss, S.; Eisner, M. X-ray Characterization of Melanins—II. Pigment Cell Res. 1994, 7, 263–273. [Google Scholar] [CrossRef]
- Zajac, G.; Gallas, J.; Cheng, J.; Eisner, M.; Moss, S.; Alvarado-Swaisgood, A. The fundamental unit of synthetic melanina verification by tunneling microscopy of X-ray-scattering results. Biochim. Biophys. Acta 1994, 1199, 271–278. [Google Scholar] [CrossRef]
- Zajac, G.; Gallas, J.; Alvarado-Swaisgood, A. Tunneling microscopy verification of an x-ray scattering-derived molecular model of tyrosine-based melanin. J. Vac. Sci. Technol. B. 1994, 12, 1512–1516. [Google Scholar] [CrossRef]
- Liu, Y.; Simon, J. The effect of preparation procedures on the morphology of melanin from the ink sac of Sepia officinalis. Pigment Cell Res. 2003, 16, 72–80. [Google Scholar] [CrossRef]
- Chen, C.T.; Martin-Martinez, F.J.; Jung, G.S.; Buehler, M.J. Polydopamine and eumelanin molecular structures investigated with ab initio calculations. Chem. Sci. 2017, 8, 1631–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, J.; Williams, S.L.; Forster, C.J.; Kansara, V.; End, P.; Serrano-Wu, M.H. High-throughput melanin-binding affinity and in silico methods to aid in the prediction of drug exposure in ocular tissue. J. Pharm. Sci. 2015, 104, 3997–4001. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, O.; Lindquist, N.G. Melanin affinity and its possible role in neurodegeneration. J. Neural Transm. 2013, 120, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, O.; Lindquist, N.G. Melanin and neuromelanin binding of drugs and chemicals: Toxicological implications. Arch. Toxicol. 2016, 90, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Nofsinger, J.; Forest, S.; Eibest, L.; Gold, K.; Simon, J. Probing the building blocks of eumelanins using scanning electron microscopy. Pigment Cell Res. 2000, 13, 179–184. [Google Scholar] [CrossRef]
- Watt, A.A.R.; Bothma, J.P.; Meredith, P. The supramolecular structure of melanin. Soft Matter. 2009, 5, 3754. [Google Scholar] [CrossRef]
- Pezzella, A.; Panzella, L.; Crescenzi, O.; Napolitano, A.; Navaratman, S.; Edge, R.; Land, E.J.; Barone, V.; d’Ischia, M. Short-Lived Quinonoid Species from 5,6-Dihydroxyindole Dimers en Route to Eumelanin Polymers: Integrated Chemical, Pulse Radiolytic, and Quantum Mechanical Investigation. J. Am. Chem. Soc. 2006, 128, 15490–15498. [Google Scholar] [CrossRef]
- Chen, C.T.; Ball, V.; de Almeida Gracio, J.J.; Singh, M.K.; Toniazzo, V.; Ruch, D.; Buehler, M.J. Self-Assembly of Tetramers of 5,6-Dihydroxyindole Explains the Primary Physical Properties of Eumelanin: Experiment, Simulation, and Design. ACS Nano 2013, 7, 1524–1532. [Google Scholar] [CrossRef]
- Micillo, R.; Panzella, L.; Koike, K.; Monfrecola, G.; Napolitano, A.; d’Ischia, M. “Fifty Shades” of Black and Red or How Carboxyl Groups Fine Tune Eumelanin and Pheomelanin Properties. Int. J. Mol. Sci. 2016, 17, 746. [Google Scholar] [CrossRef]
- Jakubiak, P.; Lack, F.; Thun, J.; Urtti, A.; Alvarez-Sánchez, R. Influence of Melanin Characteristics on Drug Binding Properties. Mol. Pharm. 2019, 16, 2549–2556. [Google Scholar] [CrossRef]
- Pezzella, A.; d’Ischia, M.; Napolitano, A.; Palumbo, A.; Prota, G. An integrated approach to the structure of Sepia melanin. Evidence for a high proportion of degraded 5,6-dihydroxyindole-2-carboxylic acid units in the pigment backbone. Tetrahedron 1997, 53, 8281–8286. [Google Scholar] [CrossRef]
- Magarelli, M.; Passamonti, P.; Renieri, C. Purification, characterization and analysis of sepia melanin from commercial sepia ink (Sepia Officinalis). Rev. CES Med. Vet. Zootec. 2010, 5, 18–28. [Google Scholar] [CrossRef]
- Liu, Y.; Hong, L.; Bowers, C.; Wakamatsu, K.; Ito, S.; Simon, J. chemical and spectroscopic properties of human black and red hair melanosomes. Photochem. Photobiol. 2005, 81, 135–144. [Google Scholar] [CrossRef]
- Terranovaa, M.L.; Tamburri, E. Understanding the way eumelanin works: A unique example of properties and skills driven by molecular heterogeneity. Polymer 2021, 229, 123952. [Google Scholar] [CrossRef]
- Supakar, S.; Banerjee, A.; Jha, T. Intermolecular association of some selected melanin monomers and their optical absorption. Comput. Theor. Chem. 2019, 1151, 43–49. [Google Scholar] [CrossRef]
- Pezzella, A.; Panzella, L.; Crescenzi, O.; Napolitano, A.; Navaratnam, S.; Edge, R.; Land, E.J.; Barone, V.; d’Ischia, M. Lack of Visible Chromophore Development in the Pulse Radiolysis Oxidation of 5,6-Dihydroxyindole-2-carboxylic Acid Oligomers: DFT Investigation and Implications for Eumelanin Absorption Properties. J. Org. Chem. 2009, 74, 3727–3734. [Google Scholar] [CrossRef]
- Panzella, L.; Napolitano, A.; d’Ischia, M. Is DHICA the key to dopachrome tautomerase and melanocyte functions? Pigment Cell Melanoma Res. 2011, 24, 248–249. [Google Scholar] [CrossRef]
- Jiang, S.; Liu, X.M.; Dai, X.; Zhou, Q.; Lei, T.C.; Beermann, F.; Wakamatsu, K.; Xu, S. Regulation of DHICA-mediated antioxidation by dopachrome tautomerase: Implication for skin photoprotection against UVA radiation. Free Radic. Biol. Med. 2010, 48, 1144–1151. [Google Scholar] [CrossRef]
- Cecchi, T.; Pezzella, A.; Di Mauro, E.; Cestola, S.; Ginsburg, D.; Luzi, M.; Rigucci, A.; Santato, C. On the antioxidant activity of eumelanin biopigments: A quantitative comparison between free radical scavenging and redox properties. Nat. Prod. Res. 2020, 34, 2465–2473. [Google Scholar] [CrossRef]
- Kaxiras, E.; Tsolakidis, A.; Zonios, G.; Meng, S. Structural model of eumelanin. Phys. Rev. Lett. 2006, 97, 218102. [Google Scholar] [CrossRef]
- Pezzella, A.; Iadonisi, A.; Valerio, S.; Panzella, L.; Napolitano, A.; Adinolfi, M.; d’Ischia, M. Disentangling eumelanin “black chromophore”: Visible absorption changes as signatures of oxidation state- and aggregation-dependent dynamic interactions in a model water-soluble 5,6-dihydroxyindole polymer. J. Am. Chem. Soc. 2009, 131, 15270–15275. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Chuang, C.; Cao, J.; Ball, V.; Ruch, D.; Buehler, M.J. Excitonic effects from geometric order and disorder explain broadband optical absorption in eumelanin. Nat. Commun. 2014, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assis Oliveira, L.B.; L Fonseca, T.; Costa Cabral, B.J.; Coutinho, K.; Canuto, S. Hydration Effects on the Electronic Properties of Eumelanin Building Blocks. J. Chem. Phys. 2016, 145, 084501. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Buehler, M.J. Polydopamine and eumelanin models in various oxidation states. Phys. Chem. Chem. Phys. 2018, 20, 28135–28143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltani, S.; Sowlati-Hashjin, S.; Feugmo, C.G.T.; Karttunen, M. Free energy and stacking of eumelanin nanoaggregates. J. Phys. Chem. B. 2022, 22, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Ghosh, D. Charge transfer in DHICA eumelanin-like oligomers: Role of hydrogen bonds. Chem. Commun. 2020, 56, 10481–10484. [Google Scholar] [CrossRef]
- Micillo, R.; Panzella, L.; Iacomino, M.; Prampolini, G.; Cacelli, I.; Ferretti, A.; Crescenzi, O.; Koike, K.; Napolitano, A.; d’Ischia, M. Eumelanin broadband absorption develops from aggregation-modulated chromophore interactions under structural and redox control. Sci. Rep. 2017, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Powell, B. 5,6-dihydroxyindole-2-carboxylic acid (DHICA): A First Principles Density-Functional Study. Chem. Phys. Lett. 2005, 402, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Kaxiras, E. Theoretical Models of Eumelanin Protomolecules and Their Optical Properties. Biophys. J. 2008, 94, 2095–2105. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, K.; Ito, S. Advanced Chemical Methods in Melanin Determination. Pigment Cell Res. 2002, 15, 174–183. [Google Scholar] [CrossRef]
- Ito, S. Reexamination of the structure of eumelanin. J. Biol. Chem. 1986, 883, 155–161. [Google Scholar] [CrossRef]
- Schroeder, R.L.; Pendleton, P.; Gerber, J. Physical factors affecting chloroquine binding to melanin. Colloids Surf. B Biointerfaces 2015, 134, 8–16. [Google Scholar] [CrossRef]
- Kirla, K.T.; Groh, K.J.; Poetzsch, M.; Banote, R.K.; Stadnicka-Michalak, J.; Eggen, R.I.L.; Schirmer, K.; Kraemer, T. Importance of Toxicokinetics to Assess the Utility of Zebrafish Larvae as Model for Psychoactive Drug Screening Using Meta-Chlorophenylpiperazine (mCPP) as Example. Front. Pharmacol. 2018, 9, 414. [Google Scholar] [CrossRef] [Green Version]
- Okuda, H.; Nakamura, A.; Wakamatsu, K.; Hanson, G.; Ito, S.; Sota, T. Mid-infrared absorption spectrum of 5,6-dihydroxyindole-2-carboxylic acid. Chem. Phys. Lett. 2007, 433, 355–359. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. A. 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Kim, K.; Jordan, K.D. Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem. 1994, 98, 10089–10094. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–5261. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Selfconsistent molecular orbital methods. XXIII. A polarizationtype basis set for secondrow elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Davidson, E.R.; Feller, D. Basis set selection for molecular calculations. Chem. Rev. 1989, 86, 681–696. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pezzella, A.; Crescenzi, O.; Panzella, L.; Napolitano, A.; Land, E.J.; Barone, V.; d’Ischia, M. Free Radical Coupling of o-Semiquinones Uncovered. J. Am. Chem. Soc. 2013, 135, 12142–12149. [Google Scholar] [CrossRef] [PubMed]
- Dodda, L.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin? J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Robertson, M.; Tirado-Rives, J.; Jorgensen, W. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [Green Version]
- Cox, S.R.; Williams, D.E. Representation of the molecular electrostatic potential by a net atomic charge model. J. Comput. Chem. 1981, 2, 304–323. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Besler, B.; KM, M.J.; Kollman, P. Atomic Charges Derived from Semiempirical Methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmüller, H. More Bang for Your Buck: Improved use of GPU Nodes for GROMACS 2018. J. Comp. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.; Postma, J.; van Gunsteren, W.; Hermans, J. Intermolecular Forces; Pullman, B., Ed.; Springer: Dordrecht, The Netherlands, 1981. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.; Darden, T.; Lee, H.; Pedersen, L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Gapsys, V.; de Groot, B.L. Comment on ‘Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size’. eLife 2019, 8, e44718. [Google Scholar] [CrossRef]
- Gapsys, V.; de Groot, B.L. On the Importance of Statistics in Molecular Simulations for Thermodynamics, Kinetics and Simulation Box Size. eLife 2020, 8, e45318. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; Version 1.8; DeLano Scientific LLC: South San Francisco, CA, USA, 2015. [Google Scholar]
- Liu, Y.; Ao, X.; Wang, Q.; Wang, J.; Ge, H. PiViewer: An open-source tool for automated detection and display of π-π interactions. Chem. Biol. Drug Des. 2018, 92, 1809–1814. [Google Scholar] [CrossRef]
- Carter-Fenk, K.; Herbert, J.M. Reinterpreting π-stacking. Phys. Chem. Chem. Phys. 2020, 22, 24870–24886. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Simon, J.D. Physical and chemical characterization of iris and choroid melanosomes isolated from newborn and mature cows. Photochem. Photobiol. 2005, 81, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Simon, J.D. Current Understanding of the Binding Sites, Capacity, Affinity, and Biological Significance of Metals in Melanin. J. Phys. Chem. B. 2007, 111, 7938–7947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, L.X.; Schenter, G.K.; Glezakou, V.; Fulton, J.L. Molecular Simulation Analysis and X-ray Absorption Measurement of Ca2+, K+ and Cl- Ions in Solution. J. Phys. Chem. B 2006, 110, 23644–23654. [Google Scholar] [CrossRef]
- Corani, A.; Huijser, A.; Gustavsson, T.; Markovitsi, D.; Malmqvist, P.; Pezzella, A.; d’Ischia, M.; Sundström, V. Superior Photoprotective Motifs and Mechanisms in Eumelanins Uncovered. J. Am. Chem. Soc. 2014, 136, 11626–11635. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Eumelanins (nm) | Number of Water Molecules | Box Size (nm) | Counterions (K) | Duration () |
|---|---|---|---|---|
| 2 uncharged | 4218 | 0 | 1.073 | |
| 4 uncharged | 5640 | 0 | 0.983 | |
| 27 uncharged | 6341 | 0 | 2.237 | |
| 50 uncharged | 20,000 | 0 | 2.050 | |
| 2 charges | 3487 | 8 | 1.000 | |
| 4 charges | 4501 | 16 | 1.100 | |
| 27 charges | 6272 | 108 | 3.802 | |
| 50 charges | 19,800 | 200 | 1.361 | |
| 25 uncharged-25 charged | 19,900 | 100 | 1.974 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soltani, S.; Sowlati-Hashjin, S.; Tetsassi Feugmo, C.G.; Karttunen, M. Structural Investigation of DHICA Eumelanin Using Density Functional Theory and Classical Molecular Dynamics Simulations. Molecules 2022, 27, 8417. https://doi.org/10.3390/molecules27238417
Soltani S, Sowlati-Hashjin S, Tetsassi Feugmo CG, Karttunen M. Structural Investigation of DHICA Eumelanin Using Density Functional Theory and Classical Molecular Dynamics Simulations. Molecules. 2022; 27(23):8417. https://doi.org/10.3390/molecules27238417
Chicago/Turabian StyleSoltani, Sepideh, Shahin Sowlati-Hashjin, Conrard Giresse Tetsassi Feugmo, and Mikko Karttunen. 2022. "Structural Investigation of DHICA Eumelanin Using Density Functional Theory and Classical Molecular Dynamics Simulations" Molecules 27, no. 23: 8417. https://doi.org/10.3390/molecules27238417
APA StyleSoltani, S., Sowlati-Hashjin, S., Tetsassi Feugmo, C. G., & Karttunen, M. (2022). Structural Investigation of DHICA Eumelanin Using Density Functional Theory and Classical Molecular Dynamics Simulations. Molecules, 27(23), 8417. https://doi.org/10.3390/molecules27238417