
Comparative Modelling of Organic Anion Transporting Polypeptides: Structural Insights and Comparison of Binding Modes

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
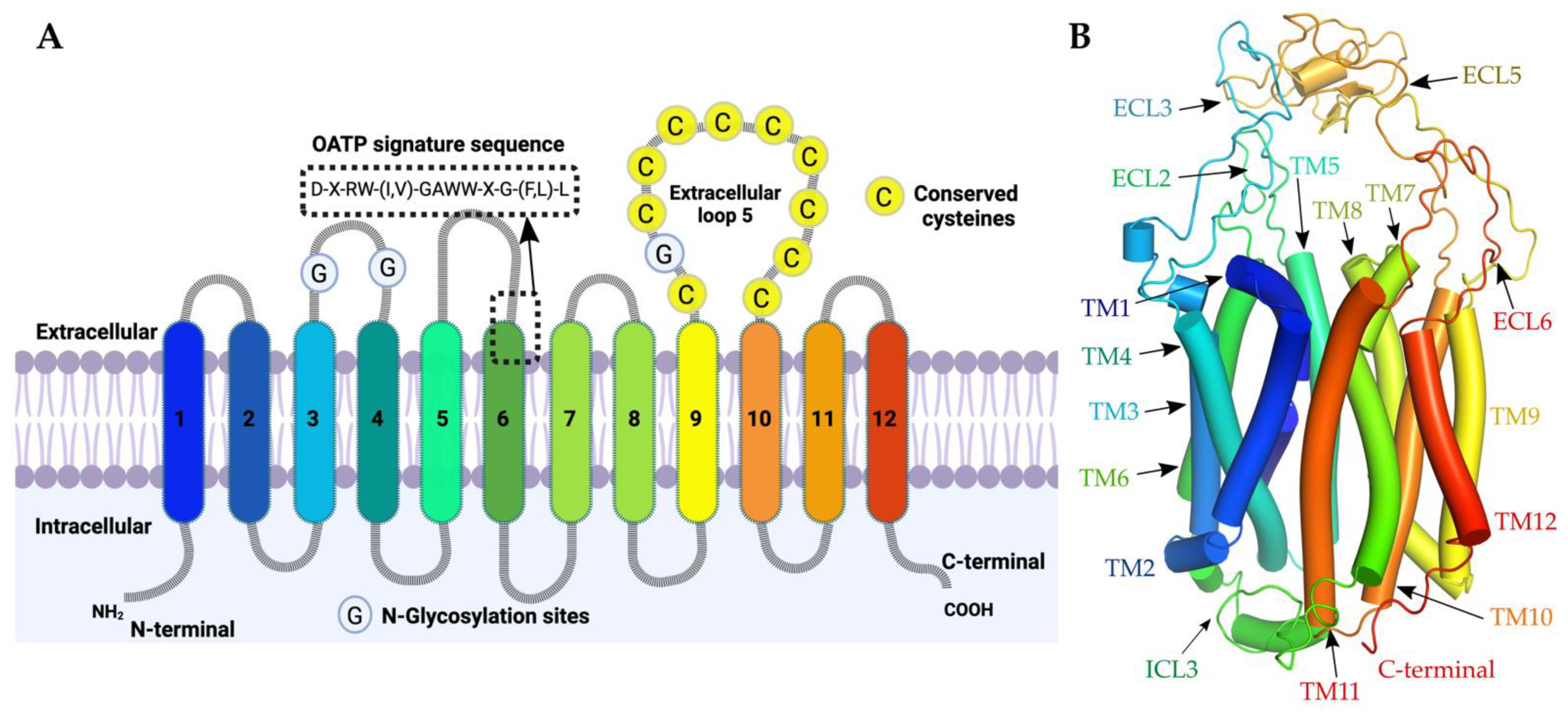
2.1. OATPs Share a Conserved Transmembrane Helix Fold Harbouring a Druggable Binding Pocket
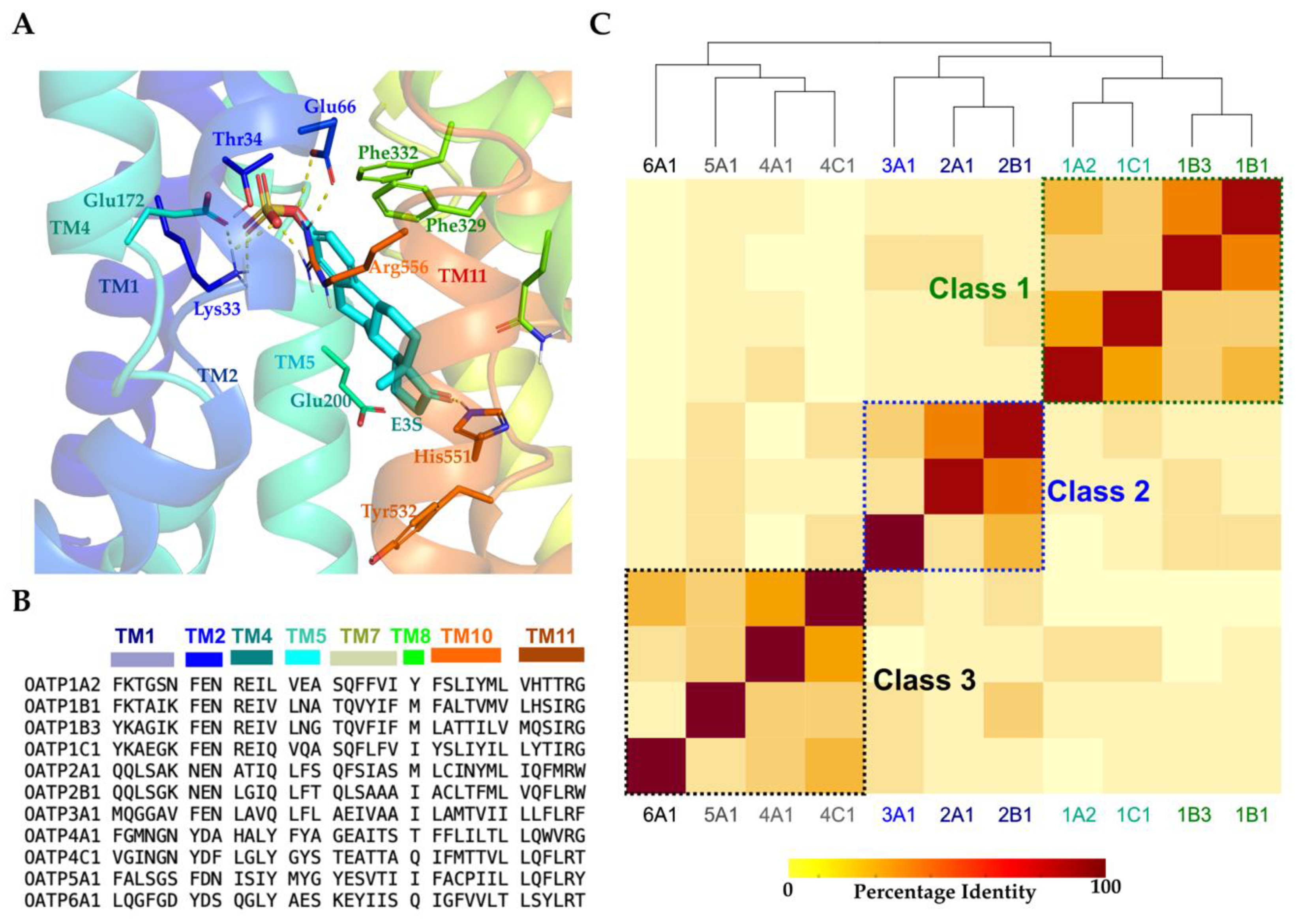
2.2. Binding Site Analysis and Classification
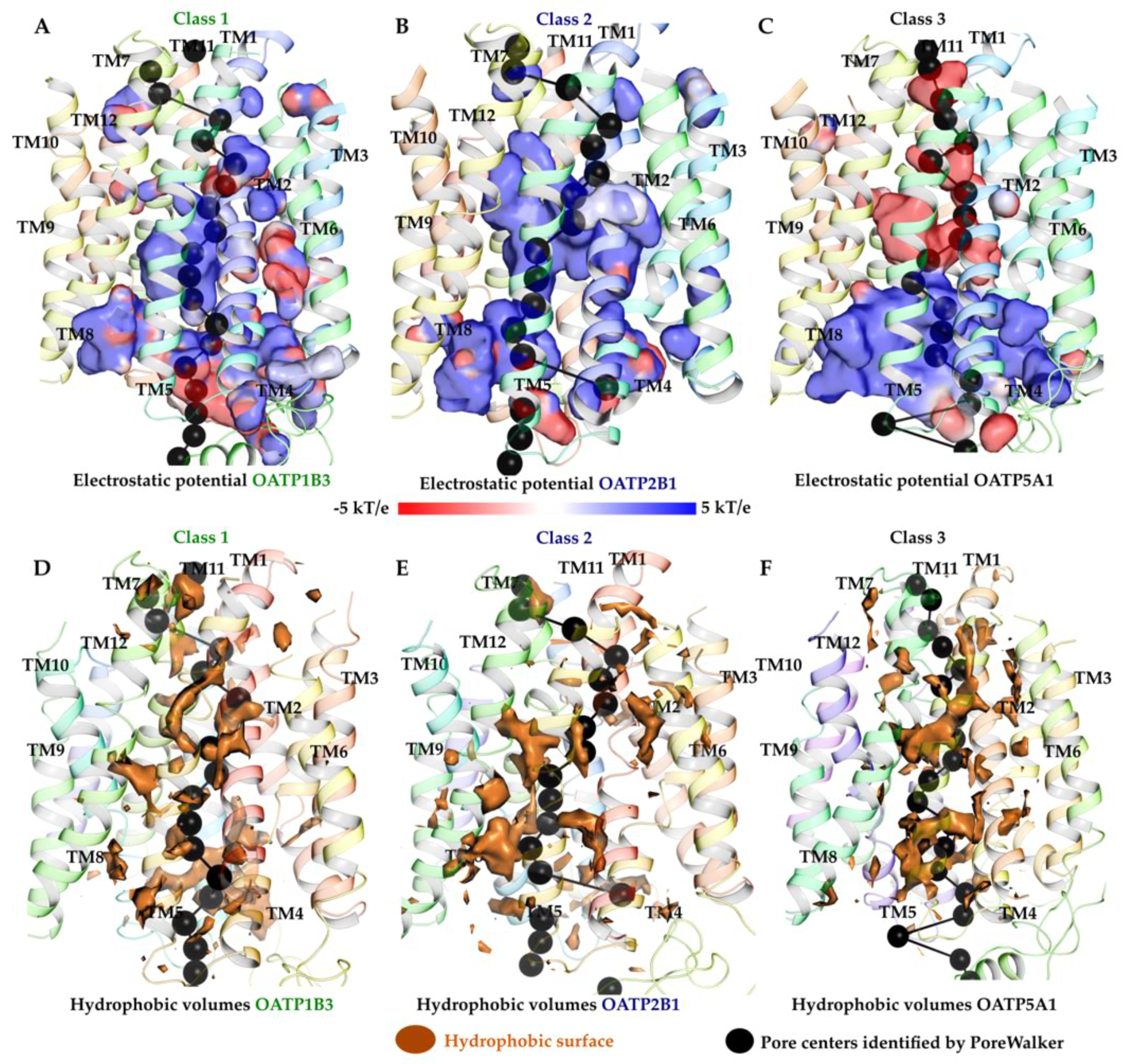
2.3. Electrostatic Potential and Hydrophobic Surface of Pore
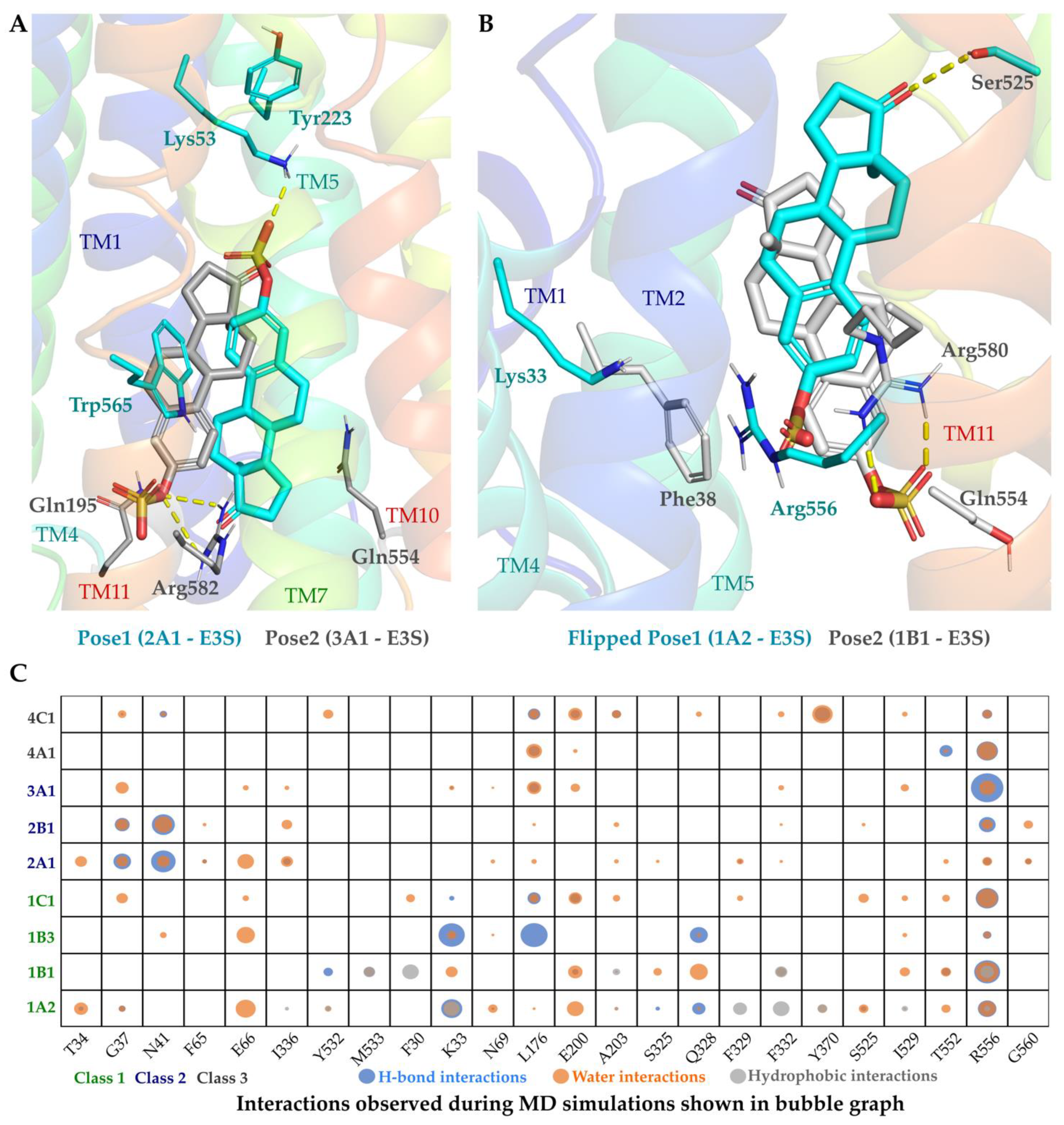
2.4. OATP Substrates Display Multiple Binding Modes with a Dynamic Interaction Profile
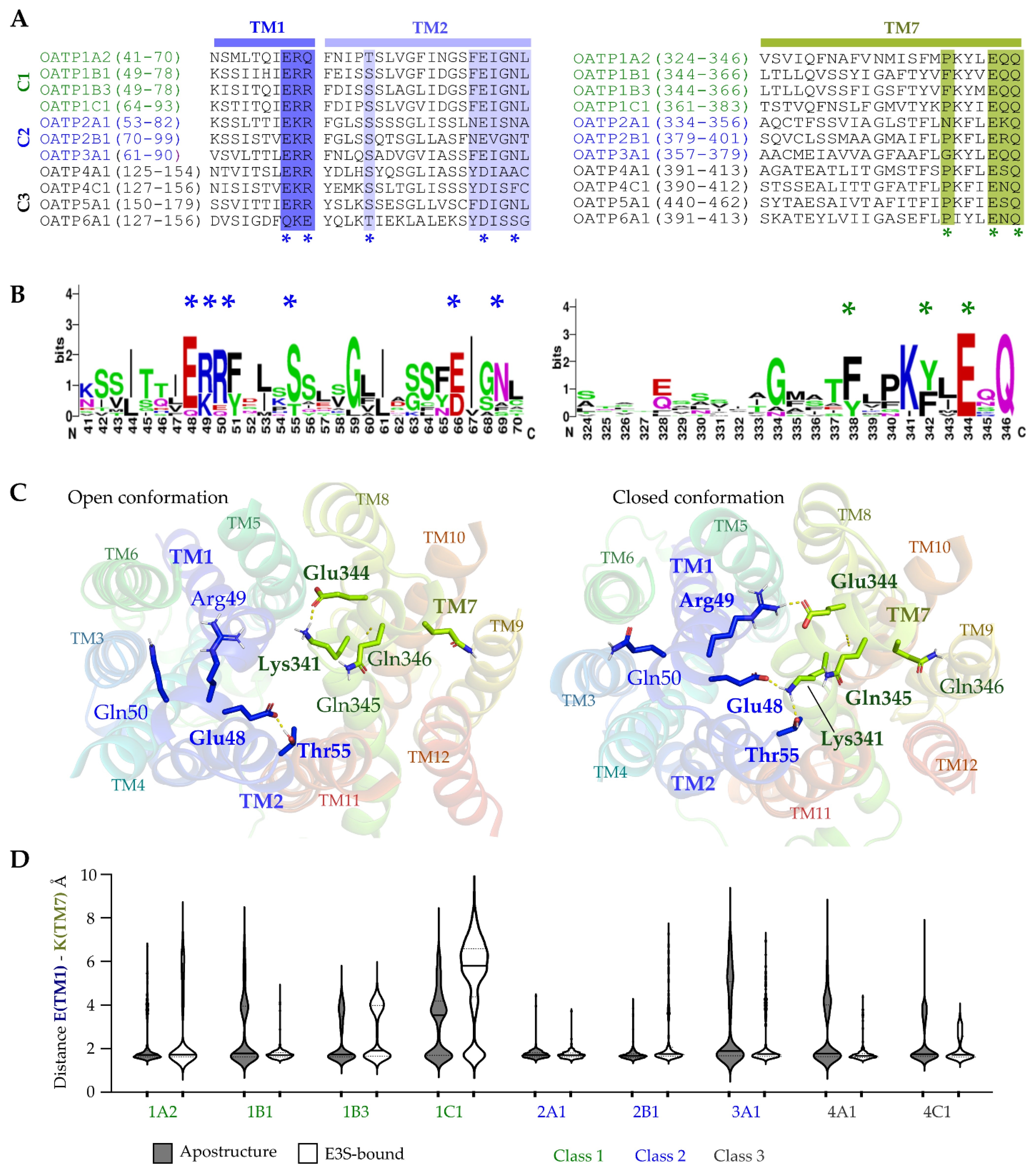
2.5. Conserved Salt–Bridge Interactions’ Potential Role in Controlling OATPs Entrance
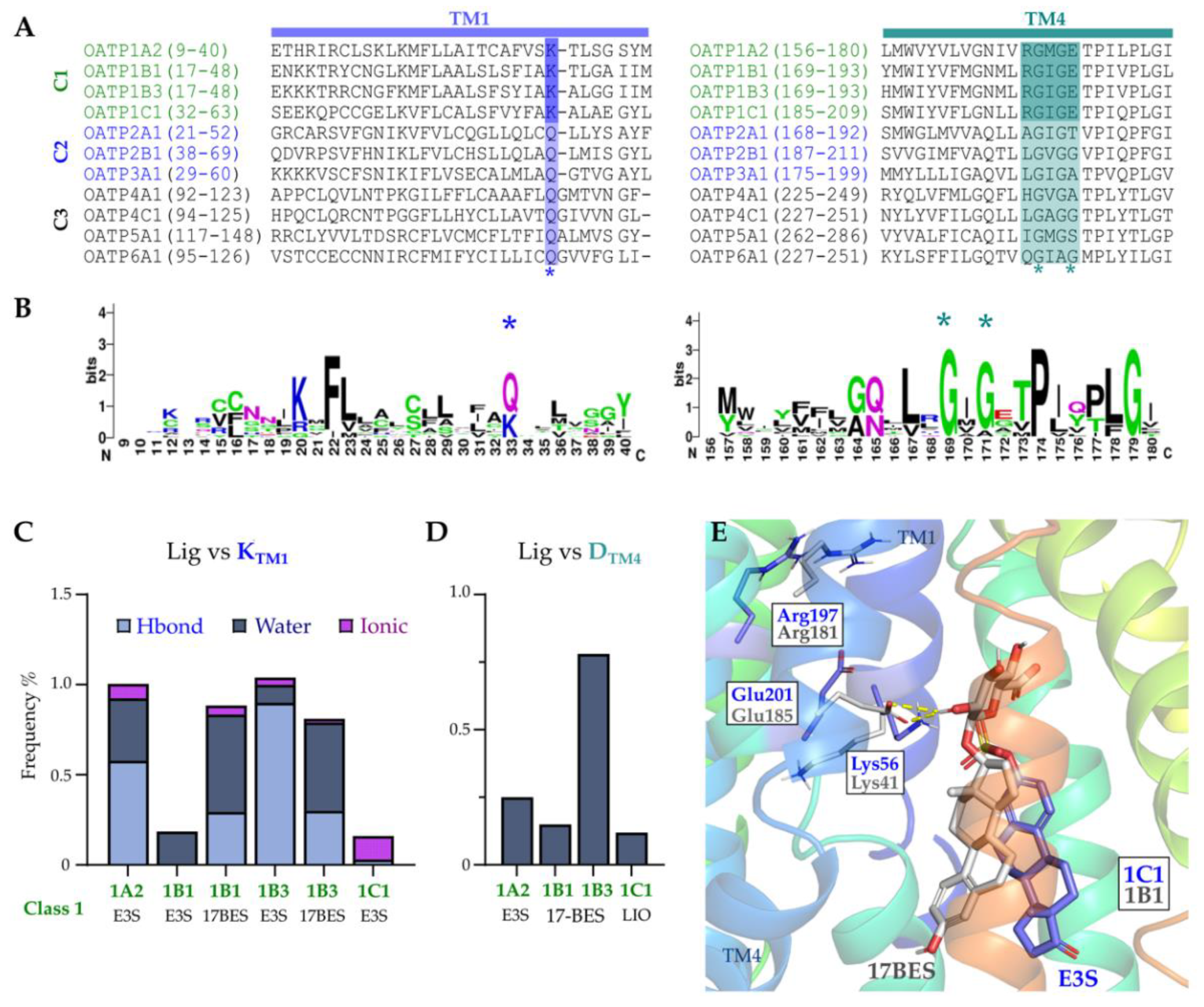
2.6. Conserved Lysine on TM1 and ‘RGI/MGE’ Motif on TM4 among OATP1 Family and Importance in the Ligand Binding
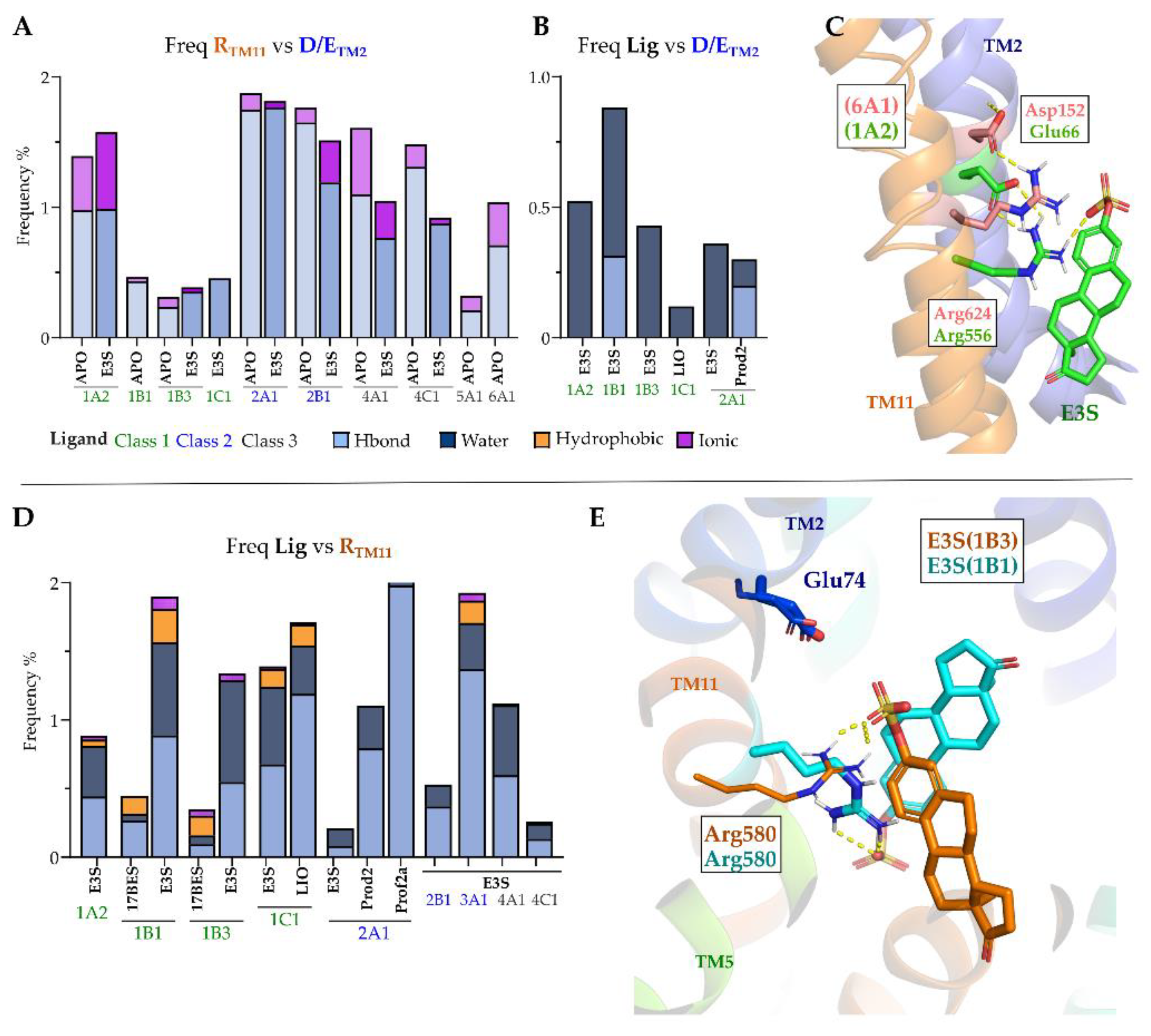
2.7. OATP1′s TM2 Conserved Motif ‘GSFEIGNL’ Displays Stable Interactions with Conserved Arginine on TM11
3. Materials and Methods
3.1. Homology Modelling and Validation
3.2. Model Evaluation
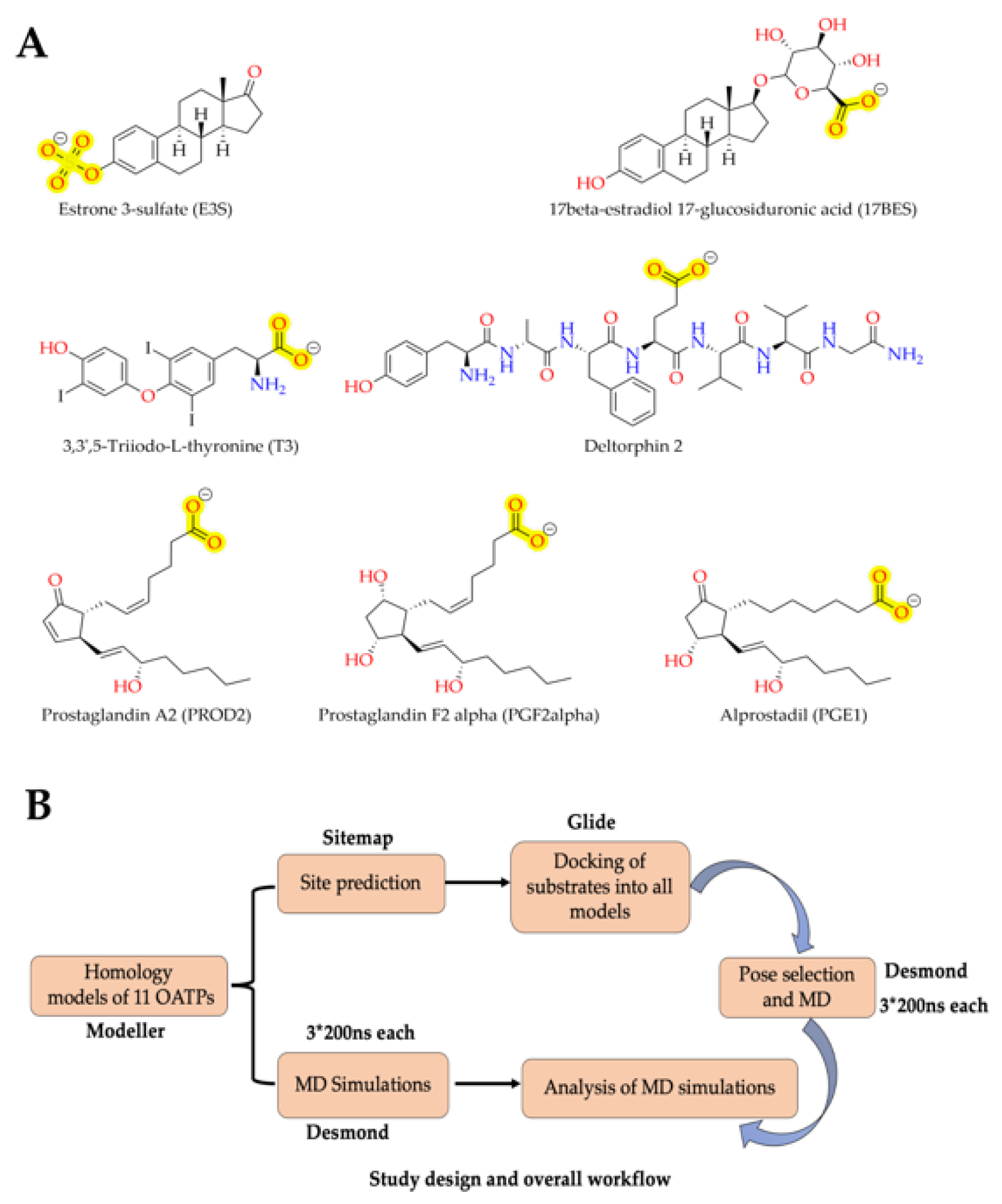
3.3. Preparation of Ligands
3.4. Preparation of Protein
3.5. Binding Site Analysis and Classification
3.6. Sitemap and Grid Generation
3.7. Docking of Substrates
3.8. Molecular Dynamics
3.9. Molecular Dynamics Analysis
3.10. Data Visualization
3.11. Electrostatic Potentials and Hydrophobic Volumes Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hagenbuch, B.; Meier, P. The superfamily of organic anion transporting polypeptides. Biochim. Biophys. Acta-Biomembr. 2003, 1609, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer zu Schwabedissen, H.E.; Tirona, R.G.; Yip, C.S.; Ho, R.H.; Kim, R.B. Interplay between the Nuclear Receptor Pregnane X Receptor and the Uptake Transporter Organic Anion Transporter Polypeptide 1A2 Selectively Enhances Estrogen Effects in Breast Cancer. Cancer Res. 2008, 68, 9338–9347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, M.; Zhou, F. Trafficking and other regulatory mechanisms for organic anion transporting polypeptides and organic anion transporters that modulate cellular drug and xenobiotic influx and that are dysregulated in disease. Br. J. Pharmacol. 2017, 174, 1908–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFeely, S.J.; Ritchie, T.K.; Yu, J.; Nordmark, A.; Levy, R.H.; Ragueneau-Majlessi, I. Identification and Evaluation of Clinical Substrates of Organic Anion Transporting Polypeptides 1B1 and 1B3. Clin. Transl. Sci. 2019, 12, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Guideline on the Investigation of Drug Interactions; Committee for Human Medicinal Products (CHMP): Amsterdam, The Netherlands, 2012.
- US Food and Drug Administration (FDA). Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations; Center for Drug Evaluation and Research (CDER), FDA: Silver Spring, MD, USA, 2012; pp. 1–75.
- Tamai, I.; Nezu, J.I.; Uchino, H.; Sai, Y.; Oku, A.; Shimane, M.; Tsuji, A. Molecular Identification and Characterization of Novel Members of the Human Organic Anion Transporter (OATP) Family. Biochem. Biophys. Res. Commun. 2000, 273, 251–260. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Meier, P.J. Organic anion transporting polypeptides of the OATP/ SLC21 family: Phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. Eur. J. Physiol. 2004, 447, 653–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullak-Ublick, G.A.; Hagenbuch, B.; Stieger, B.; Schteingart, C.D.; Hofmann, A.F.; Wolkoff, A.W.; Meier, P.J. Molecular and functional characterization of an organic anion transporting polypeptide cloned from human liver. Gastroenterology 1995, 109, 1274–1282. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Targeted drug delivery to treat pain and cerebral hypoxia. Pharmacol. Rev. 2013, 65, 291–314. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.I.; Betterton, R.D.; Davis, T.P.; Ronaldson, P.T. Transporter-mediated delivery of small molecule drugs to the brain: A critical mechanism that can advance therapeutic development for ischemic stroke. Pharmaceutics 2020, 12, 154. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Z.; Tay-Sontheimer, J.; Levy, R.H.; Ragueneau-Majlessi, I. Intestinal Drug Interactions Mediated by OATPs: A Systematic Review of Preclinical and Clinical Findings. J. Pharm. Sci. 2017, 106, 2312–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Glaeser, H.; Smith, L.H.; Roberts, R.L.; Moeckel, G.W.; Gervasini, G.; Leake, B.F.; Kim, R.B. Polymorphisms in Human Organic Anion-transporting Polypeptide 1A2 (OATP1A2): Implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 2005, 280, 9610–9617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemi, M. Role of OATP transporters in the disposition of drugs. Pharmacogenomics 2007, 8, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.F.; Acharya, M.R.; Desai, N.; Figg, W., II; Sparreboom, A. Identification of OATP1B3 as a high-affinity hepatocellular transporter of paclitaxel. Cancer Biol. Ther. 2005, 4, 815–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzi, J.; Grube, M.; Zu Schwabedissen, H.E.M. OATP2B1—The underrated member of the organic anion transporting polypeptide family of drug transporters? Biochem. Pharmacol. 2021, 188, 114534. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yuan, J.; Li, Z.; Wang, Z.; Cheng, D.; Du, Y.; Li, W.; Kan, Q.; Zhang, W. Genetic Polymorphisms and Function of the Organic Anion-Transporting Polypeptide 1A2 and Its Clinical Relevance in Drug Disposition. Pharmacology 2015, 95, 201–208. [Google Scholar] [CrossRef]
- Franke, R.M.; Scherkenbach, L.A.; Sparreboom, A. Pharmacogenetics of the organic anion transporting polypeptide 1A2,”. Pharmacogenomics 2009, 10, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Hänggi, E.; Grundschober, A.F.; Leuthold, S.; Meier, P.J.; St-Pierre, M.V. Functional Analysis of the Extracellular Cysteine Residues in the Human Organic Anion Transporting Polypeptide, OATP2B1. Mol. Pharmacol. 2006, 70, 806–817. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Wells, J.; Meredith, D. The Signature Sequence Region of the Human Drug Transporter Organic Anion Transporting Polypeptide 1B1 Is Important for Protein Surface Expression. J. Drug Deliv. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Hong, W.; Huang, J.; Zhan, K.; Huang, H.; Hong, M. N-glycosylation Dictates Proper Processing of Organic Anion Transporting Polypeptide 1B1. PLoS ONE 2012, 7, e52563. [Google Scholar] [CrossRef] [PubMed]
- Mandery, K.; Sticht, H.; Bujok, K.; Schmidt, I.; Fahrmayr, C.; Balk, B.; Fromm, M.F.; Glaeser, H. Functional and Structural Relevance of Conserved Positively Charged Lysine Residues in Organic Anion Transporting Polypeptide 1B3. Mol. Pharmacol. 2011, 80, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Strømme, P.; Groeneweg, S.; Lima de Souza, E.C.; Zevenbergen, C.; Torgersbråten, A.; Holmgren, A.; Gurcan, E.; Meima, M.E.; Peeters, R.P.; Visser, W.E.; et al. Mutated Thyroid Hormone Transporter OATP1C1 Associates with Severe Brain Hypometabolism and Juvenile Neurodegeneration. Thyroid 2018, 28, 1406–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Hong, W.; Huang, H.; Lu, H.; Lin, G.; Hong, M. Identification of Amino Acids Essential for Estrone-3-Sulfate Transport within Transmembrane Domain 2 of Organic Anion Transporting Polypeptide 1B1. PLoS ONE 2012, 7, e36647. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Wu, Z.; Fang, Z.; Huang, J.; Huang, H.; Hong, M. Amino Acid Residues in the Putative Transmembrane Domain 11 of Human Organic Anion Transporting Polypeptide 1B1 Dictate Transporter Substrate Binding, Stability, and Trafficking. Mol. Pharm. 2015, 12, 4270–4276. [Google Scholar] [CrossRef]
- Glaeser, H.; Mandery, K.; Sticht, H.; Fromm, M.; König, J. Relevance of conserved lysine and arginine residues in transmembrane helices for the transport activity of organic anion transporting polypeptide 1B3. Br. J. Pharmacol. 2010, 159, 698–708. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Ke, M.; Zhang, Y.; Yan, Z.; Wu, J. Structure of a mammalian sperm cation channel complex. Nature 2021, 595, 746–750. [Google Scholar] [CrossRef]
- Lee, W.; Ha, J.; Sugiyama, Y. Post-translational regulation of the major drug transporters in the families of organic anion transporters and organic anion–transporting polypeptides. J. Biol. Chem. 2020, 295, 17349–17364. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Meier-Abt, F.; Mokrab, Y.; Mizuguchi, K. Organic Anion Transporting Polypeptides of the OATP/SLCO Superfamily: Identification of New Members in Nonmammalian Species, Comparative Modeling and a Potential Transport Mode. J. Membr. Biol. 2006, 208, 213–227. [Google Scholar] [CrossRef]
- Tuerkova, A.; Ungvári, O.; Laczkó-Rigó, R.; Mernyák, E.; Szakács, G.; Özvegy-Laczka, C.; Zdrazil, B. Data-Driven Ensemble Docking to Map Molecular Interactions of Steroid Analogs with Hepatic Organic Anion Transporting Polypeptides. J. Chem. Inf. Model. 2021, 61, 3109–3127. [Google Scholar] [CrossRef]
- Weaver, Y.M.; Hagenbuch, B. Several Conserved Positively Charged Amino Acids in OATP1B1 are Involved in Binding or Translocation of Different Substrates. J. Membr. Biol. 2010, 236, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chen, J.; Xu, S.; Ni, C.; Fang, Z.; Hong, M. Amino-terminal region of human organic anion transporting polypeptide 1B1 dictates transporter stability and substrate interaction. Toxicol. Appl. Pharmacol. 2019, 378, 114642. [Google Scholar] [CrossRef]
- Badagnani, I.; Castro, R.A.; Taylor, T.R.; Brett, C.M.; Huang, C.C.; Stryke, D.; Kawamoto, M.; Johns, S.J.; Ferrin, T.E.; Carlson, E.J.; et al. Interaction of Methotrexate with Organic-Anion Transporting Polypeptide 1A2 and Its Genetic Variants. J. Pharmacol. Exp. Ther. 2006, 318, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Adla, S.K.; Tonduru, A.K.; Kronenberger, T.; Kudova, E.; Poso, A.; Huttunen, K.M. Neurosteroids: Structure-Uptake Relationships and Computational Modeling of Organic Anion Transporting Polypeptides (OATP)1A2. Molecules 2021, 26, 5662. [Google Scholar] [CrossRef] [PubMed]
- Tuerkova, A.; Bongers, B.J.; Norinder, U.; Ungvári, O.; Székely, V.; Tarnovskiy, A.; Szakács, G.; Özvegy-Laczka, C.; van Westen, G.J.; Zdrazil, B. Identifying Novel Inhibitors for Hepatic Organic Anion Transporting Polypeptides by Machine Learning-Based Virtual Screening. J. Chem. Inf. Model. 2022. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.R.; Won, J.; Heo, L.; Seok, C. GalaxyRefine2: Simultaneous refinement of inaccurate local regions and overall protein structure. Nucleic Acids Res. 2019, 47, W451–W455. [Google Scholar] [CrossRef] [PubMed]
- Mak, L.; Marcus, D.; Howlett, A.; Yarova, G.; Duchateau, G.; Klaffke, W.; Bender, A.; Glen, R.C. Metrabase: A cheminformatics and bioinformatics database for small molecule transporter data analysis and (Q)SAR modeling. J. Cheminform. 2015, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2019-4: LigPrep, Schrödinger; LLC: New York, NY, USA, 2019.
- Schrödinger Release 2019-4: Epik; Schrödinger; LLC: New York, NY, USA, 2019.
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2019-4: MacroModel, Schrödinger; LLC: New York, NY, USA, 2019.
- Schrödinger Release 2019-4: Protein Preparation Wizard; Epik, Schrödinger; LLC: New York, NY, USA, 2016.
- Schrödinger Release 2019-4: SiteMap, Schrödinger; LLC: New York, NY, USA, 2019.
- Halgren, T. New Method for Fast and Accurate Binding-site Identification and Analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]
- LLC. Schrödinger Release 2019-4: Glide, Schrödinger; LLC: New York, NY, USA, 2019. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2019-4: Desmond Molecular Dynamics System; Maestro-Desmond Interoperability Tools; D. E. Shaw Research: New York, NY, USA; Schrödinger: New York, NY, USA, 2019.
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini-Calace, M.; Maiwald, T.; Thornton, J.M. PoreWalker: A Novel Tool for the Identification and Characterization of Channels in Transmembrane Proteins from Their Three-Dimensional Structure. PLoS Comput. Biol. 2009, 5, e1000440. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OATP | Uniprot ID | Amino Acid Length | Gene Name | DELTA-BLAST Search | Alignment Used for Model Building | ||
|---|---|---|---|---|---|---|---|
| % Identity with 6E9N | % Identity with 1TGS | % Identity with 6E9N | % Identity with 1TGS | ||||
| OATP1A2 | P46721 | 670 | SLCO1A2 | 11.4 | 30.2 | 13 | 27.3 |
| OATP1B1 | Q9Y6L6 | 691 | SLCO1B1 | 11.9 | 21 | 15.2 | 27.3 |
| OATP1B3 | Q9NPD5 | 702 | SLCO1B3 | 11.6 | 21 | 14.4 | 27.3 |
| OATP1C1 | Q9NYB5 | 712 | SLCO1C1 | 13.5 | 27.9 | 14.2 | 25.5 |
| OATP2A1 | Q92959 | 643 | SLCO2A1 | 12.3 | 30.4 | 14.2 | 40 |
| OATP2B1 | O94956 | 709 | SLCO2B1 | 11.8 | 18.7 | 12.2 | 21.8 |
| OATP3A1 | Q9UIG8 | 710 | SLCO3A1 | 12 | 27.2 | 15 | 29.1 |
| OATP4A1 | Q96BD0 | 722 | SLCO4A1 | 9.7 | 24.1 | 11.5 | 25.5 |
| OATP4C1 | Q6ZQN7 | 724 | SLCO4C1 | 10.7 | 28 | 13.7 | 27.3 |
| OATP5A1 | Q9H2Y9 | 848 | SLCO5A1 | 11.5 | 31.5 | 13 | 32.7 |
| OATP6A1 | Q86UG4 | 719 | SLCO6A1 | 10.9 | 26 | 13.7 | 29.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonduru, A.K.; Adla, S.K.; Huttunen, K.M.; Kronenberger, T.; Poso, A. Comparative Modelling of Organic Anion Transporting Polypeptides: Structural Insights and Comparison of Binding Modes. Molecules 2022, 27, 8531. https://doi.org/10.3390/molecules27238531
Tonduru AK, Adla SK, Huttunen KM, Kronenberger T, Poso A. Comparative Modelling of Organic Anion Transporting Polypeptides: Structural Insights and Comparison of Binding Modes. Molecules. 2022; 27(23):8531. https://doi.org/10.3390/molecules27238531
Chicago/Turabian StyleTonduru, Arun Kumar, Santosh Kumar Adla, Kristiina M. Huttunen, Thales Kronenberger, and Antti Poso. 2022. "Comparative Modelling of Organic Anion Transporting Polypeptides: Structural Insights and Comparison of Binding Modes" Molecules 27, no. 23: 8531. https://doi.org/10.3390/molecules27238531
APA StyleTonduru, A. K., Adla, S. K., Huttunen, K. M., Kronenberger, T., & Poso, A. (2022). Comparative Modelling of Organic Anion Transporting Polypeptides: Structural Insights and Comparison of Binding Modes. Molecules, 27(23), 8531. https://doi.org/10.3390/molecules27238531