
Tailoring the AIE Chromogen 2-(2-Hydroxyphenyl)benzothiazole for Use in Enzyme-Triggered Molecular Brachytherapy

 , , , ,
, , , ,
Abstract
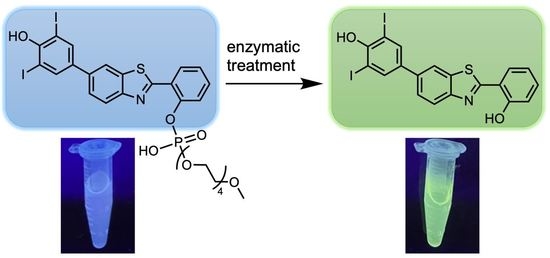
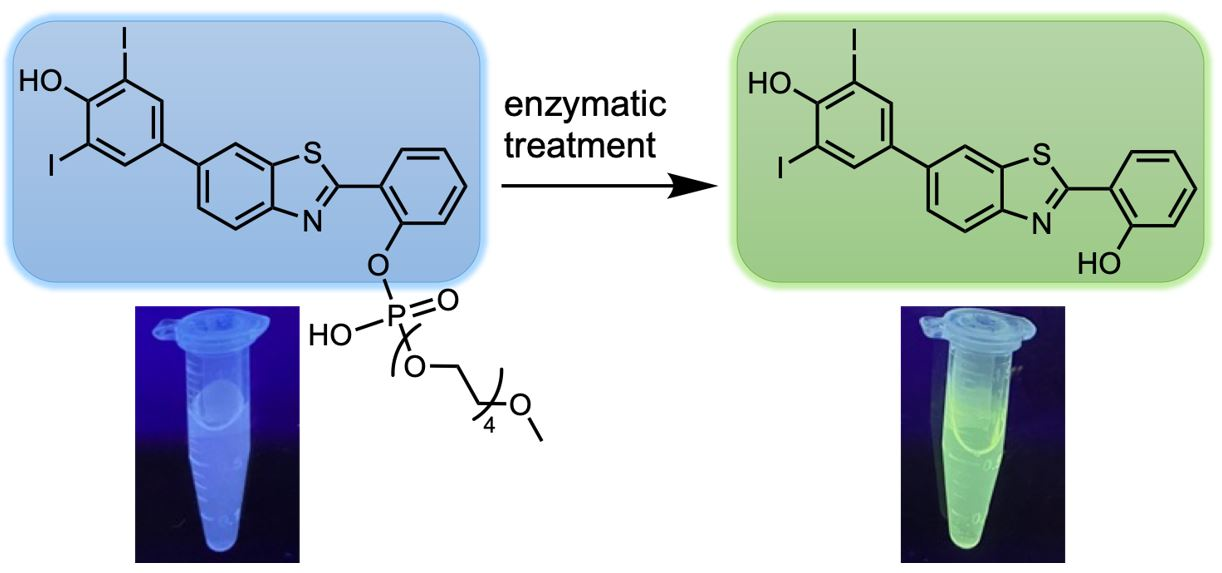
:
1. Introduction
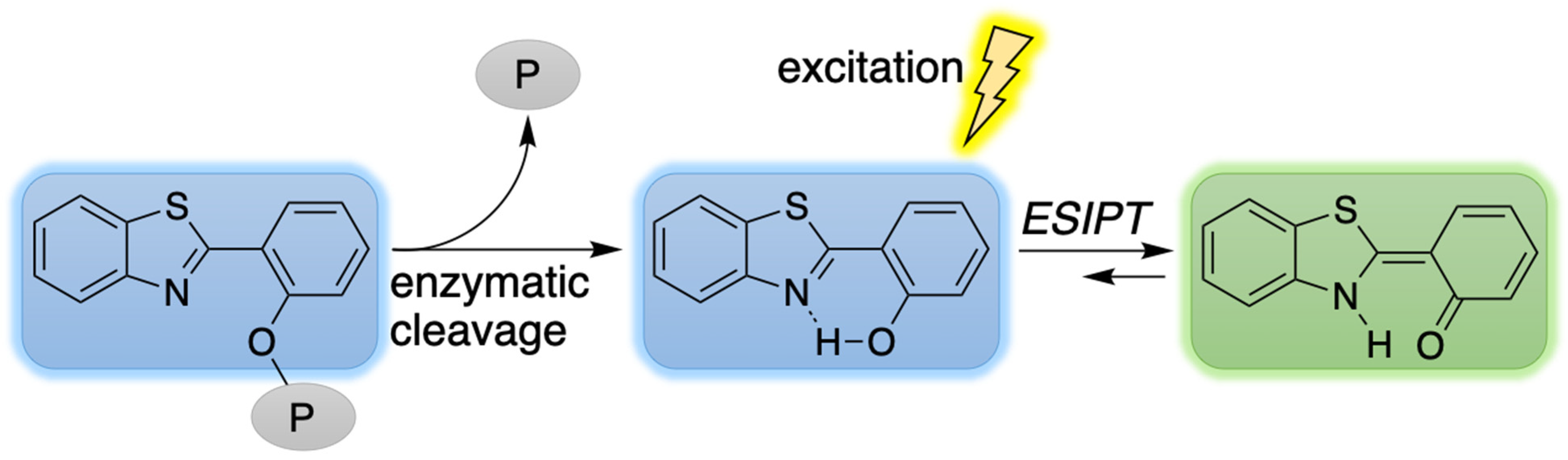
2. Reconnaissance
3. Results and Discussion
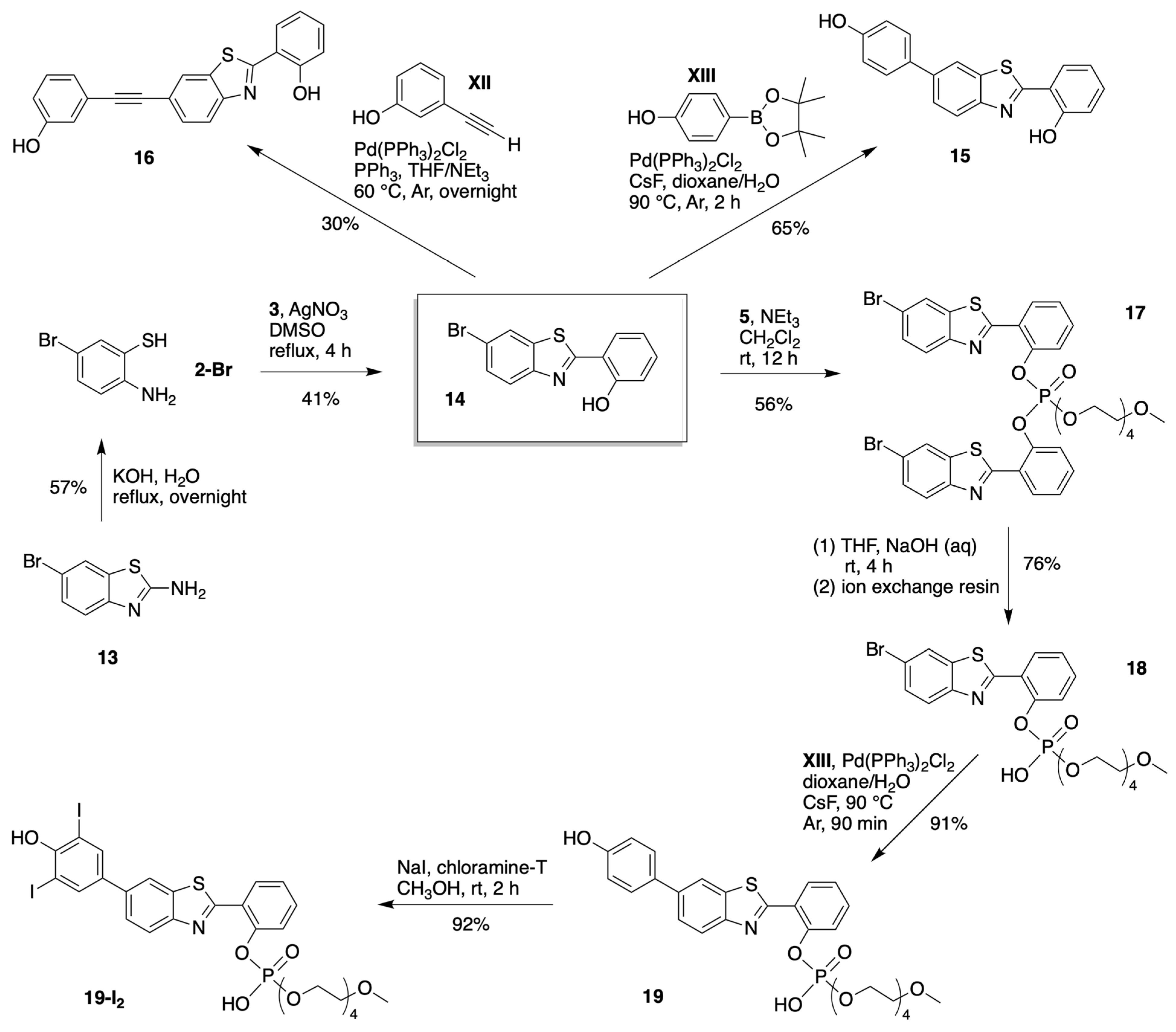
3.1. Synthesis
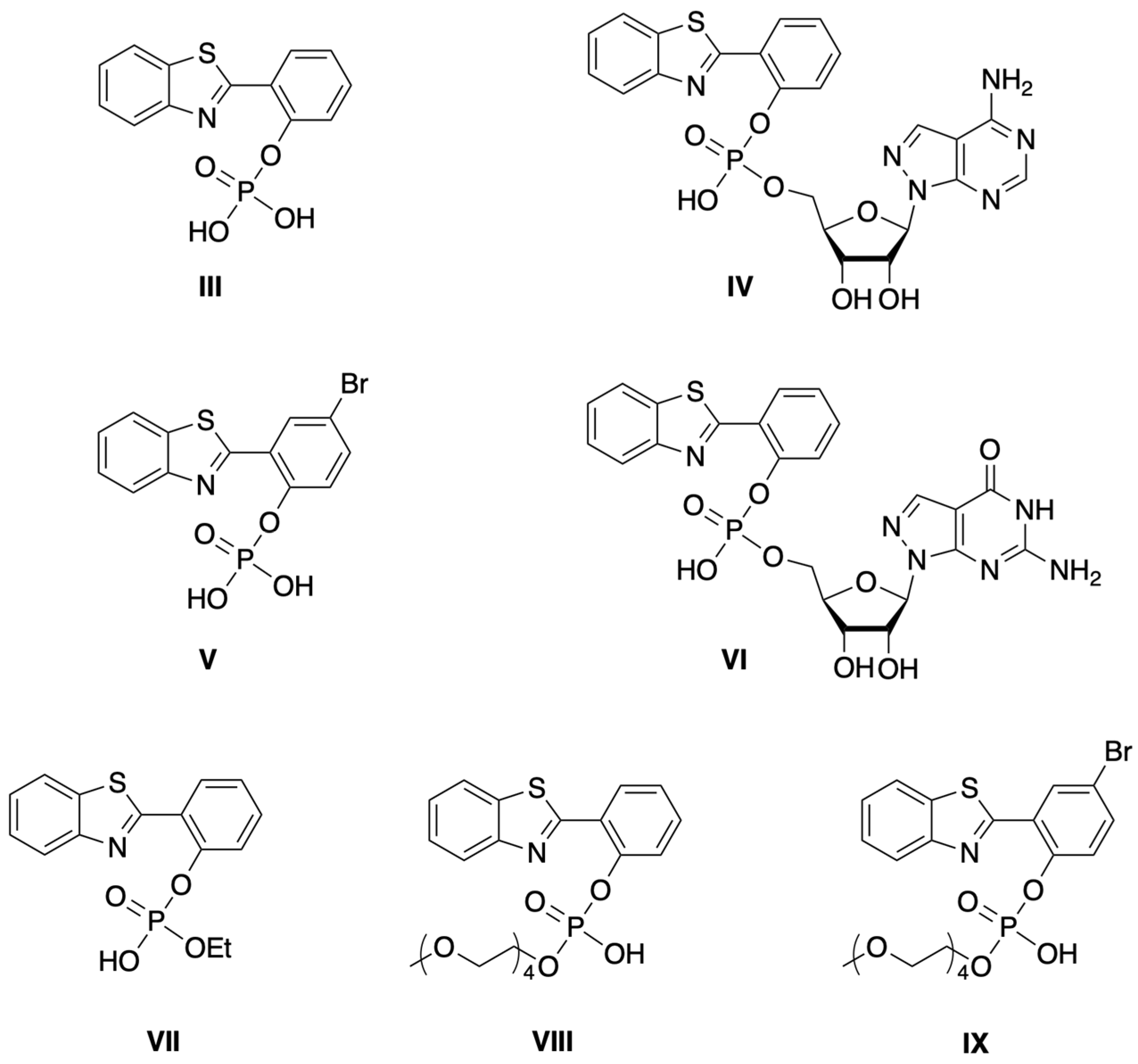
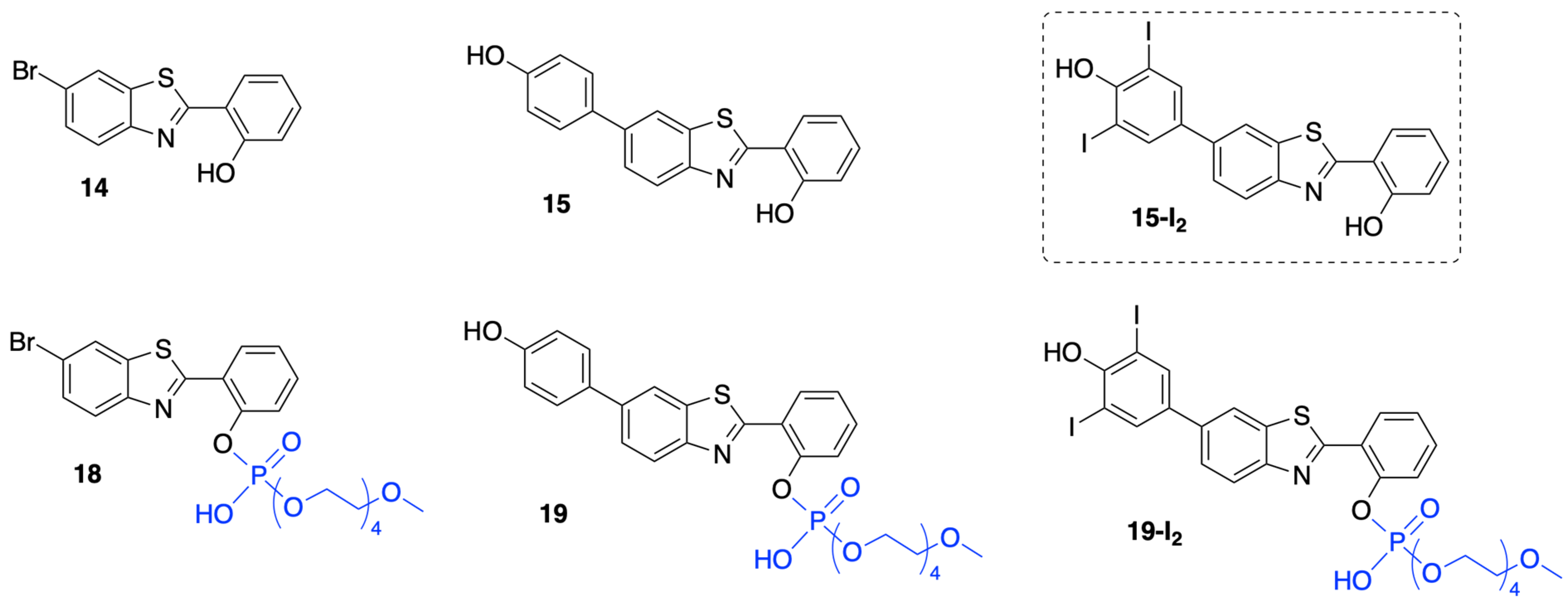
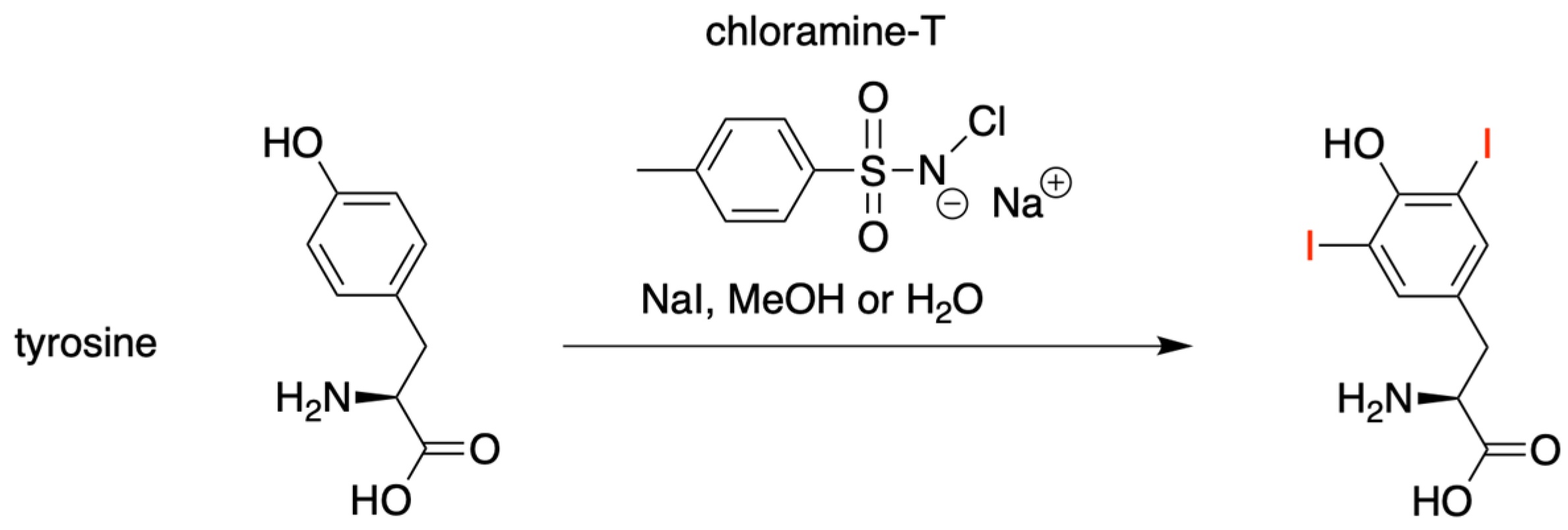
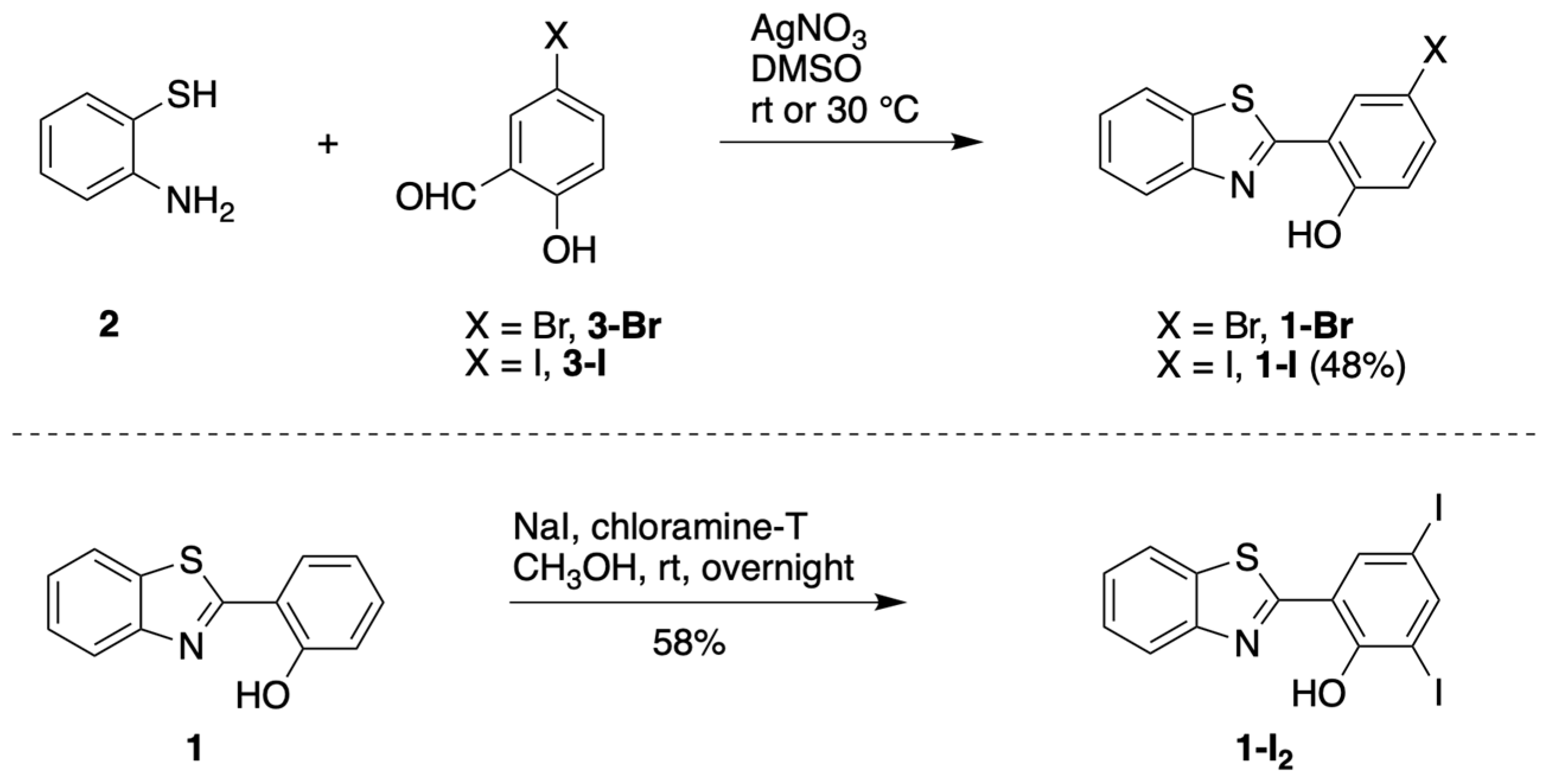
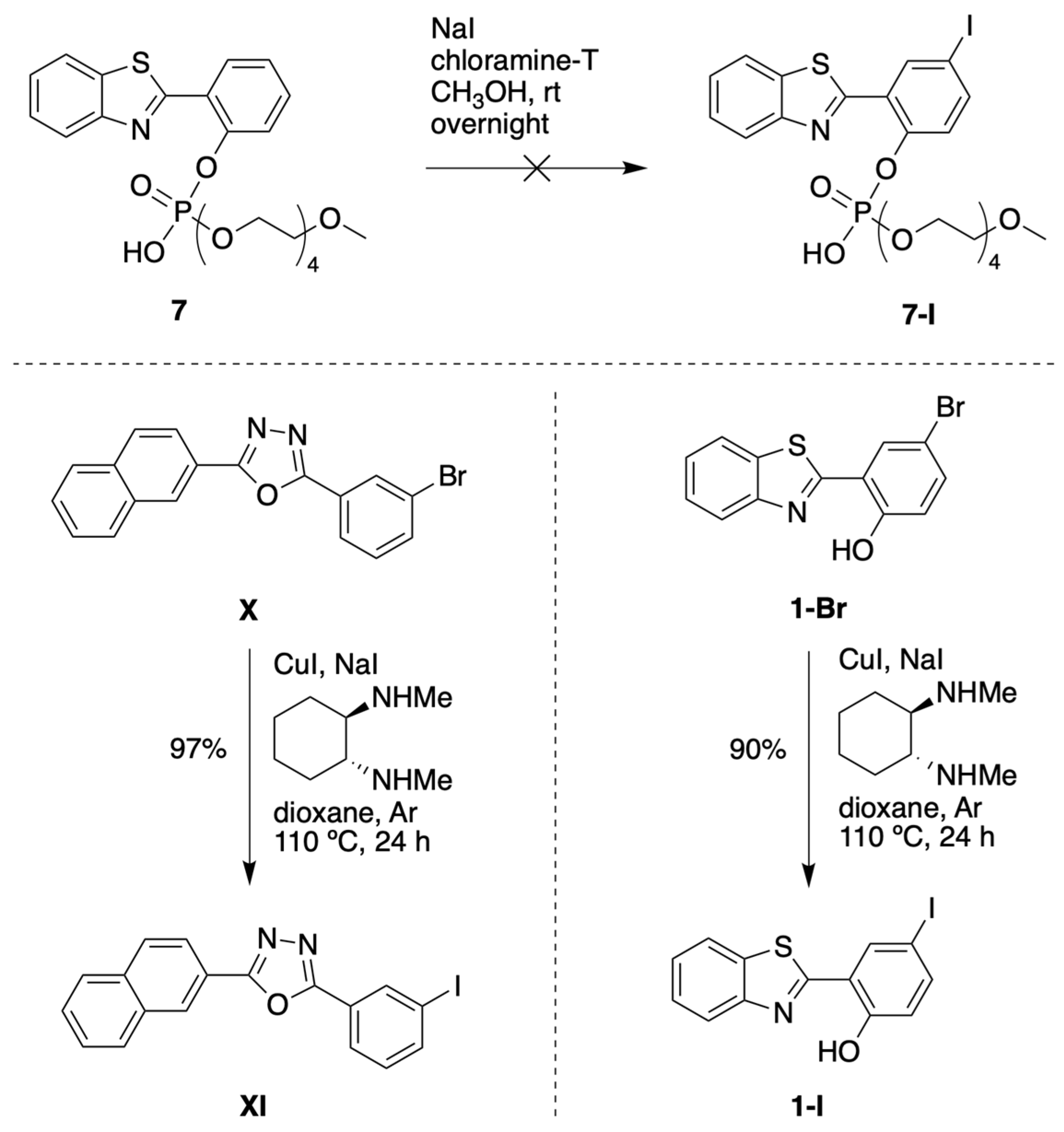
3.1.1. HBT Derivatives for Studies of Iodination
3.1.2. Introduction of Iodine with HBT Substrates
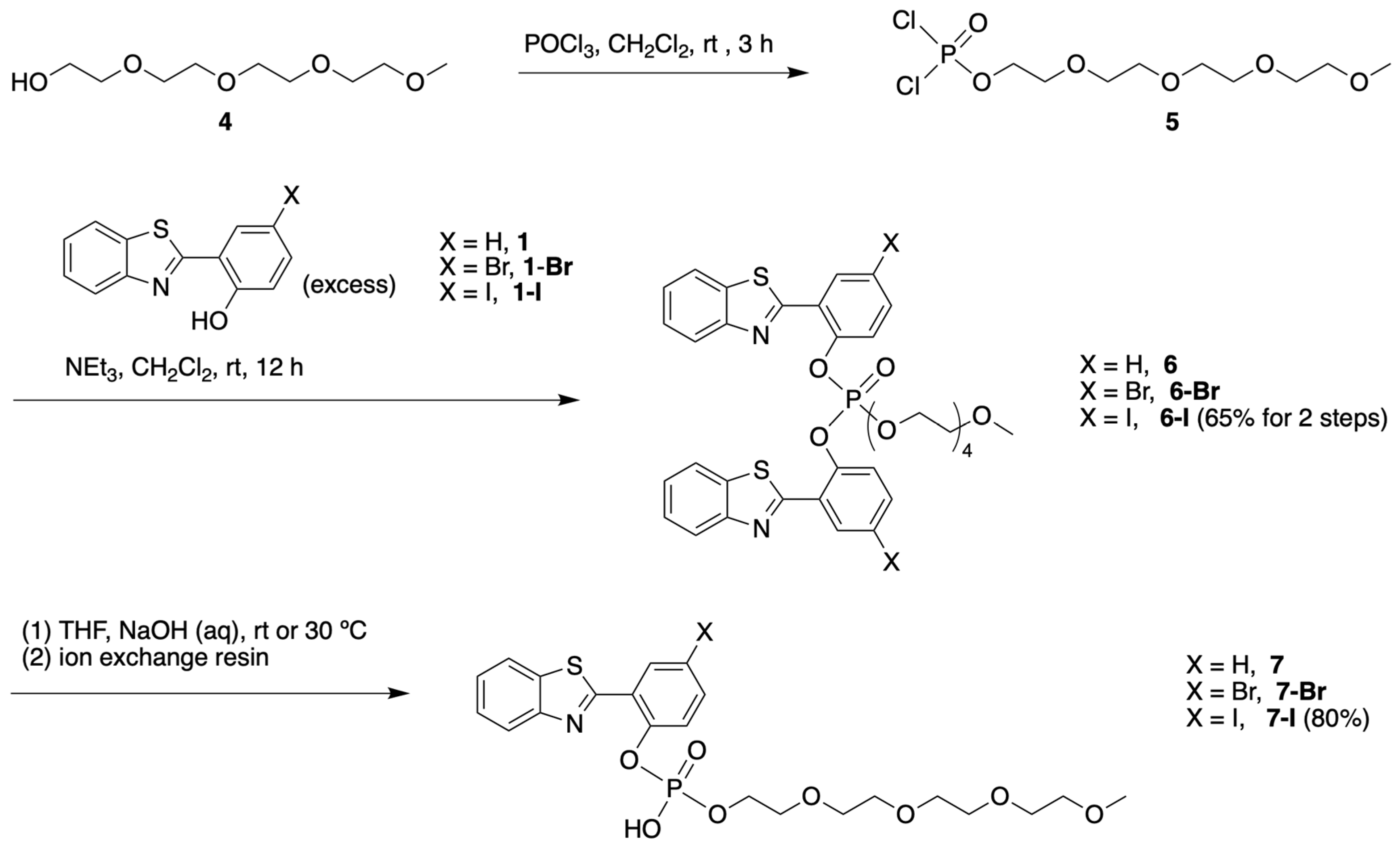
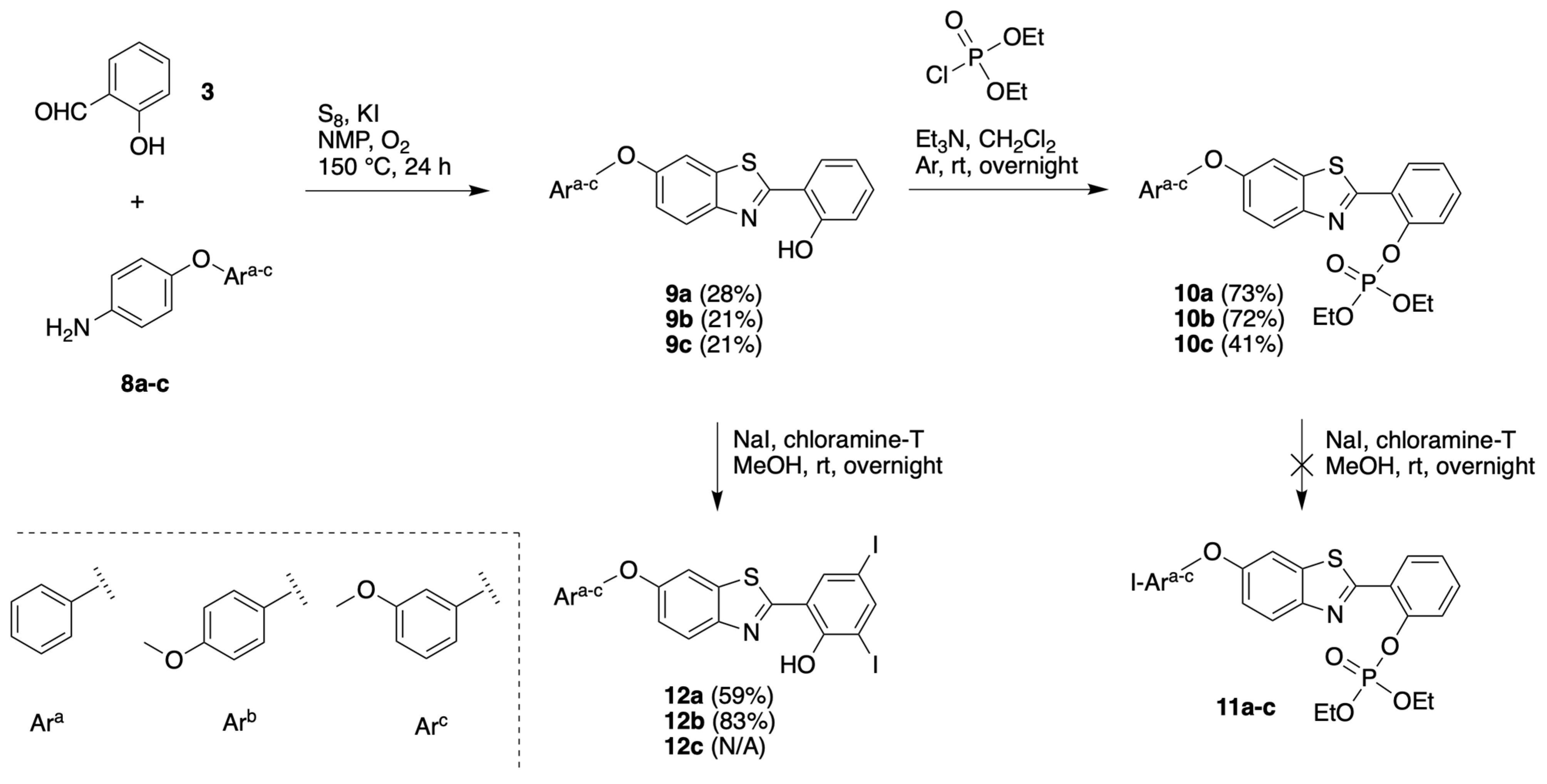
3.1.3. HBT Derivatives with an Appended Aryloxy Group for Iodination
3.1.4. HBT Derivatives with an Appended Phenol Group for Iodination
3.2. Chemical Characterization
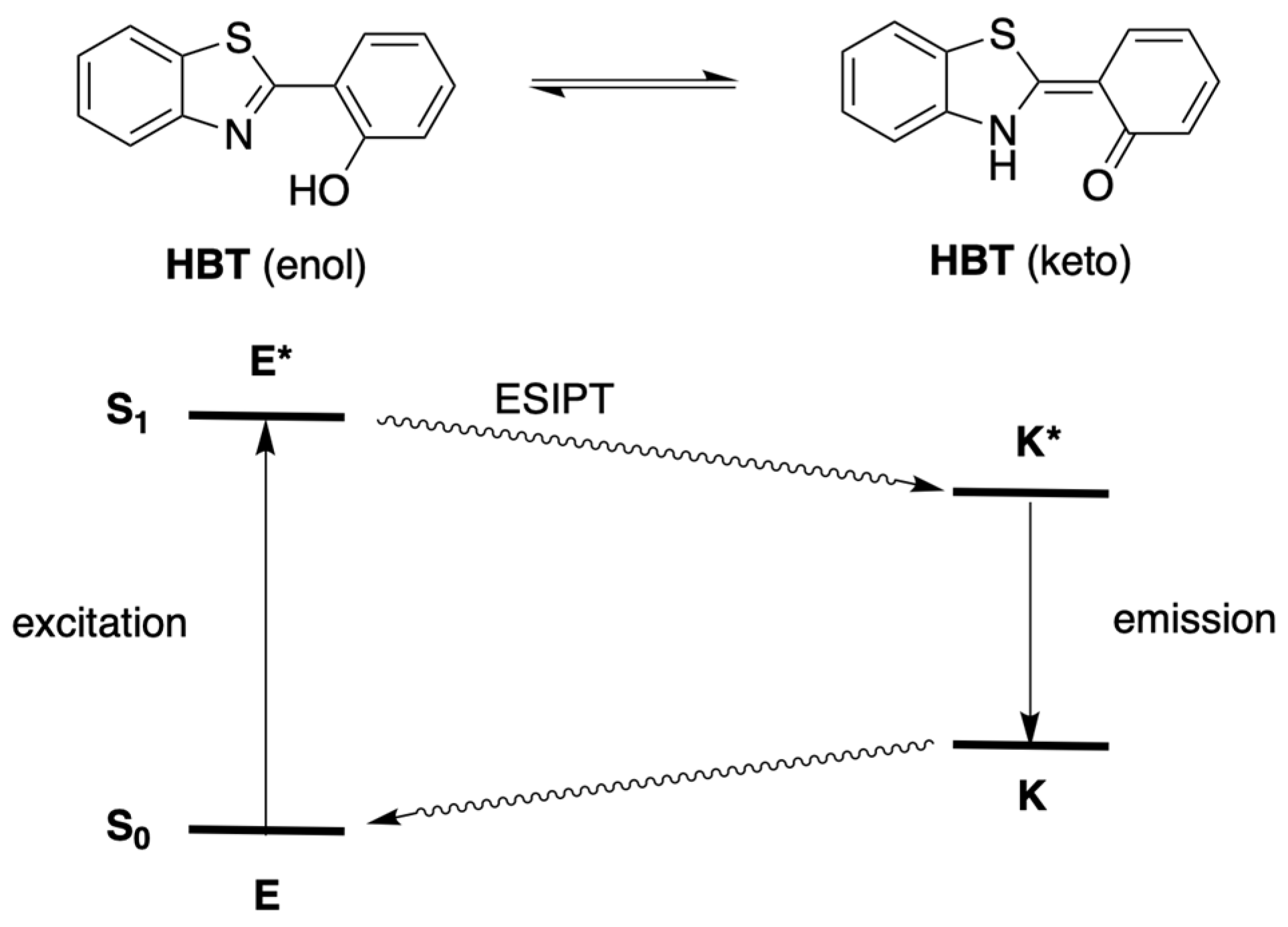
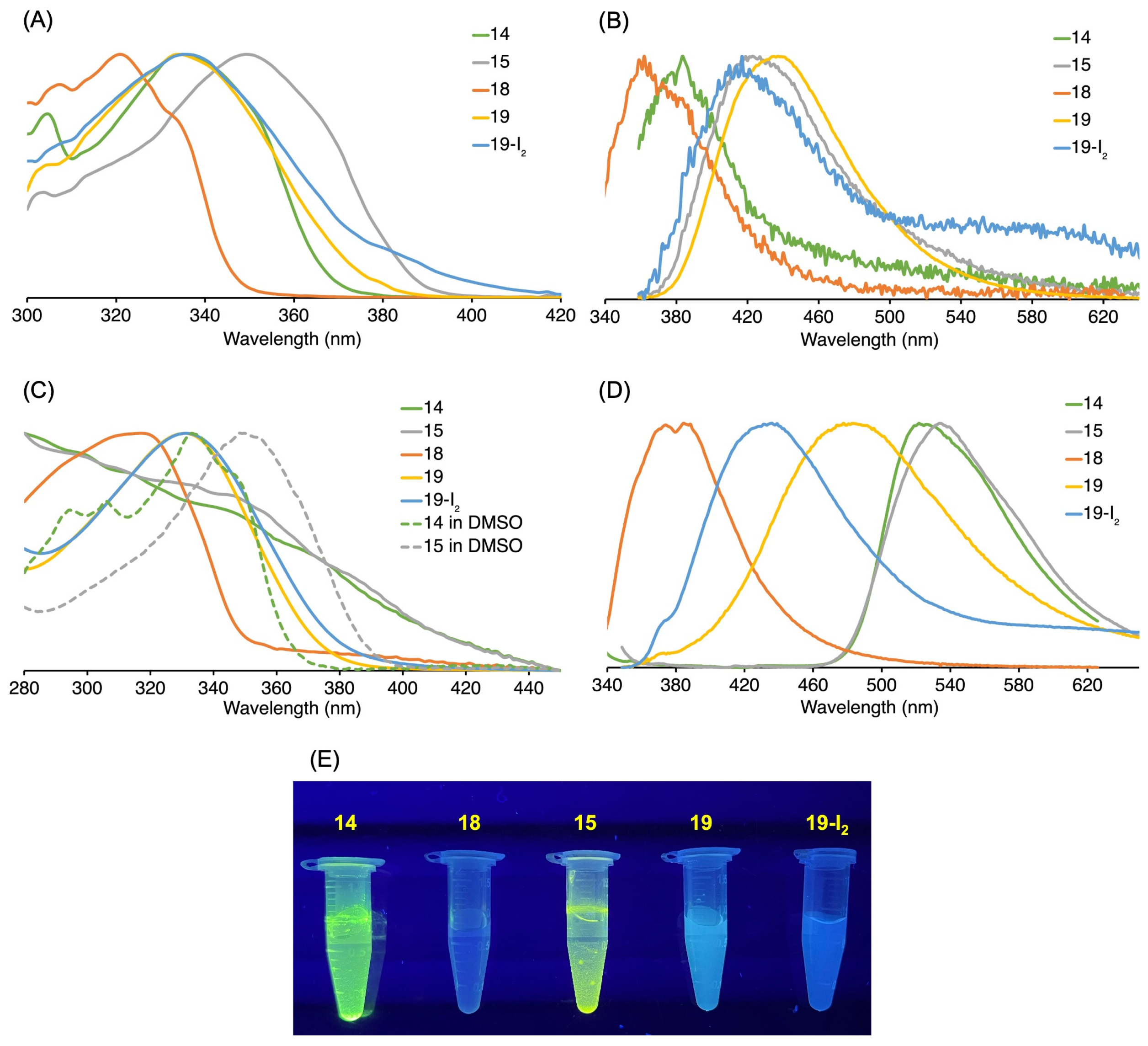
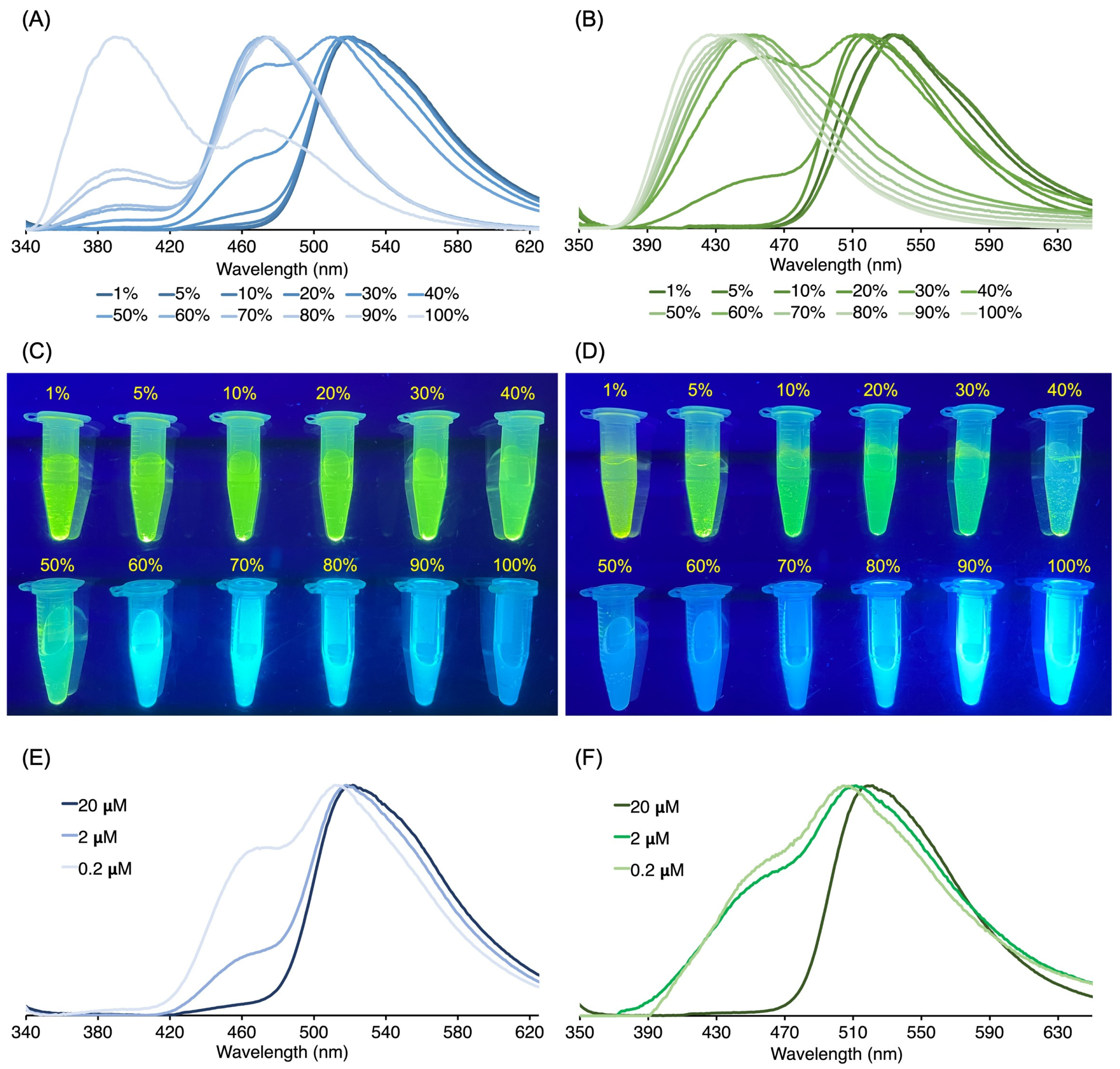
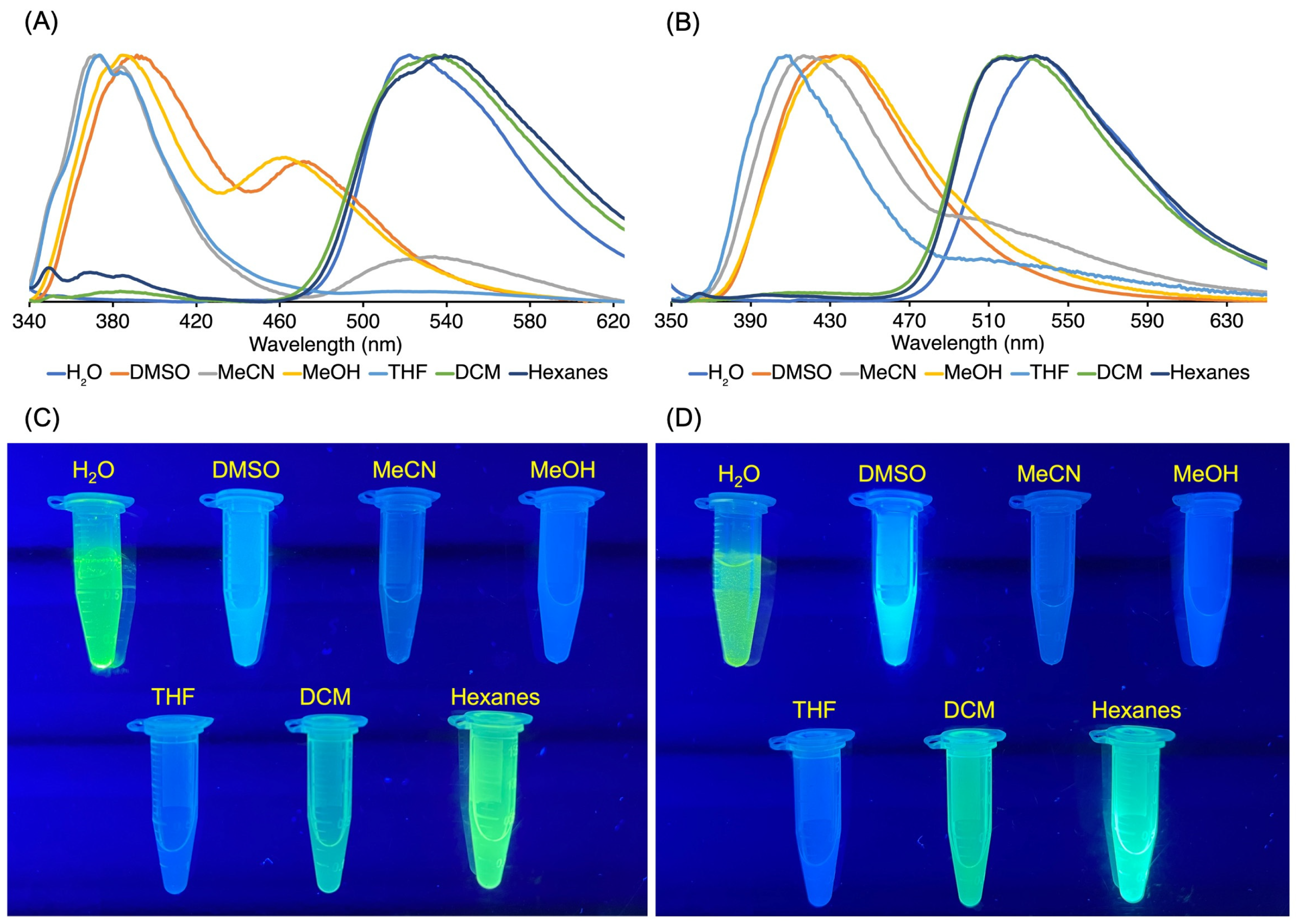
3.3. Absorption and Emission Properties
3.3.1. Homogeneous Solution

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λabs (nm) | λexc (nm) | λem (nm) b | λf c |
|---|---|---|---|---|
| 14 | 335 | 347 | 383 | 0.017 |
| 15 | 349 | 347 | 422 | 0.066 |
| 18 | 321 | 321a | 361 | 0.026 |
| 19 | 334 | 347 | 436 | 0.64 |
| 19-I2 | 336 | 347 | 416 | 0.027 |
3.3.2. Heterogeneous Samples
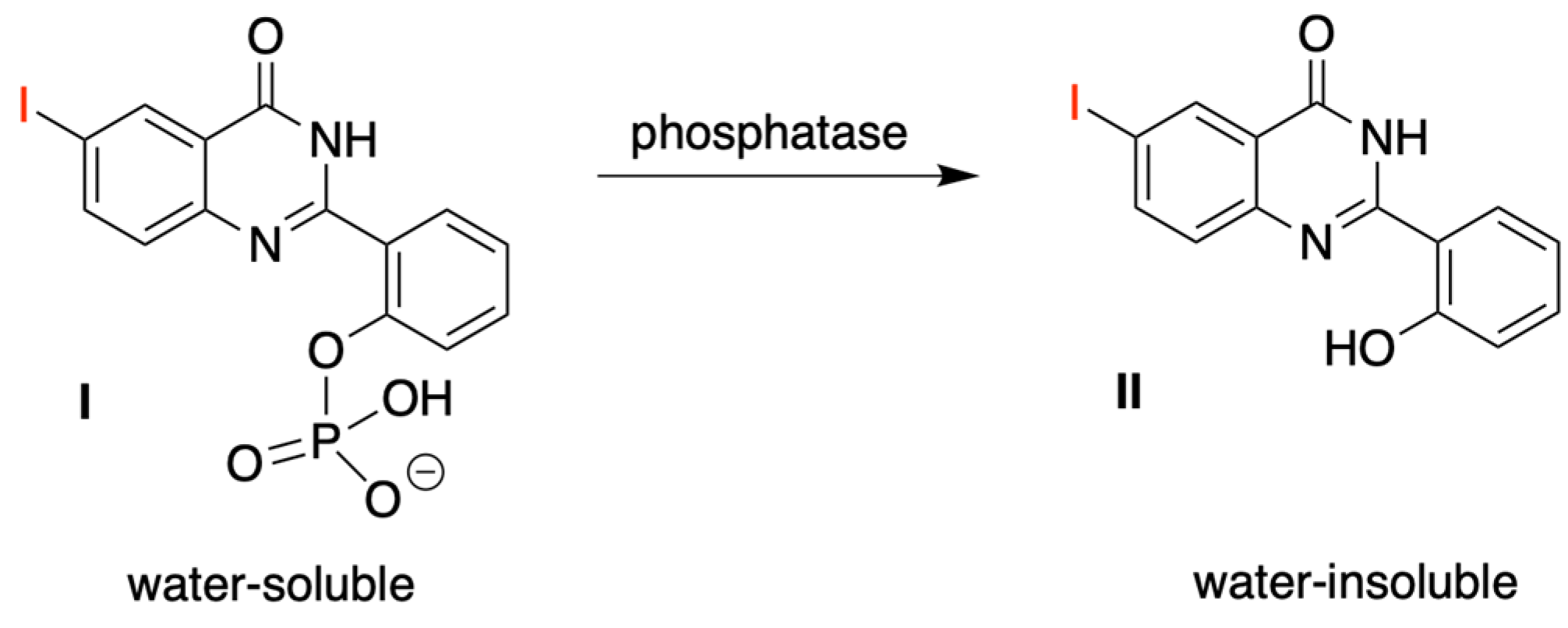
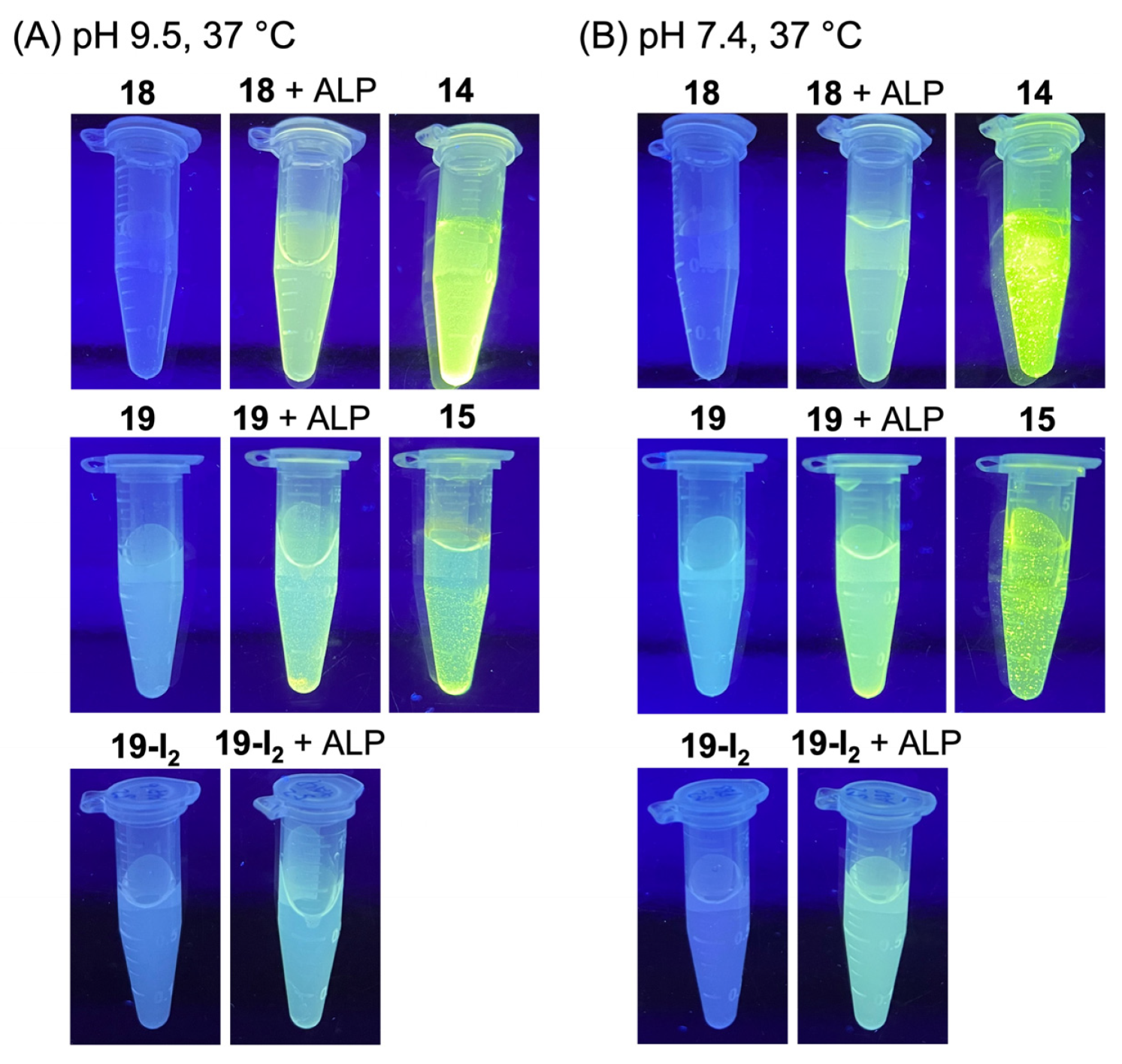
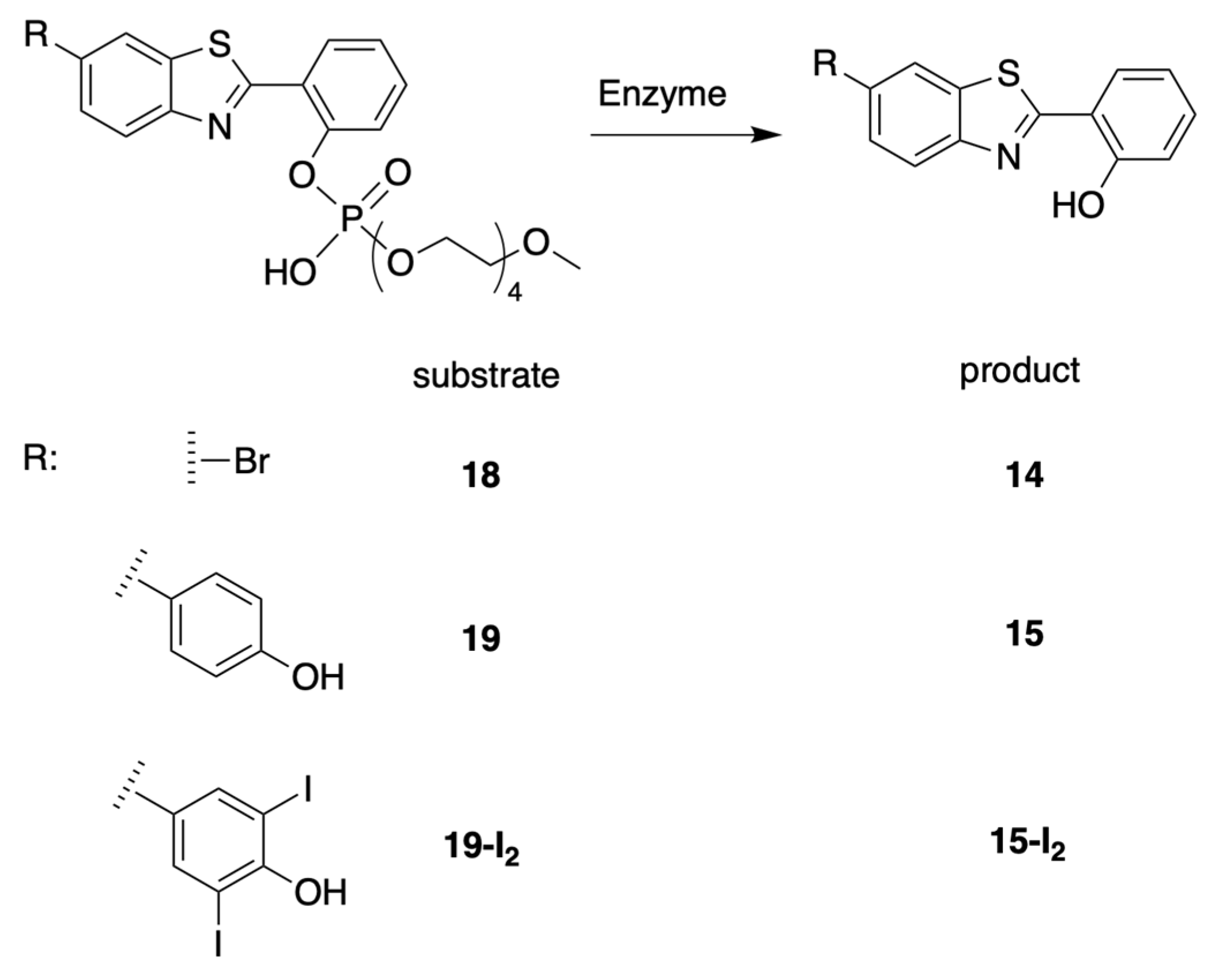
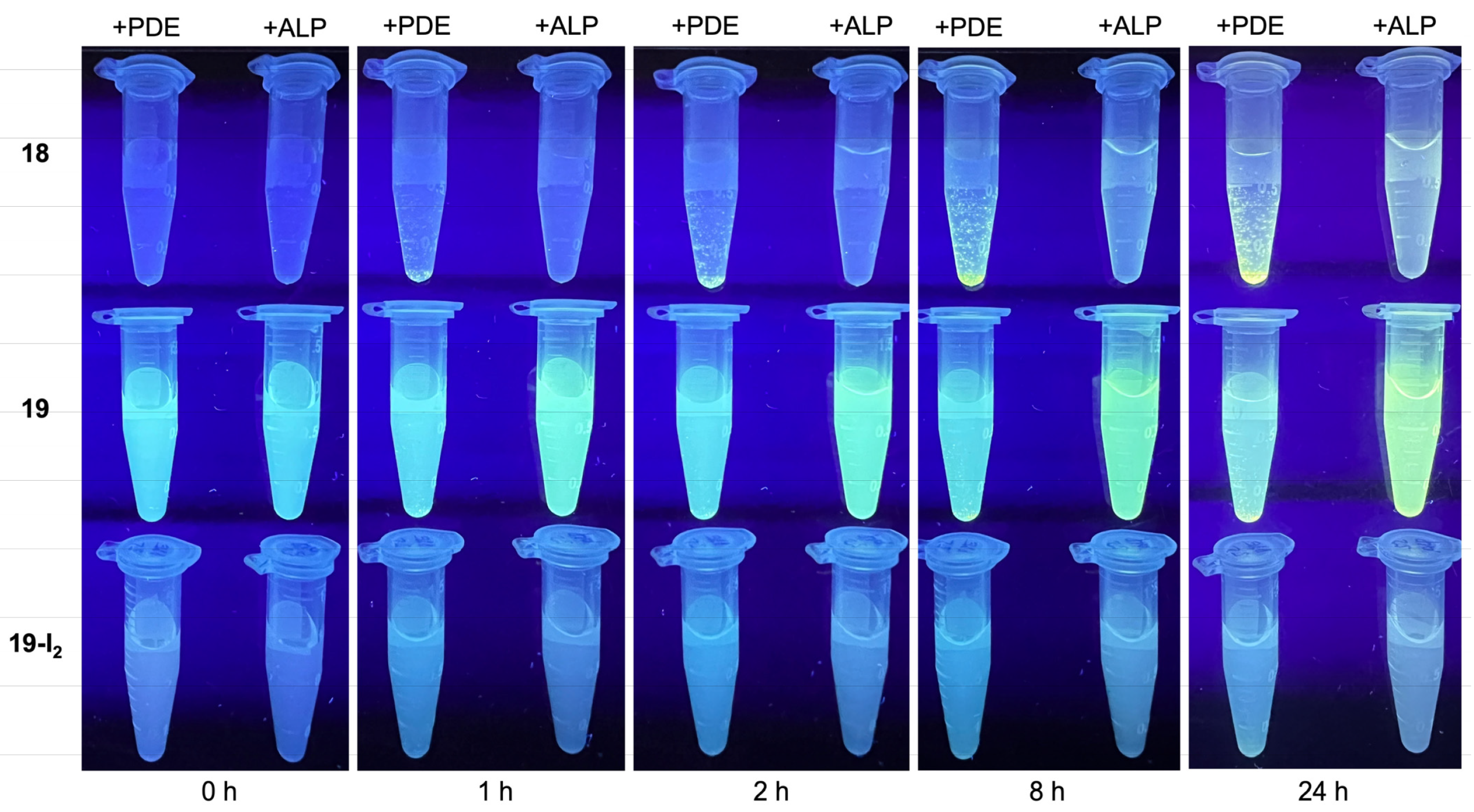
3.4. Treatment with Phosphodiesterase and Alkaline Phosphatase Enzymes
4. Materials and Methods
4.1. General Methods
4.2. Synthesis
4.3. Assays–Methods
4.3.1. Enzymes and Buffers
4.3.2. Absorption and Emission Measurements
4.3.3. AIE Visualization
4.4. Assays–Experiments
4.4.1. PDE with Target Compounds under Basic Conditions
4.4.2. PDE with Target Compounds under Physiological Conditions
4.4.3. PDE Timecourse with Target Compounds under Physiological Conditions
4.4.4. ALP with Target Compounds under Basic Conditions
4.4.5. ALP with Target Compounds under Physiological Conditions
4.4.6. PDE vs ALP with Target Compounds under Physiological Conditions–Timecourse
5. Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Williamson, J.F.; Brenner, D.J. Chapter 19. Physics and Biology of Brachytherapy. In Perez and Brady’s Principles and Practice of Radiation Oncology, 5th ed.; Halperin, E.C., Perez, C.A., Brady, L.W., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008; pp. 423–475. [Google Scholar]
- Otter, S.J.; Stewart, A.J.; Devlin, P.M. Modern Brachytherapy. Hematol. Oncol. Clin. N. Am. 2019, 33, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Bagshawe, K.D. Towards Generating Cytotoxic Agents at Cancer Sites. Br. J. Cancer 1989, 60, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Bagshawe, K.D.; Sharma, S.K.; Springer, C.J.; Rogers, G.T. Antibody Directed Enzyme Prodrug Therapy (ADEPT): A Review of Some Theoretical, Experimental and Clinical Aspects. Ann. Oncol. 1994, 5, 879–891. [Google Scholar] [CrossRef]
- Niculescu-Duvaz, I.; Springer, C.J. Antibody-Directed Enzyme Prodrug Therapy (ADEPT): A Review. Adv. Drug Deliv. Rev. 1997, 26, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Bagshawe, K.D. Antibody-Directed Enzyme Prodrug Therapy (ADEPT) for Cancer. Expert Rev. Anticancer Ther. 2006, 6, 1421–1431. [Google Scholar] [CrossRef]
- Boerman, O.C.; van Schaijk, F.G.; Oyen, W.J.G.; Corstens, F.H.M. Pretargeted Radioimmunotherapy of Cancer: Progress Step by Step. J. Nucl. Med. 2003, 44, 400–411. [Google Scholar]
- Membreno, R.; Cook, B.E.; Fung, K.; Lewis, J.S.; Zeglis, B.M. Click-Mediated Pretargeted Radioimmunotherapy of Colorectal Carcinoma. Mol. Pharm. 2018, 15, 1729–1734. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, B. Enzyme-Instructed Self-Assembly: A Multistep Process for Potential Cancer Therapy. Bioconjug. Chem. 2015, 26, 987–999. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.-H.; Harapanhalli, R.S.; Dahman, B.A.; Chen, K.; Wang, K.; Adelstein, S.J.; Kassis, A.I. Synthesis and Biologic Evaluation of Radioiodinated Quinazolinone Derivative for Enzyme-Mediated Insolubilization Therapy. Bioconjug. Chem. 2002, 13, 357–364. [Google Scholar] [CrossRef]
- Chen, K.; Wang, K.; Kirichian, A.M.; Al Aowad, A.F.; Iyer, L.K.; Adelstein, S.J.; Kassis, A.I. In silico Design, Synthesis, and Biological Evaluation of Radioiodinated Quinazolinone Derivatives for Alkaline Phosphatase-Mediated Cancer Diagnosis and Therapy. Mol. Cancer Ther. 2006, 5, 3001–3013. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Kirichian, A.M.; Al Aowad, A.F.; Adelstein, S.J.; Kassis, A.I. Effect of Chemical, Physical, and Biologic Properties of Tumor-Targeting Radioiodinated Quinazolinone Derivative. Bioconjug. Chem. 2007, 18, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Al Aowad, A.F.; Adelstein, S.J.; Kassis, A.I. Molecular-Docking-Guided Design, Synthesis, and Biologic Evaluation of Radioiodinated Quinazolinone Prodrugs. J. Med. Chem. 2007, 50, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Pospisil, P.; Wang, K.; Al Aowad, A.F.; Iyer, L.K.; Adelstein, S.J.; Kassis, A.I. Computational Modeling and Experimental Evaluation of A Novel Prodrug for Targeting the Extracellular Space of Prostate Tumors. Cancer Res. 2007; 67, 2197–2205. [Google Scholar] [CrossRef] [Green Version]
- Kassis, A.I.; Korideck, H.; Wang, K.; Pospisil, P.; Adelstein, S.J. Novel Prodrugs for Targeting Diagnostic and Therapeutic Radionuclides to Solid Tumors. Molecules 2008, 13, 391–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Palmer, M.R.; Makrigiorgos, G.M.; Kassis, A.I. Solid-Tumor Radionuclide Therapy Dosimetry: New Paradigms in View of Tumor Microenvironment and Angiogenesis. Med. Phys. 2010, 37, 2974–2984. [Google Scholar] [CrossRef] [Green Version]
- Pospisil, P.; Korideck, H.; Wang, K.; Yang, Y.; Iyer, L.K.; Kassis, A.I. Computational and Biological Evaluation of Quinazolinone Prodrug for Targeting Pancreatic Cancer. Chem. Biol. Drug Des. 2012, 79, 926–934. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Chien, M.-P.; Thompson, M.P.; Barback, C.V.; Ku, T.-H.; Hall, D.J.; Gianneschi, N.C. Enzyme-Directed Assembly of A Nanoparticle Probe in Tumor Tissue. Adv. Mater. 2013, 25, 3599–3604. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Borbas, K.E.; Lindsey, J.S. Soluble Precipitable Porphyrins for Use in Targeted Molecular Brachytherapy. New J. Chem. 2008, 32, 436–451. [Google Scholar] [CrossRef]
- Kassis, A.I. Compounds and Methods for Enzyme-Mediated Tumor Imaging and Therapy. U.S. Patent 9,320,815 B2, 26 April 2016. [Google Scholar]
- Rose, S. A Proposal for A New Direction to Treat Cancer. J. Theor. Biol. 1998, 195, 111–128. [Google Scholar] [CrossRef]
- Mayers, G.L. Targeted Molecular Brachytherapy. Drug Dev. Res. 2006, 67, 94–106. [Google Scholar] [CrossRef]
- Fujita, H.; Dou, J.; Matsumoto, N.; Wu, Z.; Lindsey, J.S. Enzymatically Triggered Chromogenic Cross-Linking Agents Under Physiological Conditions. New J. Chem. 2020, 44, 719–743. [Google Scholar] [CrossRef]
- Fujita, H.; Zhang, Y.; Wu, Z.; Lindsey, J.S. Chromogenic Agents Built Around a Multifunctional Double-Triazine Framework for Enzymatically Triggered Cross-Linking Under Physiological Conditions. New J. Chem. 2020, 44, 3856–3867. [Google Scholar] [CrossRef]
- Lee, D.L.; Chin, H.-L.M.; Knudsen, C.G.; Mayers, G.L.; Rose, D.S.; Skogstrom, R.K.; Palzkill, T.; Fujita, H.; Zhang, Y.; Wu, Z.; et al. Peptide-Based Scaffolds for in vivo Immobilization and Enzyme Attachment in Therapeutic Applications. Proc. SPIE 2020, 11477, 1147708. [Google Scholar]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient Cancer Therapy with a Nanobody-Based Conjugate. Cancer Res. 2004, 64, 2853–2857. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wu, Z.; Takashima, I.; Nguyen, K.-U.; Matsumoto, N.; Lindsey, J.S. Engineering of an Archaeal Phosphodiesterase to Trigger Aggregation-Induced Emission (AIE) of Synthetic Substrates. New J. Chem. 2020, 44, 14266–14277. [Google Scholar] [CrossRef]
- Mei, J.; Leung, N.L.C.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef]
- Hong, Y.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission. Chem. Soc. Rev. 2011, 40, 5361–5388. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-I.; Kang, H.J.; Han, G.; Chung, S.J.; Kim, Y. A Highly Selective Fluorescent ESIPT Probe for the Dual Specificity Phosphatase MKP-6. Chem. Commun. 2009, 45, 5895–5897. [Google Scholar] [CrossRef]
- Sedgwick, A.C.; Wu, L.; Han, H.-H.; Bull, S.D.; He, X.-P.; James, T.D.; Sessler, J.L.; Tang, B.Z.; Tian, H.; Yoon, J. Excited-State Intramolecular Proton-transfer (ESIPT) Based Fluorescence Sensors and Imaging Agents. Chem. Soc. Rev. 2018, 47, 8842–8880. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Otsubo, T.; Ikeda, K.; Minami, A.; Suzuki, T. Histochemical Imaging of Alkaline Phosphatase Using a Novel Fluorescent Substrate. Biol. Pharm. Bull. 2014, 37, 1668–1673. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, P.; Han, K. AMP/GMP Analogs as Affinity ESIPT Probes for Highly Selective Sensing of Alkaline Phosphatase Activity in Living Systems. Chem. Asian J. 2015, 10, 2444–2451. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.W.; Makvandi, M.; Xu, K.; Mach, R.H. Rapid Cu-Catalyzed [211At]Astatination and [125I]Iodination of Boronic Esters at Room Temperature. Org. Lett. 2018, 20, 1752–1755. [Google Scholar] [CrossRef]
- Greenwood, F.C.; Hunter, W.M.; Glover, J.S. The Preparation of 131I-Labelled Human Growth Hormone of High Specific Radioactivity. Biochem. J. 1963, 89, 114–123. [Google Scholar] [CrossRef] [PubMed]
- McConahey, P.J.; Dixon, F.J. Radioiodination of Proteins by the Use of the Chloramine-T Method. Meth. Enzymol. 1980, 70, 210–213. [Google Scholar]
- Hermanson, G.T. Chapter 12. Isotopic Labeling Techniques. In Bioconjugate Techniques, 3rd ed.; Hermanson, G.T., Ed.; Academic Press: London, UK, 2013; pp. 512–523. [Google Scholar]
- He, X.; Wu, Y.; Jin, W.; Wang, X.; Wu, C.; Shang, Y. Highly Efficient AgNO3-Catalyzed Approach to 2-(Benzo[d]azol-2-yl)phenols from Salicylaldehydes with 2-Aminothiophenol, 2-Aminophenol and Benzene-1,2-diamine. Appl. Organomet. Chem. 2018, 32, e4284. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, C.; Sánchez de Groot, N.; Rimola, A.; Alvarez-Larena, A.; Lloveras, V.; Vidal-Gancedo, J.; Ventura, S.; Vendrell, J.; Sodupe, M.; González-Duarte, P. Design, Selection, and Characterization of Thioflavin-Based Intercalation Compounds with Metal Chelating Properties for Application in Alzheimer’s Disease. J. Am. Chem. Soc. 2009, 131, 1436–1451. [Google Scholar] [CrossRef]
- Anthony, K.; Brown, R.G.; Hepworth, J.D.; Hodgson, K.W.; May, B.; West, M.A. Solid-State Fluorescent Photophysics of Some 2-Substituted Benzothiazoles. J. Chem. Soc. Perkin Trans. 2 1984, 2111–2117. [Google Scholar] [CrossRef]
- Klapars, A.; Buchwald, S.L. Copper-Catalyzed Halogen Exchange in Aryl Halides: An Aromatic Finkelstein Reaction. J. Am. Chem. Soc. 2002, 124, 14844–14845. [Google Scholar] [CrossRef]
- Che, X.; Jiang, J.; Xiao, F.; Huang, H.; Deng, G.-J. Assembly of 2-Arylbenzothiazoles through Three-Component Oxidative Annulation under Transition-Metal-Free Conditions. Org. Lett. 2017, 19, 4576–4579. [Google Scholar] [CrossRef]
- Kumar, M.; Sharma, K.; Fogla, A.K.; Sharma, K.; Rathore, M. Synthesis and Antimicrobial Activity of 2,4-Diaryl-2,3-dihydrobenzo[b][1,4]thiazepines. Res. Chem. Intermed. 2013, 39, 2555–2564. [Google Scholar] [CrossRef]
- Chou, P.-T.; Cooper, W.C.; Clements, J.H.; Studer, S.L.; Chang, C.P. A Comparative Study. The Photophysics of 2-Phenylbenzoxazoles and 2-Phenylbenzothiazoles. Chem. Phys. Lett. 1993, 216, 300–304. [Google Scholar] [CrossRef]
- Potter, C.A.S.; Brown, R.G.; Vollmer, F.; Rettig, W. Role of Twisted Intramolecular Charge-transfer States in the Decay of 2-(2′-Hydroxyphenyl)benzothiazole following Excited-state Intramolecular Proton Transfer. J. Chem. Soc. Faraday Trans. 1994, 90, 59–67. [Google Scholar] [CrossRef]
- Sakurai, T.; Kobayashi, M.; Yoshida, H.; Shimizu, M. Remarkable Increase of Fluorescence Quantum Efficiency by Cyano Substitution on an ESIPT Molecule 2-(2-Hydroxyphenyl)benzothiazole: A Highly Photoluminescent Liquid Crystal Dopant. Crystals 2021, 11, 1105. [Google Scholar] [CrossRef]
- Ren, Y.; Fan, D.; Ying, H.; Li, X. Rational Design of the Benzothiazole-based Fluorescent Scaffold for Tunable Emission. Tetrahedron Lett. 2019, 60, 1060–1065. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, X.; Ma, F.; Li, Y. Substituent Effect on ESIPT Mechanisms and Photophysical Properties of HBT Derivatives. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 250, 119375. [Google Scholar] [CrossRef]
- Fujikawa, T.; Uehara, T.; Yamaji, M.; Kanetomo, T.; Ishida, T.; Maki, S.; Hirano, T. Structure-Fluorescence Relationship of Push-Pull 2-Phenylbenzothiazole Derivatives Designed Based on the Firefly Light-emitter. Tetrahedron Lett. 2018, 59, 1431–1434. [Google Scholar] [CrossRef]
- Taniguchi, M.; Lindsey, J.S. Database of Absorption and Fluorescence Spectra of >300 Common Compounds for Use in PhotochemCAD. Photochem. Photobiol. 2018, 94, 290–327. [Google Scholar] [CrossRef] [Green Version]
- Naka, K.; Sato, H.; Higashi, M. Theoretical Study of the Mechanism of the Solvent Dependency of ESIPT in HBT. Phys. Chem. Chem. Phys. 2021, 23, 20080–20085. [Google Scholar] [CrossRef]
- Chen, S.; Yakunin, A.F.; Kuznetsova, E.; Busso, D.; Pufan, R.; Proudfoot, M.; Kim, R.; Kim, S.H. Structural and Functional Characterization of A Novel Phosphodiesterase from Methanococcus jannaschii. J. Biol. Chem. 2004, 279, 31854–31862. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Gong, W.; Wu, B.; Wang, H.; He, W.; Dai, Z.; Li, Y.; Liu, Y.; Wang, Z.; Tuo, X.; et al. ASBase: The Universal Database for Aggregate Science. Aggregate 2022, e263. [Google Scholar] [CrossRef]
- Venkatesh, Y.; Das, J.; Chaudhuri, A.; Karmakar, A.; Maiti, T.K.; Singh, N.D.P. Light Triggered Uncaging of Hydrogen Sulfide (H2S) with Real-time Monitoring. Chem. Commun. 2018, 54, 3106–3109. [Google Scholar] [CrossRef] [PubMed]
















| Compound | λabs (nm) | λexc (nm) | λem (nm) |
|---|---|---|---|
| 14 | 334a | 318 | 522 |
| 15 | 350 a | 330 | 534 |
| 18 | 318 | 318 | 374, 384 |
| 19 | 330 | 330 | 480 |
| 19-I2 | 330 | 330 | 435 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Dou, J.; Nguyen, K.-U.; Eppley, J.C.; Siwawannapong, K.; Zhang, Y.; Lindsey, J.S. Tailoring the AIE Chromogen 2-(2-Hydroxyphenyl)benzothiazole for Use in Enzyme-Triggered Molecular Brachytherapy. Molecules 2022, 27, 8682. https://doi.org/10.3390/molecules27248682
Wu Z, Dou J, Nguyen K-U, Eppley JC, Siwawannapong K, Zhang Y, Lindsey JS. Tailoring the AIE Chromogen 2-(2-Hydroxyphenyl)benzothiazole for Use in Enzyme-Triggered Molecular Brachytherapy. Molecules. 2022; 27(24):8682. https://doi.org/10.3390/molecules27248682
Chicago/Turabian StyleWu, Zhiyuan, Jinghuai Dou, Kathy-Uyen Nguyen, Jayden C. Eppley, Kittipan Siwawannapong, Yunlong Zhang, and Jonathan S. Lindsey. 2022. "Tailoring the AIE Chromogen 2-(2-Hydroxyphenyl)benzothiazole for Use in Enzyme-Triggered Molecular Brachytherapy" Molecules 27, no. 24: 8682. https://doi.org/10.3390/molecules27248682
APA StyleWu, Z., Dou, J., Nguyen, K. -U., Eppley, J. C., Siwawannapong, K., Zhang, Y., & Lindsey, J. S. (2022). Tailoring the AIE Chromogen 2-(2-Hydroxyphenyl)benzothiazole for Use in Enzyme-Triggered Molecular Brachytherapy. Molecules, 27(24), 8682. https://doi.org/10.3390/molecules27248682