

BACE1 Inhibitors for Alzheimer’s Disease: The Past, Present and Any Future?
{kind=link}
{kind=link}
Abstract
:1. Introduction
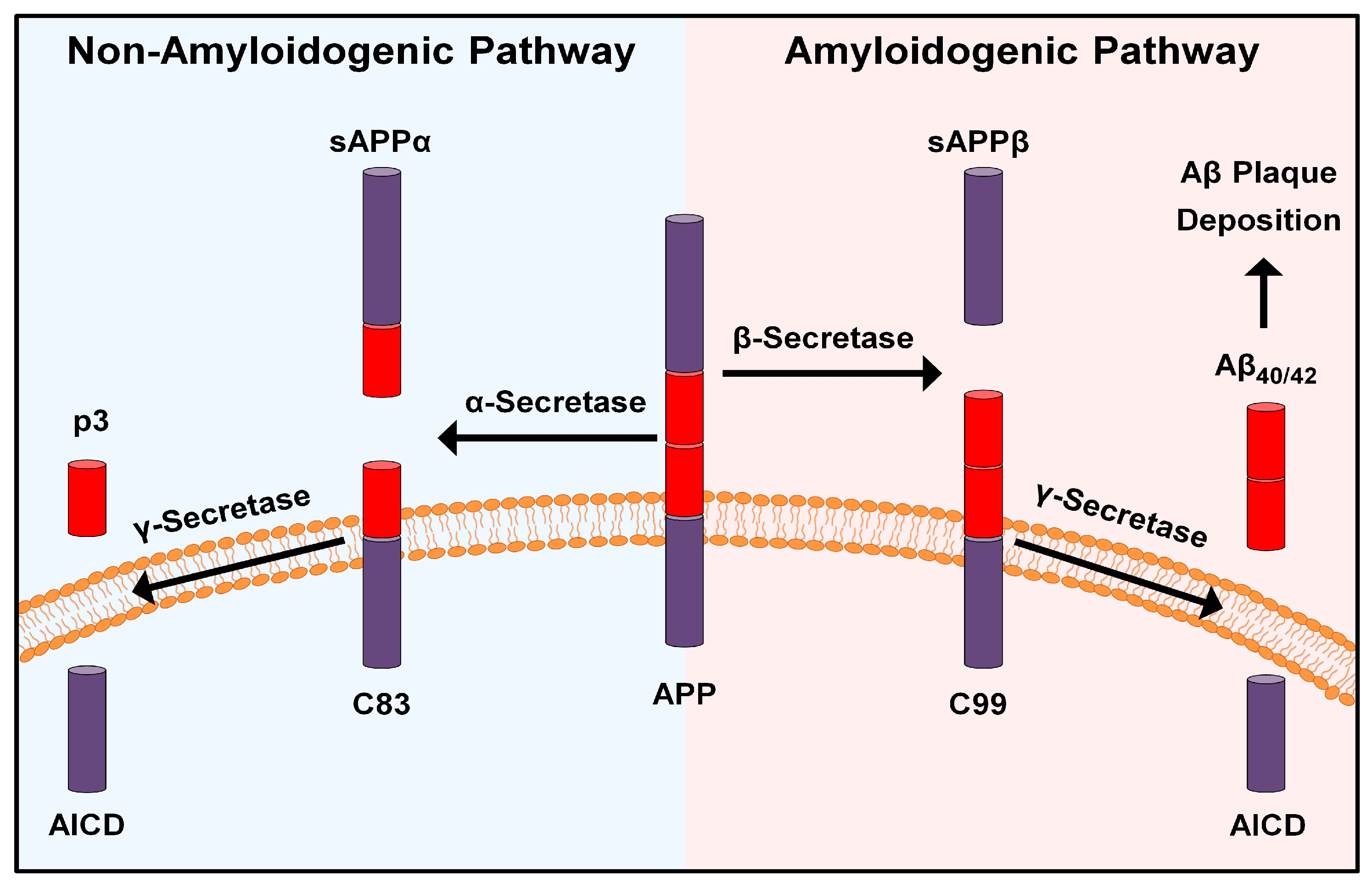
2. The “Amyloid Cascade Hypothesis” and Beyond
3. BACE1 Inhibitors in Clinical Trials: What Happened?
3.1. LY2811376
3.2. LY2886721
3.3. RG7129 (RO5508887)
3.4. BI 1181181
3.5. JNJ-54861911 (Atabecestat)
3.6. LY3314814 (AZD3293, Lanabecestat)
3.7. MK-8931 (MK-8931-009, Verubecestat)
3.8. E2609 (Elenbecestat)
3.9. CNP-520 (Umibecestat)
3.10. LY3202626
3.11. PF-06751979
3.12. CTS21166
3.13. HPP854
4. Discussion and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- World Alzheimer Report 2022. Life after Diagnosis: Navigating Treatment, Care and Support. Available online: https://www.alzint.org/resource/world-alzheimer-report-2022/ (accessed on 4 November 2022).
- Hippius, H.; Neundörfer, G. The discovery of Alzheimer’s disease. Dialogues Clin. Neurosci. 2003, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bazzari, F.H.; Abdallah, D.M.; El-Abhar, H.S. Pharmacological interventions to attenuate Alzheimer’s disease progression: The story so far. Curr. Alzheimer Res. 2019, 16, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Tam, K.Y. Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2022, 17, 543–549. [Google Scholar] [PubMed]
- Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Bian, Z.; Chen, X.; Lu, A.; Yang, Z. Review of current strategies for delivering Alzheimer’s disease drugs across the blood-brain barrier. Int. J. Mol. Sci. 2019, 20, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, B.; Gao, F.; Wu, R.; Dong, T.; Gu, C.; Lin, Q.; Zhang, Y. Vitamin D deficiency as a risk factor for dementia and Alzheimer’s disease: An updated meta-analysis. BMC Neurol. 2019, 19, 284. [Google Scholar] [CrossRef]
- Dolatshahi, M.; Salehipour, A.; Saghazadeh, A.; Sanjeari Moghaddam, H.; Aghamollaii, V.; Fotouhi, A.; Tafakhori, A. Thyroid hormone levels in Alzheimer disease: A systematic review and meta-analysis. Endocrine 2022. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.; Jansen, E.N.; Bratzke, H.; Braak, E. Neuropathological hallmarks of Alzheimer’s and Parkinson’s diseases. Prog. Brain Res. 1998, 117, 267–285. [Google Scholar]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Yan, R.; Vassar, R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Fahrenholz, F. The non-amyloidogenic pathway: Structure and function of α-secretases. Subcell Biochem. 2005, 38, 105–127. [Google Scholar] [PubMed]
- Sun, X.; Chen, W.D.; Wang, Y.D. β-Amyloid: The key peptide in the pathogenesis of Alzheimer’s disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathya, M.; Premkumar, P.; Karthick, C.; Moorthi, P.; Jayachandran, K.S.; Anusuyadevi, M. BACE1 in Alzheimer’s disease. Clin. Chim. Acta 2012, 414, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Tong, Y.; Qing, H.; Chen, C.H.; Song, W. Increased BACE1 maturation contributes to the pathogenesis of Alzheimer’s disease in Down syndrome. FASEB J. 2006, 20, 1361–1368. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Szaruga, M. Mechanisms of neurodegeneration—Insights from familial Alzheimer’s disease. Semin. Cell Dev. Biol. 2020, 105, 75–85. [Google Scholar]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory markers: Key indicators in the pathology of neurodegenerative diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef]
- Bazzari, F.H.; Abdallah, D.M.; El-Abhar, H.S. Chenodeoxycholic acid ameliorates AlCl3-induced Alzheimer’s disease neurotoxicity and cognitive deterioration via enhanced insulin signaling in rats. Molecules 2019, 24, 1992. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.P.; de Castro, A.A.; Soares, F.V.; da Cunha, E.F.; Ramalho, T.C. Future therapeutic perspectives into the Alzheimer’s disease targeting the oxidative stress hypothesis. Molecules 2019, 24, 4410. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Hong, F.; Yang, S. Amyloidosis in Alzheimer’s disease: Pathogeny, etiology, and related therapeutic directions. Molecules 2022, 27, 1210. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.A.; Gordon, B.A.; Mishra, S.; Su, Y.; Perrin, R.J.; Cairns, N.J.; Morris, J.C.; Ances, B.M.; Benzinger, T.L. Widespread distribution of tauopathy in preclinical Alzheimer’s disease. Neurobiol. Aging 2018, 72, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bazzari, A.H.; Parri, H.R. Neuromodulators and long-term synaptic plasticity in learning and memory: A steered-glutamatergic perspective. Brain Sci. 2019, 9, 300. [Google Scholar] [CrossRef] [Green Version]
- Qiao, A.; Li, J.; Hu, Y.; Wang, J.; Zhao, Z. Reduction BACE1 expression via suppressing NF-κB mediated signaling by Tamibarotene in a mouse model of Alzheimer’s disease. IBRO Neurosci. Rep. 2021, 10, 153–160. [Google Scholar] [CrossRef]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Keogh, C.E.; Rude, K.M.; Gareau, M.G. Role of pattern recognition receptors and the microbiota in neurological disorders. J. Physiol. 2021, 599, 1379–1389. [Google Scholar] [CrossRef]
- Cao, W.; Zheng, H. Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 51. [Google Scholar] [CrossRef]
- Ortí-Casañ, N.; Wu, Y.; Naudé, P.J.; De Deyn, P.P.; Zuhorn, I.S.; Eisel, U.L. Targeting TNFR2 as a novel therapeutic strategy for Alzheimer’s disease. Front. Neurosci. 2019, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Sun, E.; Motolani, A.; Campos, L.; Lu, T. The pivotal role of NF-KB in the pathogenesis and therapeutics of Alzheimer’s disease. Int. J. Mol. Sci. 2022, 23, 8972. [Google Scholar] [CrossRef]
- Gali, C.C.; Fanaee-Danesh, E.; Zandl-Lang, M.; Albrecher, N.M.; Tam-Amersdorfer, C.; Stracke, A.; Sachdev, V.; Reichmann, F.; Sun, Y.; Avdili, A.; et al. Amyloid-beta impairs insulin signaling by accelerating autophagy-lysosomal degradation of LRP-1 and IR-β in blood-brain barrier endothelial cells in vitro and in 3XTg-AD mice. Mol. Cell. Neurosci. 2019, 99, 103390. [Google Scholar] [CrossRef]
- Rahman, S.O.; Panda, B.P.; Parvez, S.; Kaundal, M.; Hussain, S.; Akhtar, M.; Najmi, A.K. Neuroprotective role of astaxanthin in hippocampal insulin resistance induced by Aβ peptides in animal model of Alzheimer’s disease. Biomed. Pharmacother. 2019, 110, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wang, P. Role of insulin receptor substance-1 modulating PI3K/Akt insulin signaling pathway in Alzheimer’s disease. 3 Biotech 2021, 11, 179. [Google Scholar] [CrossRef] [PubMed]
- Toral-Rios, D.; Pichardo-Rojas, P.S.; Alonso-Vanegas, M.; Campos-Peña, V. GSK3β and tau protein in Alzheimer’s Disease and epilepsy. Front. Cell. Neurosci. 2020, 14, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, N.; Paliwal, S.; Jain, S.; Verma, K.; Paliwal, S.; Sharma, S. GSK-3β and its Inhibitors in Alzheimer’s Disease: A Recent Update. Mini Rev. Med. Chem. 2022, 22, 2881–2895. [Google Scholar]
- Wang, S.; Mims, P.N.; Roman, R.J.; Fan, F. Is beta-amyloid accumulation a cause or consequence of Alzheimer’s disease? J. Alzheimer’s Park. Dement. 2016, 1, 007. [Google Scholar]
- Maccioni, R.B.; Farías, G.; Morales, I.; Navarrete, L. The revitalized tau hypothesis on Alzheimer’s disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padurariu, M.; Ciobica, A.; Lefter, R.; Lacramioara Serban, I.; Stefanescu, C.; Chirita, R. The oxidative stress hypothesis in Alzheimer’s disease. Psychiatr. Danub. 2013, 25, 401–409. [Google Scholar]
- Bivona, G.; Iemmolo, M.; Piccoli, T.; Agnello, L.; Lo Sasso, B.; Ciaccio, M.; Ghersi, G. High Cerebrospinal Fluid CX3CL1 Levels in Alzheimer’s Disease Patients but Not in Non-Alzheimer’s Disease Dementia. J. Clin. Med. 2022, 11, 5498. [Google Scholar] [CrossRef]
- Gong, C.X.; Liu, F.; Iqbal, K. Multifactorial hypothesis and multi-targets for Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef]
- Martenyi, F.; Lowe, S.; Dean, R.A.; Monk, S.A.; Gonzales, C.R.; Friedrich, S.; May, P.C.; Audia, J.E.; Citron, M.; LaBell, E.S.; et al. P4-088: Central and Peripheral Pharmacokinetic and Pharmacodynamic Effects of the β-site APP Cleavage Enzyme (BACE1) Inhibitor LY2811376 In Humans. Alzheimer’s Dement. 2010, 6, e48. [Google Scholar] [CrossRef]
- LY2886721. Available online: https://www.alzforum.org/therapeutics/ly2886721 (accessed on 4 November 2022).
- Reising, N.C.; Day, T.A.; Hole, J.T.; Tingley III, F.D.; Gonzalez-DeWhitt, P.A.; Mergott, D.J.; McKinzie, D.L.; Demattos, R.B.; Hayashi, M.L.; Riddell, D.R. P1-114: Measurement of endogenous mouse tau in cerebrospinal fluid from aged Pdapp mice following treatment with Ab-lowering compounds. Alzheimer’s Dement. 2018, 14, P314. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, F.; Zhao, J.; Wang, P.; Sun, X. Cleavage of Kv2. 1 by BACE1 decreases potassium current and reduces neuronal apoptosis. Neurochem. Int. 2022, 155, 105310. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=LY2886721&term=&cntry=&state=&city=&dist= (accessed on 4 November 2022).
- Martenyi, F.; Dean, R.A.; Lowe, S.; Nakano, M.; Monk, S.; Willis, B.A.; Gonzales, C.; Mergott, D.; Leslie, D.; May, P.; et al. BACE inhibitor LY2886721 safety and central and peripheral PK and PD in healthy subjects (HSs). Alzheimer’s Dement. 2012, 8, P583–P584. [Google Scholar] [CrossRef]
- Study of LY2886721 in Mild Cognitive Impairment Due to Alzheimer’s Disease or Mild Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT01561430?cond=LY2886721&draw=1&rank=6 (accessed on 4 November 2022).
- Dong, Y.; Li, X.; Cheng, J.; Hou, L. Drug development for Alzheimer’s disease: Microglia induced neuroinflammation as a target? Int. J. Mol. Sci. 2019, 20, 558. [Google Scholar] [CrossRef] [PubMed]
- May, P.C.; Willis, B.A.; Lowe, S.L.; Dean, R.A.; Monk, S.A.; Cocke, P.J.; Audia, J.E.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; et al. The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 2015, 35, 1199–1210. [Google Scholar] [CrossRef] [Green Version]
- Dekeryte, R.; Franklin, Z.; Hull, C.; Croce, L.; Kamli-Salino, S.; Helk, O.; Hoffmann, P.A.; Yang, Z.; Riedel, G.; Delibegovic, M.; et al. The BACE1 inhibitor LY2886721 improves diabetic phenotypes of BACE1 knock-in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166149. [Google Scholar] [CrossRef]
- RG7129. Available online: https://www.alzforum.org/therapeutics/rg7129 (accessed on 4 November 2022).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=RO5508887&term=&cntry=&state=&city=&dist= (accessed on 4 November 2022).
- Kumar, D.; Ganeshpurkar, A.; Kumar, D.; Modi, G.; Gupta, S.K.; Singh, S.K. Secretase inhibitors for the treatment of Alzheimer’s disease: Long road ahead. Eur. J. Med. Chem. 2018, 148, 436–452. [Google Scholar] [CrossRef] [PubMed]
- BI 1181181. Available online: https://www.alzforum.org/therapeutics/bi-1181181 (accessed on 4 November 2022).
- Hobson, S.; Lenter, M.C.; Sauer, A.; Fuchs, K.; Lala, D.S.; Dillard, L.W.; Dorner-Ciossek, C. P4-168: Effects of the bace1 inhibitor bi 1181181 and the anti-abeta antibody m266 on abeta in rat brain homogenate and CSF. Alzheimer’s Dement. 2015, 11, P843. [Google Scholar] [CrossRef]
- Dorner-Ciossek, C.; Hobson, S.; Fuchs, K.; Sauer, A.; Bauer, M.; Morales-Ramos, A.; Venkatraman, S.; Dillard, L.W.; Kruk, B.; Howard, L.; et al. P1-314: Pharmacological characterization of the new bace1 inhibitor bi 1181181. Alzheimer’s Dement. 2015, 11, P477. [Google Scholar] [CrossRef]
- CliniclTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=BI+1181181&term=&cntry=&state=&city=&dist= (accessed on 4 November 2022).
- Borta, A.; Nicolas, L.; Kleiner, O.; Kammerer, K.P.; Schaible, J.; Pietzko, K.; Podhorna, J.; Dorner-Ciossek, C.; Scholpp, J. P3-282: Single oral doses of the novel bace inhibitor bi 1181181 significantly reduce concentrations of cerebrospinal fluid amyloid-beta peptides in healthy subjects. Alzheimer’s Dement. 2015, 11, P740. [Google Scholar] [CrossRef]
- Nicolas, L.; Kammerer, K.P.; Schaible, J.; Link, J.; Kleiner, O.; Borta, A.; Podhorna, J.; Scholpp, J. P3-283: Pharmacokinetics, pharmacodynamics, and safety of the novel bace inhibitor bi1181181 after oral administration of single ascending doses in healthy subjects. Alzheimer’s Dement. 2015, 11, P740. [Google Scholar] [CrossRef]
- Atabecestat. Available online: https://www.alzforum.org/therapeutics/atabecestat (accessed on 4 November 2022).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=JNJ-54861911&term=&cntry=&state=&city=&dist= (accessed on 4 November 2022).
- Timmers, M.; Van Broeck, B.; Ramael, S.; Slemmon, J.; De Waepenaert, K.; Russu, A.; Bogert, J.; Stieltjes, H.; Shaw, L.M.; Engelborghs, S.; et al. Profiling the dynamics of CSF and plasma Aβ reduction after treatment with JNJ-54861911, a potent oral BACE inhibitor. Alzheimer’s Dement. 2016, 2, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmers, M.; Barão, S.; Van Broeck, B.; Tesseur, I.; Slemmon, J.; De Waepenaert, K.; Bogert, J.; Shaw, L.M.; Engelborghs, S.; Moechars, D.; et al. BACE1 dynamics upon inhibition with a BACE inhibitor and correlation to downstream Alzheimer’s disease markers in elderly healthy participants. J. Alzheimer’s Dis. 2017, 56, 1437–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmers, M.; Streffer, J.R.; Russu, A.; Tominaga, Y.; Shimizu, H.; Shiraishi, A.; Tatikola, K.; Smekens, P.; Börjesson-Hanson, A.; Andreasen, N.; et al. Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: Randomized, double-blind, placebo-controlled study. Alzheimer’s Res. Ther. 2018, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Liver Tox Ends Janssen BACE Program. Available online: https://www.alzforum.org/news/research-news/liver-tox-ends-janssen-bace-program (accessed on 4 November 2022).
- Henley, D.; Raghavan, N.; Sperling, R.; Aisen, P.; Raman, R.; Romano, G. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1483–1485. [Google Scholar] [CrossRef] [PubMed]
- Novak, G.; Streffer, J.R.; Timmers, M.; Henley, D.; Brashear, H.R.; Bogert, J.; Russu, A.; Janssens, L.; Tesseur, I.; Tritsmans, L.; et al. Long-term safety and tolerability of atabecestat (JNJ-54861911), an oral BACE1 inhibitor, in early Alzheimer’s disease spectrum patients: A randomized, double-blind, placebo-controlled study and a two-period extension study. Alzheimer’s Res. Ther. 2020, 12, 58. [Google Scholar] [CrossRef]
- Sperling, R.; Henley, D.; Aisen, P.S.; Raman, R.; Donohue, M.C.; Ernstrom, K.; Rafii, M.S.; Streffer, J.; Shi, Y.; Karcher, K.; et al. Findings of efficacy, safety, and biomarker outcomes of atabecestat in preclinical Alzheimer disease: A truncated randomized phase 2b/3 clinical trial. JAMA Neurol. 2021, 78, 293–301. [Google Scholar] [CrossRef]
- De Jonghe, S.; Weinstock, D.; Aligo, J.; Washington, K.; Naisbitt, D. Biopsy Pathology and Immunohistochemistry of a Case of Immune-Mediated Drug-Induced Liver Injury With Atabecestat. Hepatology 2021, 73, 452–455. [Google Scholar] [CrossRef]
- Thomson, P.J.; Kafu, L.; Meng, X.; Snoeys, J.; De Bondt, A.; De Maeyer, D.; Wils, H.; Leclercq, L.; Vinken, P.; Naisbitt, D.J. Drug-specific T-cell responses in patients with liver injury following treatment with the BACE inhibitor atabecestat. Allergy 2021, 76, 1825–1835. [Google Scholar] [CrossRef]
- Lanabecestat. Available online: https://www.alzforum.org/therapeutics/azd3293 (accessed on 4 November 2022).
- Eketjäll, S.; Janson, J.; Kaspersson, K.; Bogstedt, A.; Jeppsson, F.; Fälting, J.; Haeberlein, S.B.; Kugler, A.R.; Alexander, R.C.; Cebers, G. AZD3293: A novel, orally active BACE1 inhibitor with high potency and permeability and markedly slow off-rate kinetics. J. Alzheimer’s Dis. 2016, 50, 1109–1123. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.R.; Selzler, K.J.; Downing, A.M.; Willis, B.A.; Aluise, C.D.; Zimmer, J.; Bragg, S.; Andersen, S.; Ayan-Oshodi, M.; Liffick, E.; et al. Development review of the BACE1 inhibitor lanabecestat (AZD3293/LY3314814). J. Prev. Alzheimer’s Dis. 2017, 4, 247–254. [Google Scholar]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=AZD3293&term=&cntry=&state=&city=&dist= (accessed on 4 November 2022).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=Lanabecestat&term=&cntry=&state=&city=&dist= (accessed on 15 November 2022).
- Cebers, G.; Alexander, R.C.; Haeberlein, S.B.; Han, D.; Goldwater, R.; Ereshefsky, L.; Olsson, T.; Ye, N.; Rosen, L.; Russell, M.; et al. AZD3293: Pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 55, 1039–1053. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Matsuki, S.; Matsuguma, K.; Yoshihara, T.; Uchida, N.; Azuma, F.; Russell, M.; Hughes, G.; Haeberlein, S.B.; Alexander, R.C.; et al. BACE1 inhibitor lanabecestat (AZD3293) in a phase 1 study of healthy Japanese subjects: Pharmacokinetics and effects on plasma and cerebrospinal fluid Aβ peptides. J. Clin. Pharmacol. 2017, 57, 1460–1471. [Google Scholar] [CrossRef]
- Ye, N.; Monk, S.A.; Daga, P.; Bender, D.M.; Rosen, L.B.; Mullen, J.; Minkwitz, M.C.; Kugler, A.R. Clinical bioavailability of the novel BACE1 inhibitor lanabecestat (AZD3293): Assessment of tablet formulations versus an oral solution and the impact of gastric pH on pharmacokinetics. Clin. Pharmacol. Drug Dev. 2018, 7, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An Efficacy and Safety Study of Lanabecestat (LY3314814) in Early Alzheimer’s Disease (AMARANTH). Available online: https://clinicaltrials.gov/ct2/show/NCT02245737?cond=Lanabecestat&phase=12&draw=2&rank=1 (accessed on 15 November 2022).
- A Study of Lanabecestat (LY3314814) in Early Alzheimer’s Disease Dementia. Available online: https://clinicaltrials.gov/ct2/show/NCT02972658?cond=Lanabecestat&phase=12&draw=2&rank=2 (accessed on 15 November 2022).
- A Study of Lanabecestat (LY3314814) in Participants With Mild Alzheimer’s Disease Dementia (DAYBREAK-ALZ). Available online: https://clinicaltrials.gov/ct2/show/NCT02783573?cond=Lanabecestat&phase=12&draw=2&rank=3 (accessed on 15 November 2022).
- Wessels, A.M.; Tariot, P.N.; Zimmer, J.A.; Selzler, K.J.; Bragg, S.M.; Andersen, S.W.; Landry, J.; Krull, J.H.; Downing, A.M.; Willis, B.A.; et al. Efficacy and safety of lanabecestat for treatment of early and mild Alzheimer disease: The AMARANTH and DAYBREAK-ALZ randomized clinical trials. JAMA Neurol. 2020, 77, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Wessels, A.M.; Lines, C.; Stern, R.A.; Kost, J.; Voss, T.; Mozley, L.H.; Furtek, C.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; et al. Cognitive outcomes in trials of two BACE inhibitors in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 1483–1492. [Google Scholar] [CrossRef]
- Zimmer, J.A.; Shcherbinin, S.; Devous Sr, M.D.; Bragg, S.M.; Selzler, K.J.; Wessels, A.M.; Shering, C.; Mullen, J.; Landry, J.; Andersen, S.W.; et al. Lanabecestat: Neuroimaging results in early symptomatic Alzheimer’s disease. Alzheimer’s Dement. 2021, 7, e12123. [Google Scholar] [CrossRef] [PubMed]
- Verubecestat. Available online: https://www.alzforum.org/therapeutics/verubecestat (accessed on 15 November 2022).
- Chris Min, K.; Dockendorf, M.F.; Palcza, J.; Tseng, J.; Ma, L.; Stone, J.A.; Kleijn, H.J.; Hodsman, P.; Masuo, K.; Tanen, M.; et al. Pharmacokinetics and Pharmacodynamics of the BACE 1 Inhibitor Verubecestat (MK-8931) in Healthy Japanese Adults: A Randomized, Placebo-Controlled Study. Clin. Pharmacol. Ther. 2019, 105, 1234–1243. [Google Scholar] [CrossRef]
- Forman, M.; Palcza, J.; Tseng, J.; Stone, J.A.; Walker, B.; Swearingen, D.; Troyer, M.D.; Dockendorf, M.F. Safety, Tolerability, and Pharmacokinetics of the β-Site Amyloid Precursor Protein-Cleaving Enzyme 1 Inhibitor Verubecestat (MK-8931) in Healthy Elderly Male and Female Subjects. Clin. Transl. Sci. 2019, 12, 545–555. [Google Scholar] [CrossRef] [PubMed]
- An Efficacy and Safety Trial of Verubecestat (MK-8931) in Mild to Moderate Alzheimer’s Disease (P07738) (EPOCH). Available online: https://clinicaltrials.gov/ct2/show/NCT01739348?cond=MK-8931&draw=2&rank=4 (accessed on 15 November 2022).
- Efficacy and Safety Trial of Verubecestat (MK-8931) in Participants with Prodromal Alzheimer’s Disease (MK-8931-019) (APECS). Available online: https://clinicaltrials.gov/ct2/show/NCT01953601?cond=MK-8931&draw=2&rank=3 (accessed on 15 November 2022).
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Egan, M.F.; Mukai, Y.; Voss, T.; Kost, J.; Stone, J.; Furtek, C.; Mahoney, E.; Cummings, J.L.; Tariot, P.N.; Aisen, P.S.; et al. Further analyses of the safety of verubecestat in the phase 3 EPOCH trial of mild-to-moderate Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 68. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- Sur, C.; Kost, J.; Scott, D.; Adamczuk, K.; Fox, N.C.; Cummings, J.L.; Tariot, P.N.; Aisen, P.S.; Vellas, B.; Voss, T.; et al. BACE inhibition causes rapid, regional, and non-progressive volume reduction in Alzheimer’s disease brain. Brain 2020, 143, 3816–3826. [Google Scholar] [CrossRef]
- Sergott, R.C.; Raji, A.; Kost, J.; Sur, C.; Jackson, S.; Locco, A.; Patel, A.; Furtek, C.; Mattson, B.; Egan, M.F. Retinal Optical Coherence Tomography Metrics Are Unchanged in Verubecestat Alzheimer’s Disease Clinical Trial but Correlate with Baseline Regional Brain Atrophy. J. Alzheimer’s Dis. 2021, 79, 275–287. [Google Scholar] [CrossRef]
- Elenbecestat. Available online: https://www.alzforum.org/therapeutics/elenbecestat (accessed on 15 November 2022).
- Moriyama, T.; Fukushima, T.; Kokate, T.; Albala, B. [P3–037]: Preclinical studies with elenbecestat, a novel bace1 inhibitor, show no evidence of hypopigmentation. Alzheimer’s Dement. 2017, 13, P944. [Google Scholar] [CrossRef]
- Albala, B.; Lai, R.Y.; Aluri, J.; Boyd, P.; Chang, M.K.; Dayal, S.; Ferry, J.; Rege, B. [P2–003]: Elenbecestat pharmacokinetic drug-drug interactions indicated no dosage adjustments required for most concomitant treatments. Alzheimer’s Dement. 2017, 13, P605–P6066. [Google Scholar] [CrossRef]
- Lai, R.Y.; Darpo, B.; Dayal, S.; Hall, N.; Chang, M.K.; Albala, B.; Ferry, J.; Rege, B. [P1–043]: Elenbecestat, a novel oral bace inhibitor, has no clinically meaningful effect on qtc interval up to a supratherapeutic dose of 200 mg. Alzheimer’s Dement. 2017, 13, P250–P251. [Google Scholar] [CrossRef]
- Hayata, N.; Yasuda, S.; Kanekiyo, M.; Ito, S.; Yoshida, M.; Kawaguchi, H.; Lai, R.Y.; Kaplow, J.; Albala, B.; Luthman, J.; et al. P1-040: Elenbecestat, a novel bace inhibitor, demonstrates similar pharmacokinetics and tolerability in japanese subjects with multiple dosings. Alzheimer’s Dement. 2018, 14, P282. [Google Scholar] [CrossRef]
- Dose-Finding Study to Evaluate Safety, Tolerability, and Efficacy of E2609 in Participants with Mild Cognitive Impairment Due to Alzheimer’s Disease (Prodromal Alzheimer’s Disease) and Mild to Moderate Dementia Due to Alzheimer’s Disease. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02322021?term=E2609&draw=3&rank=10 (accessed on 16 November 2022).
- Lynch, S.Y.; Kaplow, J.; Zhao, J.; Dhadda, S.; Luthman, J.; Albala, B. P4-389: Elenbecestat, E2609, a bace inhibitor: Results from a phase-2 study in subjects with mild cognitive impairment and mild-to-moderate dementia due to Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, P1623. [Google Scholar] [CrossRef]
- A 24-Month Study to Evaluate the Efficacy and Safety of Elenbecestat (E2609) in Participants with Early Alzheimer’s Disease (MissionAD1). Available online: https://www.clinicaltrials.gov/ct2/show/NCT02956486?term=E2609&draw=3&rank=9 (accessed on 16 November 2022).
- A Placebo-Controlled, Double-Blind, Parallel-Group, 24-Month Study with an Open-Label Extension Phase to Evaluate the Efficacy and Safety of Elenbecestat (E2609) in Subjects With Early Alzheimer’s Disease. Available online: https://fdaaa.trialstracker.net/trial/NCT03036280/ (accessed on 16 November 2022).
- Irizarry, M.C.; Gee, M.; Roberts, C.; Giorgi, L.; Kanekiyo, M.; LeBlanc, M.; Putti, K.; Kaplow, J.; Dhadda, S. Cognitive Outcomes in the Very Mild Subgroup in the Phase 3 Studies of Elenbecestat in Early Ad (Mission ad Program). In 2021 Alzheimer’s Association International Conference. 2021. Available online: https://alz.confex.com/alz/2021/meetingapp.cgi/Paper/57910 (accessed on 16 November 2022).
- Umibecestat. Available online: https://www.alzforum.org/therapeutics/umibecestat (accessed on 16 November 2022).
- Neumann, U.; Ufer, M.; Jacobson, L.H.; Rouzade-Dominguez, M.L.; Huledal, G.; Kolly, C.; Lüönd, R.M.; Machauer, R.; Veenstra, S.J.; Hurth, K.; et al. The BACE-1 inhibitor CNP 520 for prevention trials in Alzheimer’s disease. EMBO Mol. Med. 2018, 10, e9316. [Google Scholar] [CrossRef]
- A Study of CAD106 and CNP520 Versus Placebo in Participants at Risk for the Onset of Clinical Symptoms of Alzheimer’s Disease (GS1). Available online: https://www.clinicaltrials.gov/ct2/show/NCT02565511?term=CNP520&draw=2&rank=3 (accessed on 16 November 2022).
- A Study of CNP520 Versus Placebo in Participants at Risk for the Onset of Clinical Symptoms of Alzheimer’s Disease (GS2). Available online: https://www.clinicaltrials.gov/ct2/show/NCT03131453?term=CNP520&draw=2&rank=1 (accessed on 16 November 2022).
- Umibecestat-Driven Cognitive Decline Is Reversible. Available online: https://www.alzforum.org/news/conference-coverage/umibecestat-driven-cognitive-decline-reversible (accessed on 16 November 2022).
- LY3202626. Available online: https://www.alzforum.org/therapeutics/ly3202626 (accessed on 16 November 2022).
- McKinzie, D.L.; May, P.C.; Boggs, L.N.; Yang, Z.; Brier, R.A.; Monk, S.A.; Willis, B.A.; Borders, A.R.; Winneroski, L.L.; Green, S.J.; et al. P1-080: Nonclinical Pharmacological Characterization of the Bace1 Inhibitor LY3202626. Alzheimer’s Dement. 2016, 12, P432–P433. [Google Scholar] [CrossRef]
- Boggs, L.N.; May, P.C.; Yang, Z.; Brier, R.A.; Monk, S.A.; Borders, A.R.; Winneroski, L.L.; Green, S.J.; Mergott, D.J.; McKinzie, D.L. P3-035: A Correlational Analysis of Exposure and Pharmacodynamic Effects of the Bace1 Inhibitor LY3202626 in PDAPP Mice Following Acute Oral Dosing. Alzheimer’s Dement. 2016, 12, P831. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=LY3202626&cntry=&state=&city=&dist= (accessed on 17 November 2022).
- Willis, B.A.; Lowe, S.L.; Daugherty, L.L.; Dean, R.A.; English, B.; Ereshefsky, L.; Gevorkyan, H.; James, D.E.; Jhee, S.; Lin, Q.; et al. P1-044: Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of LY3202626, a Novel Bace1 Inhibitor, in Healthy Subjects and Patients with Alzheimer’s Disease. Alzheimer’s Dement. 2016, 12, P418. [Google Scholar] [CrossRef]
- Katyayan, K.; Yi, P.; Monk, S.; Cassidy, K. Excretion, mass balance, and metabolism of [14C] LY3202626 in humans: An interplay of microbial reduction, reabsorption, and aldehyde oxidase oxidation that leads to an extended excretion profile. Drug Metab. Dispos. 2020, 48, 698–707. [Google Scholar] [CrossRef]
- A Study of LY3202626 on Disease Progression in Participants With Mild Alzheimer’s Disease Dementia (NAVIGATE-AD). Available online: https://clinicaltrials.gov/ct2/show/NCT02791191?term=LY3202626&draw=2&rank=2 (accessed on 17 November 2022).
- Lo, A.C.; Evans, C.D.; Mancini, M.; Wang, H.; Shcherbinin, S.; Lu, M.; Natanegara, F.; Willis, B.A. Phase II (NAVIGATE-AD study) results of LY3202626 effects on patients with mild Alzheimer’s disease dementia. J. Alzheimer’s Dis. Rep. 2021, 5, 321–336. [Google Scholar] [CrossRef] [PubMed]
- A Study of LY3002813 in Participants With Early Symptomatic Alzheimer’s Disease (TRAILBLAZER-ALZ). Available online: https://clinicaltrials.gov/ct2/show/NCT03367403?term=LY3202626&draw=2&rank=5 (accessed on 17 November 2022).
- PF-06751979. Available online: https://www.alzforum.org/therapeutics/pf-06751979 (accessed on 17 November 2022).
- O’Neill, B.T.; Beck, E.M.; Butler, C.R.; Nolan, C.E.; Gonzales, C.; Zhang, L.; Doran, S.D.; Lapham, K.; Buzon, L.M.; Dutra, J.K.; et al. Design and synthesis of clinical candidate PF-06751979: A potent, brain penetrant, β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitor lacking hypopigmentation. J. Med. Chem. 2018, 61, 4476–4504. [Google Scholar] [CrossRef]
- Qiu, R.; Ahn, J.E.; Alexander, R.; Brodney, M.A.; He, P.; Leurent, C.; Mancuso, J.; Margolin, R.A.; Tankisheva, E.; Chen, D. Safety, tolerability, pharmacokinetics, and pharmacodynamic effects of PF-06751979, a potent and selective oral BACE1 inhibitor: Results from phase I studies in healthy adults and healthy older subjects. J. Alzheimer’s Dis. 2019, 71, 581–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikulca, J.A.; Nguyen, V.; Gajdosik, D.A.; Teklu, S.G.; Giunta, E.A.; Lessa, E.A.; Tran, C.H.; Terak, E.C.; Raffa, R.B. Potential novel targets for A lzheimer pharmacotherapy: II. Update on secretase inhibitors and related approaches. J. Clin. Pharm. Ther. 2014, 39, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Safety Study of CTS21166 to Treat Alzheimer Disease (CTS). Available online: https://clinicaltrials.gov/ct2/show/NCT00621010?cond=CTS-21166&draw=2&rank=1 (accessed on 17 November 2022).
- Safety Study of HPP854 in Subjects with Mild Cognitive Impairment or a Diagnosis of Mild Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT01482013?cond=HPP-854&draw=2&rank=1 (accessed on 17 November 2022).
- De Strooper, B.; Chávez Gutiérrez, L. Learning by failing: Ideas and concepts to tackle γ-secretases in Alzheimer’s disease and beyond. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 419–437. [Google Scholar] [CrossRef] [PubMed]
- Luczkowski, M. “No screams and cries will convince us that white is white and black is black”, an ode to the defenders of amyloid cascade hypothesis of Alzheimer’s disease. Coord. Chem. Rev. 2016, 327, 35–42. [Google Scholar] [CrossRef]
- Dunn, B.; Stein, P.; Cavazzoni, P. Approval of aducanumab for Alzheimer disease—The FDA’s perspective. JAMA Intern. Med. 2021, 181, 1276–1278. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.; Apostolova, L.G.; Atri, A.; Salloway, S.; Weiner, M. Aducanumab: Appropriate use recommendations. J. Prev. Alzheimer’s Dis. 2021, 8, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.Y.; Fu, W.M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 2017, 24, 47. [Google Scholar] [CrossRef]
- Bazzari, A.H.; Bazzari, F.H. Medicinal plants for Alzheimer’s disease: An updated review. J. Med. Plant. Stud. 2018, 6, 81–85. [Google Scholar]
- Zhu, L.; Xu, L.; Wu, X.; Deng, F.; Ma, R.; Liu, Y.; Huang, F.; Shi, L. Tau-targeted multifunctional nanoinhibitor for Alzheimer’s disease. ACS Appl. Mater. Interfaces 2021, 13, 23328–23338. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Therapeutics of neurotransmitters in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef]
- Bazzari, A.H.; Bazzari, F.H. BDNF Therapeutic Mechanisms in Neuropsychiatric Disorders. Int. J. Mol. Sci. 2022, 23, 8417. [Google Scholar] [CrossRef]
- Yang, J.J. Brain insulin resistance and the therapeutic value of insulin and insulin-sensitizing drugs in Alzheimer’s disease neuropathology. Acta Neurol. Belg. 2022, 122, 1135–1142. [Google Scholar] [CrossRef]
- Chauhan, P.S.; Yadav, D.; Arukha, A.P. Dietary Nutrients and Prevention of Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2022, 21, 217–227. [Google Scholar] [CrossRef]
- Sagud, M.; Tudor, L.; Pivac, N. Personalized treatment interventions: Nonpharmacological and natural treatment strategies in Alzheimer’s disease. Expert Rev. Neurother. 2021, 21, 571–589. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzari, F.H.; Bazzari, A.H. BACE1 Inhibitors for Alzheimer’s Disease: The Past, Present and Any Future? Molecules 2022, 27, 8823. https://doi.org/10.3390/molecules27248823
Bazzari FH, Bazzari AH. BACE1 Inhibitors for Alzheimer’s Disease: The Past, Present and Any Future? Molecules. 2022; 27(24):8823. https://doi.org/10.3390/molecules27248823
Chicago/Turabian StyleBazzari, Firas H., and Amjad H. Bazzari. 2022. "BACE1 Inhibitors for Alzheimer’s Disease: The Past, Present and Any Future?" Molecules 27, no. 24: 8823. https://doi.org/10.3390/molecules27248823
APA StyleBazzari, F. H., & Bazzari, A. H. (2022). BACE1 Inhibitors for Alzheimer’s Disease: The Past, Present and Any Future? Molecules, 27(24), 8823. https://doi.org/10.3390/molecules27248823