

Privileged Scaffold Decoration for the Identification of the First Trisubstituted Triazine with Anti-SARS-CoV-2 Activity


 ,
,  , ,
, ,  ,
,  , and
, and 
Abstract
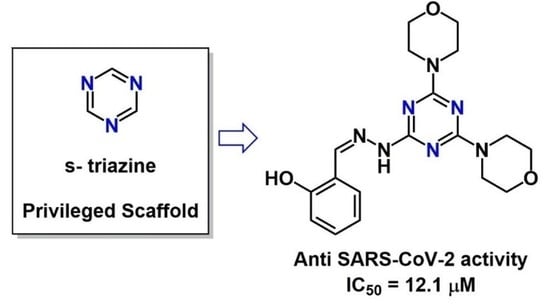
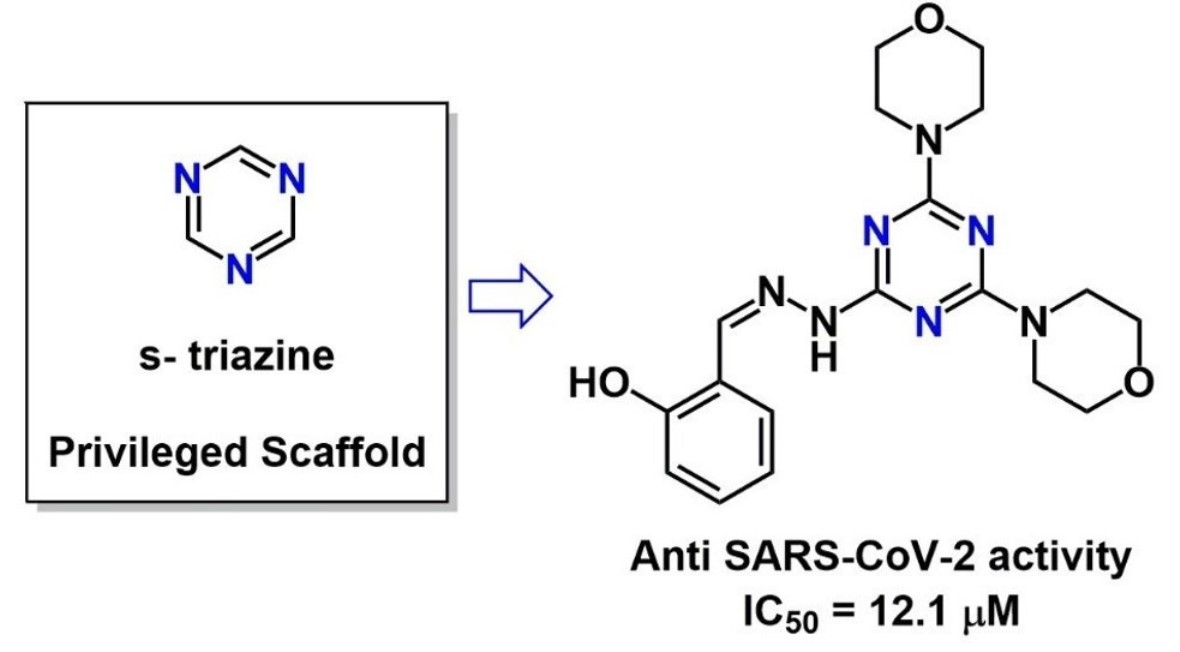
:
1. Introduction
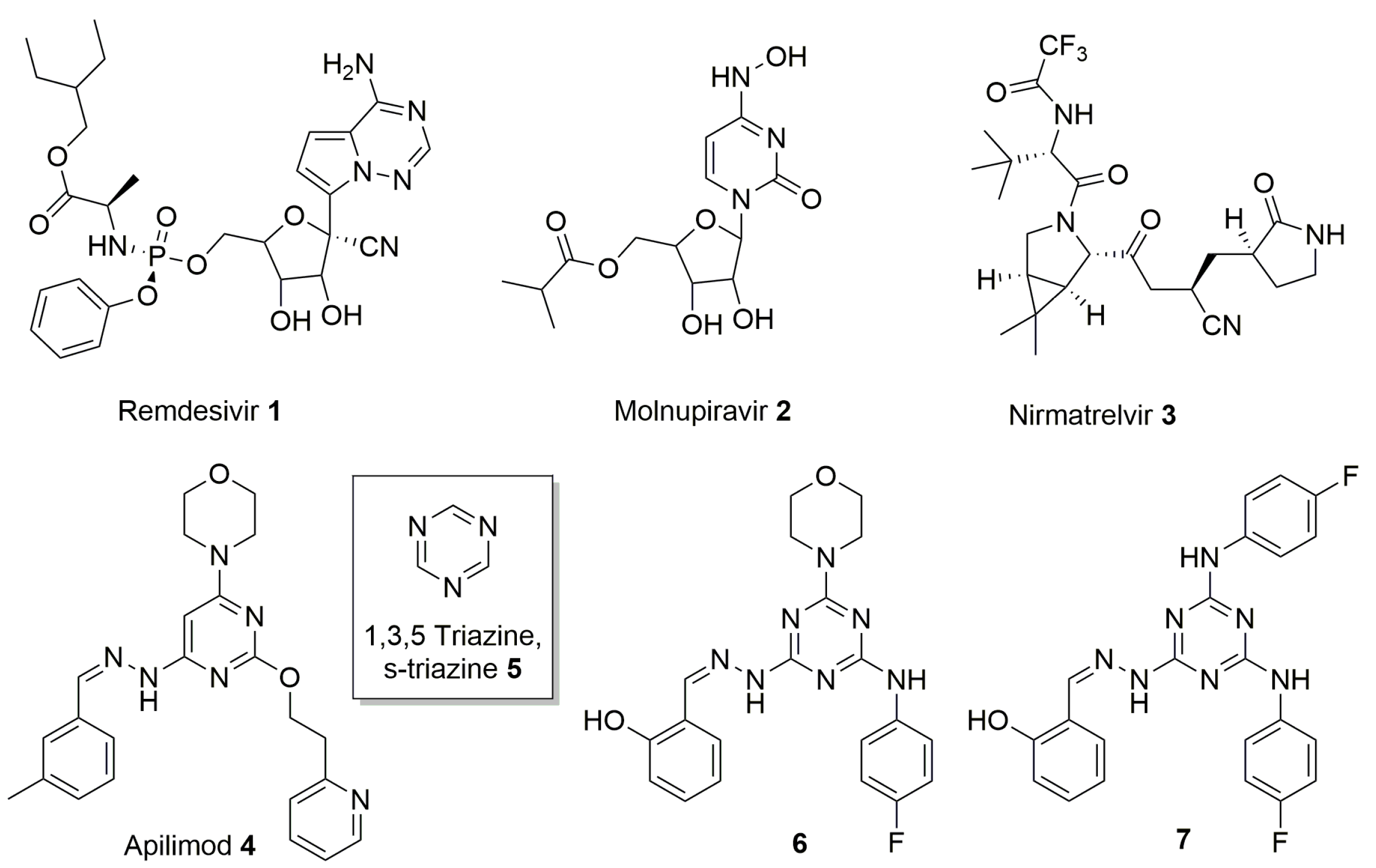
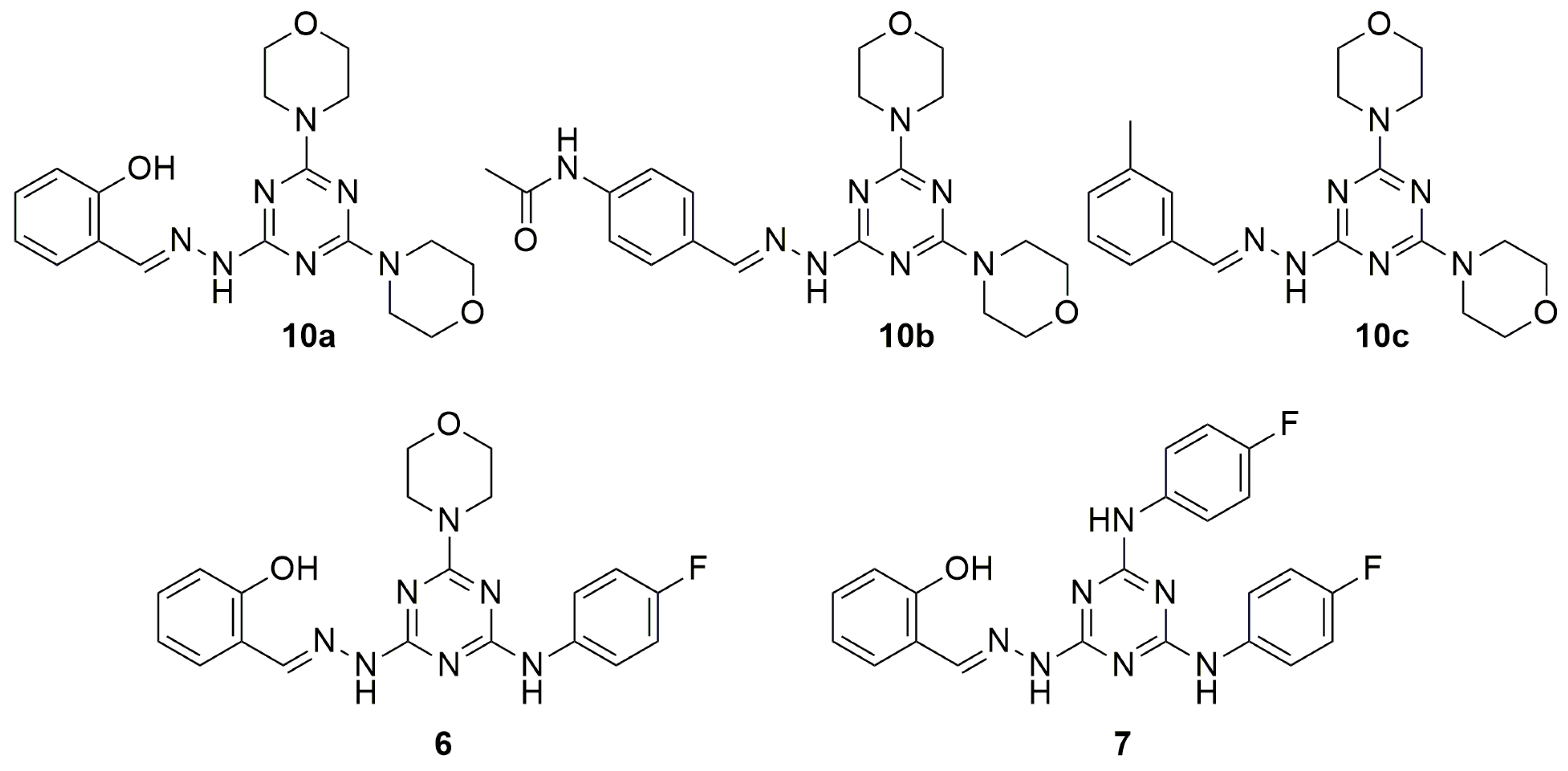
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry-General Part
3.2. Biology
3.2.1. Cells and Viruses
3.2.2. Drugs and Cytotoxicity Assay
3.2.3. Antiviral Assays-SARS-CoV-2
3.3. Enzymatic Assay
3.4. In Vitro ADME
3.4.1. HPLC/UV-MS Method
3.4.2. Aqueous Solubility
3.4.3. Parallel Artificial Membrane Permeability Assay (PAMPA)
3.4.4. Metabolic Stability Assay
3.4.5. Plasma Stability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Machhi, J.; Herskovitz, J.; Senan, A.M.; Dutta, D.; Nath, B.; Oleynikov, M.D.; Blomberg, W.R.; Meigs, D.D.; Hasan, M.; Patel, M.; et al. The Natural History, Pathobiology, and Clinical Manifestations of SARS-CoV-2 Infections. J. Neuroimmune. Pharmacol. 2020, 15, 359–386. [Google Scholar] [CrossRef] [PubMed]
- WHO. Situation Reports; World Health Organization: Geneva, Switzerland, 2020; Volume 180. [Google Scholar]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: www.covid19treatmentguidelines.nih.gov (accessed on 30 October 2022).
- Majumdar, M.; Singh, V.; Misra, T.K.; Roy, D.N. In silico studies on structural inhibition of SARS-CoV-2 main protease Mpro by major secondary metabolites of Andrographis paniculata and Cinchona officinalis. Biologia 2022, 77, 1373–1389. [Google Scholar] [CrossRef] [PubMed]
- Saravolatz, L.D.; Depcinski, S.; Sharma, M. Molnupiravir and Nirmatrelvir-Ritonavir: Oral COVID Antiviral Drugs. Clin. Infect. Dis. 2022, ciac180. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sheyi, R.; de la Torre, B.G.; El-Faham, A.; Albericio, F. s-Triazine: A Privileged Structure for Drug Discovery and Bioconjugation. Molecules 2021, 26, 864. [Google Scholar] [CrossRef]
- Shah, D.R.; Modh, R.P.; Chikhalia, K.H. Privileged s-triazines: Structure and pharmacological applications. Future Med. Chem. 2014, 6, 463–477. [Google Scholar] [CrossRef]
- Singla, P.; Luxami, V.; Paul, K. Triazine as a promising scaffold for its versatile biological behavior. Eur. J. Med. Chem. 2015, 102, 39–57. [Google Scholar] [CrossRef]
- Maga, G.; Falchi, F.; Radi, M.; Botta, L.; Casaluce, G.; Bernardini, M.; Irannejad, H.; Manetti, F.; Garbelli, A.; Samuele, A.; et al. Toward the discovery of novel anti-HIV drugs. Second-generation inhibitors of the cellular ATPase DDX3 with improved anti-HIV activity: Synthesis, structure-activity relationship analysis, cytotoxicity studies, and target validation. ChemMedChem 2011, 6, 1371–1389. [Google Scholar] [CrossRef]
- Brai, A.; Fazi, R.; Tintori, C.; Zamperini, C.; Bugli, F.; Sanguinetti, M.; Stigliano, E.; Esté, J.; Badia, R.; Franco, S.; et al. Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents. Proc. Natl. Acad. Sci. USA 2016, 113, 5388–5393. [Google Scholar] [CrossRef] [Green Version]
- Secchi, M.; Lodola, C.; Garbelli, A.; Bione, S.; Maga, G. DEAD-Box RNA Helicases DDX3X and DDX5 as Oncogenes or Oncosuppressors: A Network Perspective. Cancers 2022, 14, 3820. [Google Scholar] [CrossRef]
- Brai, A.; Trivisani, C.I.; Poggialini, F.; Pasqualini, C.; Vagaggini, C.; Dreassi, E. DEAD-Box Helicase DDX3X as a Host Target against Emerging Viruses: New Insights for Medicinal Chemical Approaches. J. Med. Chem. 2022, 65, 10195–10216. [Google Scholar] [CrossRef]
- Radi, M.; Botta, L.; Casaluce, G.; Bernardini, M.; Botta, M. Practical one-pot two-step protocol for the microwave-assisted synthesis of highly functionalized rhodanine derivatives. J. Comb. Chem. 2010, 12, 200–205. [Google Scholar] [CrossRef]
- Kourounakis, A.P.; Xanthopoulos, D.; Tzara, A. Morpholine as a privileged structure: A review on the medicinal chemistry and pharmacological activity of morpholine containing bioactive molecules. Med. Res. Rev. 2020, 40, 709–752. [Google Scholar] [CrossRef]
- Baranov, M.V.; Bianchi, F.; van den Bogaart, G. The PIKfyve Inhibitor Apilimod: A Double-Edged Sword against COVID-19. Cells 2020, 10, 30. [Google Scholar] [CrossRef]
- Mautner, L.; Hoyos, M.; Dangel, A.; Berger, C.; Ehrhardt, A.; Baiker, A. Replication kinetics and infectivity of SARS-CoV-2 variants of concern in common cell culture models. Virol. J. 2022, 19, 76. [Google Scholar] [CrossRef]
- Grazia Martina, M.; Vicenti, I.; Bauer, L.; Crespan, E.; Rango, E.; Boccuto, A.; Olivieri, N.; Incerti, M.; Zwaagstra, M.; Allodi, M.; et al. Bithiazole Inhibitors of Phosphatidylinositol 4-Kinase (PI4KIIIβ) as Broad-Spectrum Antivirals Blocking the Replication of SARS-CoV-2, Zika Virus, and Human Rhinoviruses. ChemMedChem 2021, 16, 3548–3552. [Google Scholar] [CrossRef]
- Vicenti, I.; Martina, M.G.; Boccuto, A.; De Angelis, M.; Giavarini, G.; Dragoni, F.; Marchi, S.; Trombetta, C.M.; Crespan, E.; Maga, G.; et al. System-oriented optimization of multi-target 2,6-diaminopurine derivatives: Easily accessible broad-spectrum antivirals active against flaviviruses, influenza virus and SARS-CoV-2. Eur. J. Med. Chem. 2021, 224, 113683. [Google Scholar] [CrossRef]
- Saladini, F.; Giannini, A.; Boccuto, A.; Vicenti, I.; Zazzi, M. Agreement between an in-house replication competent and a reference replication defective recombinant virus assay for measuring phenotypic resistance to HIV-1 protease, reverse transcriptase, and integrase inhibitors. J. Clin. Lab. Anal. 2018, 32, e22206. [Google Scholar] [CrossRef] [Green Version]
- Tintori, C.; Brai, A.; Dasso Lang, M.C.; Deodato, D.; Greco, A.M.; Bizzarri, B.M.; Cascone, L.; Casian, A.; Zamperini, C.; Dreassi, E.; et al. Development and in Vitro Evaluation of a Microbicide Gel Formulation for a Novel Non-Nucleoside Reverse Transcriptase Inhibitor Belonging to the N-Dihydroalkyloxybenzyloxopyrimidines (N-DABOs) Family. J. Med. Chem. 2016, 59, 2747–2759. [Google Scholar] [CrossRef]
- Brai, A.; Riva, V.; Saladini, F.; Zamperini, C.; Trivisani, C.I.; Garbelli, A.; Pennisi, C.; Giannini, A.; Boccuto, A.; Bugli, F.; et al. DDX3X inhibitors, an effective way to overcome HIV-1 resistance targeting host proteins. Eur. J. Med. Chem. 2020, 200, 112319. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | IC50 μM 2 | CC50 μM 3 | SI |
|---|---|---|---|---|
| 1 | 10a | 12.1 | >400 | >33.1 |
| 2 | 10b | NA | >200 | - |
| 3 | 10c | NA | 184 | - |
| 4 | 7 | NT | 0.8 | - |
| 5 | 13a | NA | >200 | - |
| 6 | 13b | NA | >200 | - |
| 7 | 6 | NT | 3 | - |
| 8 | 14a | NT | 70 | - |
| 9 | 14b | NA | 22 | - |
| 10 | Remdesivir | 0.07 ± 0.04 | 94.9 | 1356 |
| Cpd | µg/mL | LogS 1 |
|---|---|---|
| 6 | <0.1 | - |
| 7 | <0.1 | - |
| 10a | 3.23 | −5.07 |
| 10b | 3.46 | −5.09 |
| 10c | 4.50 | −4.93 |
| Cpd | Papp a | MR b (%) |
|---|---|---|
| 6 | 4.74 | 28.7 |
| 7 | 4.58 | 15.5 |
| 10a | 7.89 | 4.3 |
| 10b | 9.70 | 9.8 |
| 10c | 9.78 | 4.9 |
| Plasma Stability % ± SD | |||||
|---|---|---|---|---|---|
| Time (h) | 6 | 7 | 10a | 10b | 10c |
| 0 | 100.00 ± 0.19 | 100.00 ± 0.28 | 100.00 ± 0.28 | 100.00 ± 0.24 | 100.00 ± 0.16 |
| 24 | 98.78 ± 4.05 | 99.56 ± 3.78 | 98.48 ± 4.02 | 98.41 ± 4.69 | 99.52 ± 3.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cesarini, S.; Vicenti, I.; Poggialini, F.; Secchi, M.; Giammarino, F.; Varasi, I.; Lodola, C.; Zazzi, M.; Dreassi, E.; Maga, G.; et al. Privileged Scaffold Decoration for the Identification of the First Trisubstituted Triazine with Anti-SARS-CoV-2 Activity. Molecules 2022, 27, 8829. https://doi.org/10.3390/molecules27248829
Cesarini S, Vicenti I, Poggialini F, Secchi M, Giammarino F, Varasi I, Lodola C, Zazzi M, Dreassi E, Maga G, et al. Privileged Scaffold Decoration for the Identification of the First Trisubstituted Triazine with Anti-SARS-CoV-2 Activity. Molecules. 2022; 27(24):8829. https://doi.org/10.3390/molecules27248829
Chicago/Turabian StyleCesarini, Silvia, Ilaria Vicenti, Federica Poggialini, Massimiliano Secchi, Federica Giammarino, Ilenia Varasi, Camilla Lodola, Maurizio Zazzi, Elena Dreassi, Giovanni Maga, and et al. 2022. "Privileged Scaffold Decoration for the Identification of the First Trisubstituted Triazine with Anti-SARS-CoV-2 Activity" Molecules 27, no. 24: 8829. https://doi.org/10.3390/molecules27248829
APA StyleCesarini, S., Vicenti, I., Poggialini, F., Secchi, M., Giammarino, F., Varasi, I., Lodola, C., Zazzi, M., Dreassi, E., Maga, G., Botta, L., & Saladino, R. (2022). Privileged Scaffold Decoration for the Identification of the First Trisubstituted Triazine with Anti-SARS-CoV-2 Activity. Molecules, 27(24), 8829. https://doi.org/10.3390/molecules27248829