
Impact of Dispersion Force Schemes on Liquid Systems: Comparing Efficiency and Drawbacks for Well-Targeted Test Cases

 , ,
, ,  ,
,
Abstract
:1. Introduction
1.1. Motivation: Why Two Targeted Cases?
1.2. Focusing on Liquid GeSe4 as Main Structural Target: Reviewing Previous Results
2. Liquid GeSe4 by FPMD: Calculation Methodology
Dispersion Forces within First-Principles Molecular Dynamics
3. Liquid GeSe4: Results
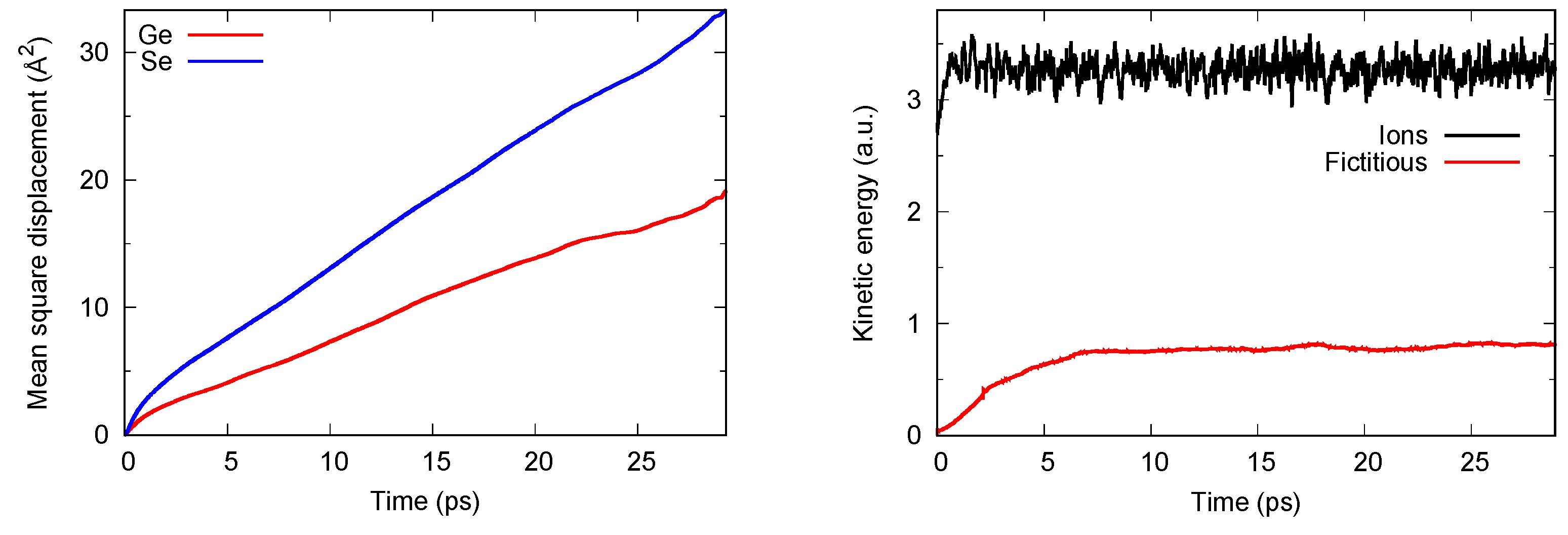
3.1. Real-Space Results
3.2. Reciprocal-Space Results
4. Discussion
4.1. The Case of an Ionic Liquid Interacting with A Solid Substrate
5. Conclusions
- (1)
- Globally, the inclusion of dispersion forces does not alter the basic structural features of liquid GeSe4 that can be described as a network in which GeSe4 tetrahedra coexist with Sen chains.
- (2)
- On the quantitative point of view, considerations of a larger size allowed recovering a higher percentage of corner-sharing tetrahedra, in line with previous results collected for other disordered chalcogenide systems [26].
- (3)
- We found that some notable differences appear in the case of D3 when considering the partial and total pair correlation functions and the bond angle distributions, as if the tetrahedral order typical of the GeSe4 network were somewhat altered.
- (4)
- These observations are confirmed by reciprocal space data and a comparison with the experimental counterpart revealing a striking, unexpected feature (i.e., an increasing spike) at very low wavevectors in the D3 case. Snapshots of the corresponding structure feature a phase separation taking place within the network in which void regions are clearly noticeable.
- (5)
- In view of these findings, we have included in this paper a second example of intriguing structural behavior that arises when employing the D3 dispersion scheme (a complex ionic liquid supported by a WSe2 layer). The action of a three-body force built in D3 is likely to be at the origin of a possible unphysical contribution.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Klimeš, J.; Michaelides, A. Perspective: Advances and challenges in treating van der Waals dispersion forces in density functional theory. J. Chem. Phys. 2012, 137, 120901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micoulaut, M. Van der Waals corrections for an improved structural description of telluride based melts. J. Chem. Phys. 2013, 138, 061103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micoulaut, M.; Coulet, M.-V.; Piarristeguy, A.; Johnson, M.R.; Cuello, G.J.; Bichara, C.; Raty, J.-Y.; Flores-Ruiz, H.; Pradel, A. Effect of concentration in Ge-Te liquids: A combined density functional and neutron scattering study. Phys. Rev. B 2014, 89, 174205. [Google Scholar] [CrossRef]
- Micoulaut, M.; Piarristeguy, A.; Flores-Ruiz, H.; Pradel, A. Towards accurate models for amorphous GeTe: Crucial effects of dispersive van der Waals corrections on the structural properties involved in the phase change mechanism. Phys. Rev. B 2017, 96, 184204. [Google Scholar] [CrossRef]
- Raty, J.-Y.; Zhang, W.; Luckas, J.; Chen, C.; Mazzarello, R.; Bichara, C.; Wuttig, M. Aging mechanism in amorphous phase-change materials. Nat. Comm. 2015, 6, 7467. [Google Scholar] [CrossRef] [Green Version]
- Lampin, E.; Bouzid, A.; Ori, G.; Boero, M.; Massobrio, C. Impact of dispersion forces on the atomic structure of a prototypical network-forming disordered system: The case of liquid GeSe2. J. Chem. Phys. 2017, 147, 044504. [Google Scholar] [CrossRef]
- Bouzid, A.; Massobrio, C.; Boero, M.; Ori, G.; Sykina, K.; Furet, E. Role of the van der Waals interactions and impact of the exchange-correlation functional in determining the structure of glassy GeTe4. Phys. Rev. B 2015, 92, 134208. [Google Scholar] [CrossRef]
- Massobrio, C.; Martin, E.; Chaker, Z.; Boero, M.; Bouzid, A.; Le Roux, S.; Ori, G. Sensitivity to dispersion forces in first-principles modeling of disordered chalcogenides. Front. Mater. 2018, 5, 78. [Google Scholar] [CrossRef] [Green Version]
- Silvestrelli, P.L.; Martin, E.; Boero, M.; Bouzid, A.; Ori, G.; Massobrio, C. Atomic structure of glassy GeTe4 as a playground to assess the performances of density functional schemes accounting for dispersion forces. J. Phys. Chem. B 2020, 124, 11273. [Google Scholar] [CrossRef]
- Marzari, N.; Vanderbilt, D. Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 1997, 56, 12847. [Google Scholar] [CrossRef]
- Silvestrelli, P.L.; Ambrosetti, A. van der Waals interactions in DFT using Wannier functions without empirical parameters. J. Chem. Phys. 2019, 150, 164109. [Google Scholar] [CrossRef] [PubMed]
- Silvestrelli, P.L. van der Waals interactions in DFT made easy by Wannier functions. Phys. Rev. Lett. 2008, 100, 053002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, T.; Boero, M. Role of van der Waals corrections in first principles simulations of alkali metal ions in aqueous solutions. J. Chem. Phys. 2015, 143, 194510. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 2006, 27, 1787. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab-initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haye, M.; Massobrio, C.; Pasquarello, A.; De Vita, A.; De Leeuw, S.W.; Car, R. Structure of Liquid GexSe(1-x) at the stiffness threshold composition. Phys. Rev. B 1998, 58, R14661–R14664. [Google Scholar] [CrossRef]
- Thorpe, M. Continuous deformations in random networks. J. Non-Cryst. Solids 1983, 57, 355. [Google Scholar] [CrossRef]
- Chen, G.; Inam, F.; Drabold, D.A. Structural origin of the intermediate phase in Ge-Se glasses. App. Phys. Lett. 2010, 97, 131901. [Google Scholar] [CrossRef]
- Sartbaeva, A.; Wells, S.A.; Huerta, A.; Thorpe, M. Local structural variability and the intermediate phase window in network glasses. Phys. Rev. B 2007, 75, 224204. [Google Scholar] [CrossRef] [Green Version]
- Bouzid, A.; Le Roux, S.; Ori, G.; Boero, M.; Massobrio, C. Origin of structural analogies and differences between the atomic structures of GeS4 and GeSe4 glasses: A first-principles study. J. Chem. Phys. 2015, 143, 034504. [Google Scholar] [CrossRef]
- Massobrio, C.; Celino, M.; Salmon, P.S.; Martin, R.A.; Micoulaut, M.; Pasquarello, A. Atomic structure of the two intermediate phase glasses SiSe4 and GeSe4. Phys. Rev. B 2009, 79, 174201. [Google Scholar] [CrossRef]
- Sykina, K.; Furet, E.; Bureau, B.; Le Roux, S.; Massobrio, C. Network connectivity and extended Se chains in the atomic structure of glassy GeSe4. Chem. Phys. Lett. 2012, 547, 30. [Google Scholar] [CrossRef]
- Lucas, P.; King, E.A.; Gulbiten, O.; Yarger, J.L.; Soignard, E.; Bureau, B. Bimodal phase percolation model for the structure of Ge-Se glasses and the existence of the intermediate phase. Phys. Rev. B 2009, 80, 214114. [Google Scholar] [CrossRef]
- Gjersing, E.L.; Sen, S.; Aitken, B.G. Structure, connectivity and configurational entropy of GexSe100−x glasses: Results from 77Se MAS NMR spectroscopy. J. Phys. Chem. C 2010, 114, 8601. [Google Scholar] [CrossRef]
- CPMD Developers Version 4.3, Copyright MPI-FKF (1997–2001). Copyright IBM Corp. (1990–2022). Available online: www.cpmd.org (accessed on 1 January 2020).
- Massobrio, C. The Structure of Amorphous Materials Using Molecular Dynamics; IOP Publishing: Bristol, UK, 2022. [Google Scholar] [CrossRef]
- Maruyama, K.; Misawa, M.; Inui, M.; Takeda, S.; Kawakita, Y.; Tamaki, S. Structural investigation of liquid Ge-chalcogen mixtures. J. Non-Cryst. Sol. 1996, 205–207, 106. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electronic density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E.; Parrinello, M. Adiabaticity in first-principles molecular dynamics. Phys. Rev. B 1992, 45, 9413. [Google Scholar] [CrossRef]
- Chaker, Z.; Ori, G.; Tugène, C.; Le Roux, S.; Boero, M.; Massobrio, C.; Martin, E.; Bouzid, A. The role of dispersion forces on the atomic structure of glassy chalcogenides: The case of GeSe4 and GeS4. J. Non-Cryst. Sol. 2018, 499, 167. [Google Scholar] [CrossRef]
- Chaker, Z.; Ori, G.; Boero, M.; Massobrio, C.; Furet, E.; Bouzid, A. First-principles study of the atomic structure of glassy Ga10Ge15Te75. J. Non-Cryst. Sol. 2018, 498, 338. [Google Scholar] [CrossRef]
- Dubbeldam, D.; Calero, S.; Vlugt, T.J.H. iRASPA: GPU-accelerated visualization software for materials scientists. Mol. Simul. 2018, 44, 653. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.T.; Zhang, Y.J.; Akashi, R.; Bahramy, M.S.; Arita, R.; Iwasa, Y. Superconducting Dome in a Gate-Tuned Band Insulator. Science 2012, 338, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, D.; Jo, S.; Berger, H.; Morpurgo, A.F. Gate-induced superconductivity in atomically thin MoS2 crystals. Nat. Nanotech. 2016, 11, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Nam, D.; Jeon, D.-Y.; Kim, G.-T. Transport-map analysis of ionic liquid-gated ambipolar WSe2 field-effect transistors. Semicond. Sci. Technol. 2019, 34, 075022. [Google Scholar] [CrossRef]
- Huang, J.-K.; Pu, J.; Hsu, C.-L.; Chiu, M.-H.; Juang, Z.-Y.; Chang, Y.-H.; Chang, W.-H.; Iwasa, Y.; Takenobu, T.; Li, L.-J. Large-Area Synthesis of Highly Crystalline WSe2 Monolayers and Device Applications. ACS Nano 2014, 8, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Ishisone, K.; Ori, G.; Boero, M. Structural, dynamical, and electronic properties of the ionic liquid 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide. Phys. Chem. Chem. Phys. 2022, 24, 9597–9607. [Google Scholar] [CrossRef]
- Zhao, W.; Ghorannevis, Z.; Chu, L.; Toh, M.; Kloc, C.; Tan, P.-H.; Eda, G. Evolution of electronic structure in atomically thin sheets of WS2 and WSe2. ACS Nano 2012, 7, 791. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Klement, P.; Jones, A.M.; Ghimire, N.J.; Yan, J.; Mandrus, D.; Taniguchi, T.; Watanabe, K.; Kitamura, K.; Yao, W.; et al. Electrically tunable excitonic light-emitting diodes based on monolayer WSe2 p–n junctions. Nat. Nanotechnol. 2014, 9, 268. [Google Scholar] [CrossRef]
- Braga, D.; Gutiérrez Lezama, I.; Berger, H.; Morpurgo, A.F. Quantitative determination of the band gap of WS2 with ambipolar ionic liquid-gated transistors. Nano Lett. 2012, 12, 5218. [Google Scholar] [CrossRef] [Green Version]
- Andreussi, O.; Marzari, N. Transport properties of room-temperature ionic liquids from classical molecular dynamics. J. Chem. Phys. 2012, 137, 044508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dommert, F.; Wendler, K.; Berger, R.; Delle Site, L.; Holm, C. Force Fields for Studying the Structure and Dynamics of Ionic Liquids: A Critical Review of Recent Developments. ChemPhysChem 2012, 13, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- Adi Kurnia, K.; Zunita, M.; Coutinho, J.A.P.; Santoso, D. Development of quantitative structure-property relationship to predict the viscosity of deep eutectic solvent for CO2 capture using molecular descriptor. J. Mol. Liquids 2022, 347, 118239. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No-vdW | 0.19 | 3.65 | 3.84 | 0.91 | 1.09 | 2.00 |
| D2 | 0.19 | 3.65 | 3.84 | 0.91 | 1.09 | 2.00 |
| D3(A) | 0.09 | 3.56 | 3.65 | 0.89 | 1.18 | 2.07 |
| D3(B) | 0.03 | 3.68 | 3.71 | 0.92 | 1.10 | 2.02 |
| MLWF | 0.19 | 3.68 | 3.87 | 0.92 | 1.08 | 2.00 |
| Ge(%) | No-vdW | D2 | D3(A) | D3(B) | MLWF | |
|---|---|---|---|---|---|---|
| l = 1 | Se | 1.1 | ||||
| l = 2 | Se2 | 3.2 | 3.5 | 8.8 | 7.2 | 2.8 |
| l = 3 | Se3 | 9.4 | 7.9 | 16.3 | 15.5 | 8.3 |
| GeSe2 | 1.1 | 1.2 | ||||
| l = 4 | Se4 | 69.3 | 70.9 | 62.7 | 70.6 | 71.4 |
| GeSe3 | 11.9 | 12.7 | 6.1 | 2.5 | 11.7 | |
| Ge2Se2 | 2.4 | 1.9 | 2.5 | |||
| l = 5 | GeSe4 | 1.1 | 2.6 | 2.9 | 1.3 | |
| Se(%) | No-vdW | D2 | D3(A) | D3(B) | MLWF | |
|---|---|---|---|---|---|---|
| l = 1 | Se | 3.7 | 3.2 | 4.9 | 5.8 | 3.3 |
| Ge | 2.4 | 2.1 | 3.2 | 3.6 | 2.3 | |
| l = 2 | Se2 | 29.4 | 30.1 | 27.0 | 26.5 | 29.5 |
| Ge2 | 23.3 | 23.7 | 18.9 | 22.0 | 23.9 | |
| GeSe | 35.0 | 35.5 | 31.9 | 31.5 | 35.6 | |
| l = 3 | GeSe2 | 2.5 | 2.3 | 5.3 | 4.0 | 2.0 |
| Ge2Se | 1.5 | 1.2 | 3.2 | 2.8 | 1.3 | |
| Se3 | 1.6 | 1.4 | 3.6 | 2.4 | 1.4 | |
| Ge3 | 1.1 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, E.; Amiehe Essomba, I.B.; Ishisone, K.; Boero, M.; Ori, G.; Massobrio, C. Impact of Dispersion Force Schemes on Liquid Systems: Comparing Efficiency and Drawbacks for Well-Targeted Test Cases. Molecules 2022, 27, 9034. https://doi.org/10.3390/molecules27249034
Martin E, Amiehe Essomba IB, Ishisone K, Boero M, Ori G, Massobrio C. Impact of Dispersion Force Schemes on Liquid Systems: Comparing Efficiency and Drawbacks for Well-Targeted Test Cases. Molecules. 2022; 27(24):9034. https://doi.org/10.3390/molecules27249034
Chicago/Turabian StyleMartin, Evelyne, Iréné Bérenger Amiehe Essomba, Kana Ishisone, Mauro Boero, Guido Ori, and Carlo Massobrio. 2022. "Impact of Dispersion Force Schemes on Liquid Systems: Comparing Efficiency and Drawbacks for Well-Targeted Test Cases" Molecules 27, no. 24: 9034. https://doi.org/10.3390/molecules27249034
APA StyleMartin, E., Amiehe Essomba, I. B., Ishisone, K., Boero, M., Ori, G., & Massobrio, C. (2022). Impact of Dispersion Force Schemes on Liquid Systems: Comparing Efficiency and Drawbacks for Well-Targeted Test Cases. Molecules, 27(24), 9034. https://doi.org/10.3390/molecules27249034