
Photo- and Radiation-Induced One-Electron Oxidation of Methionine in Various Structural Environments Studied by Time-Resolved Techniques



Abstract
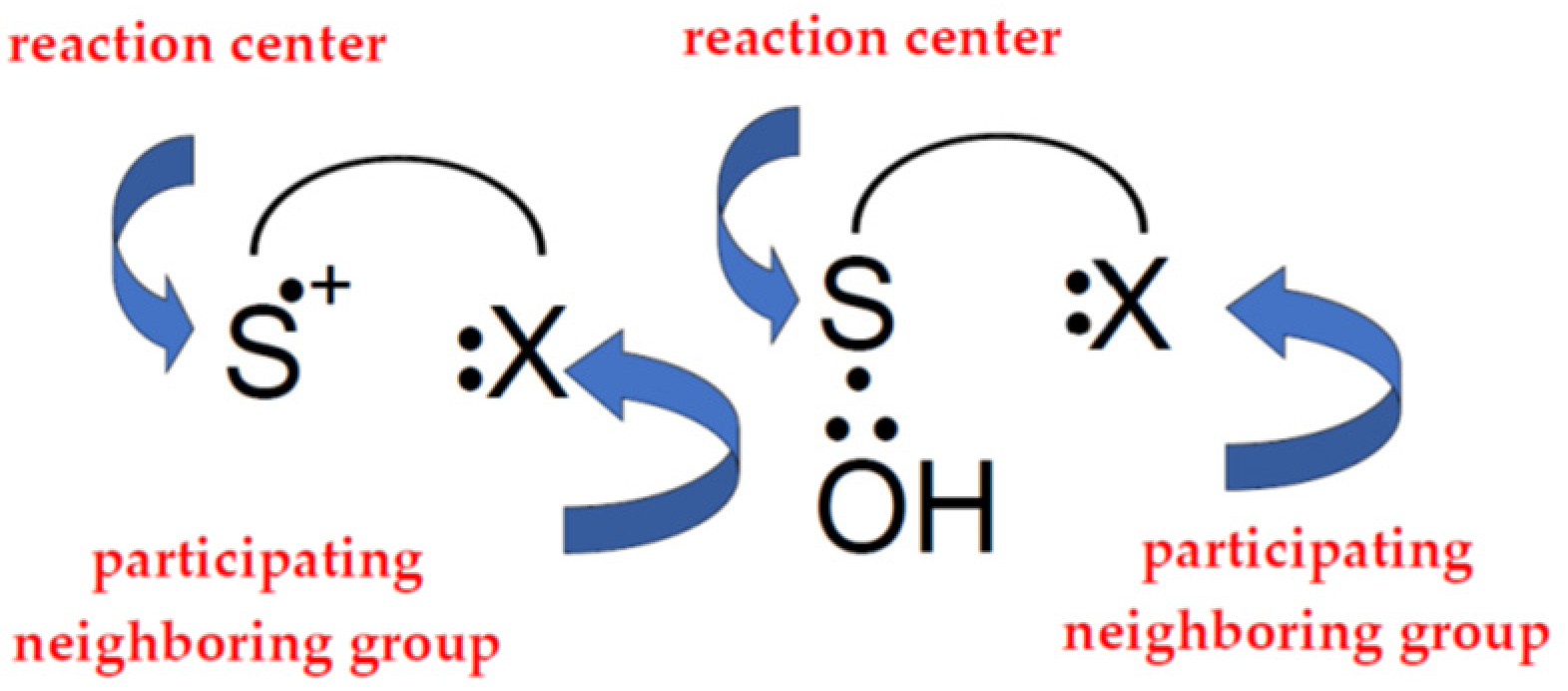
:1. Introduction
2. Methionine and Methionine Derivatives
2.1. Radiation-Induced Oxidation
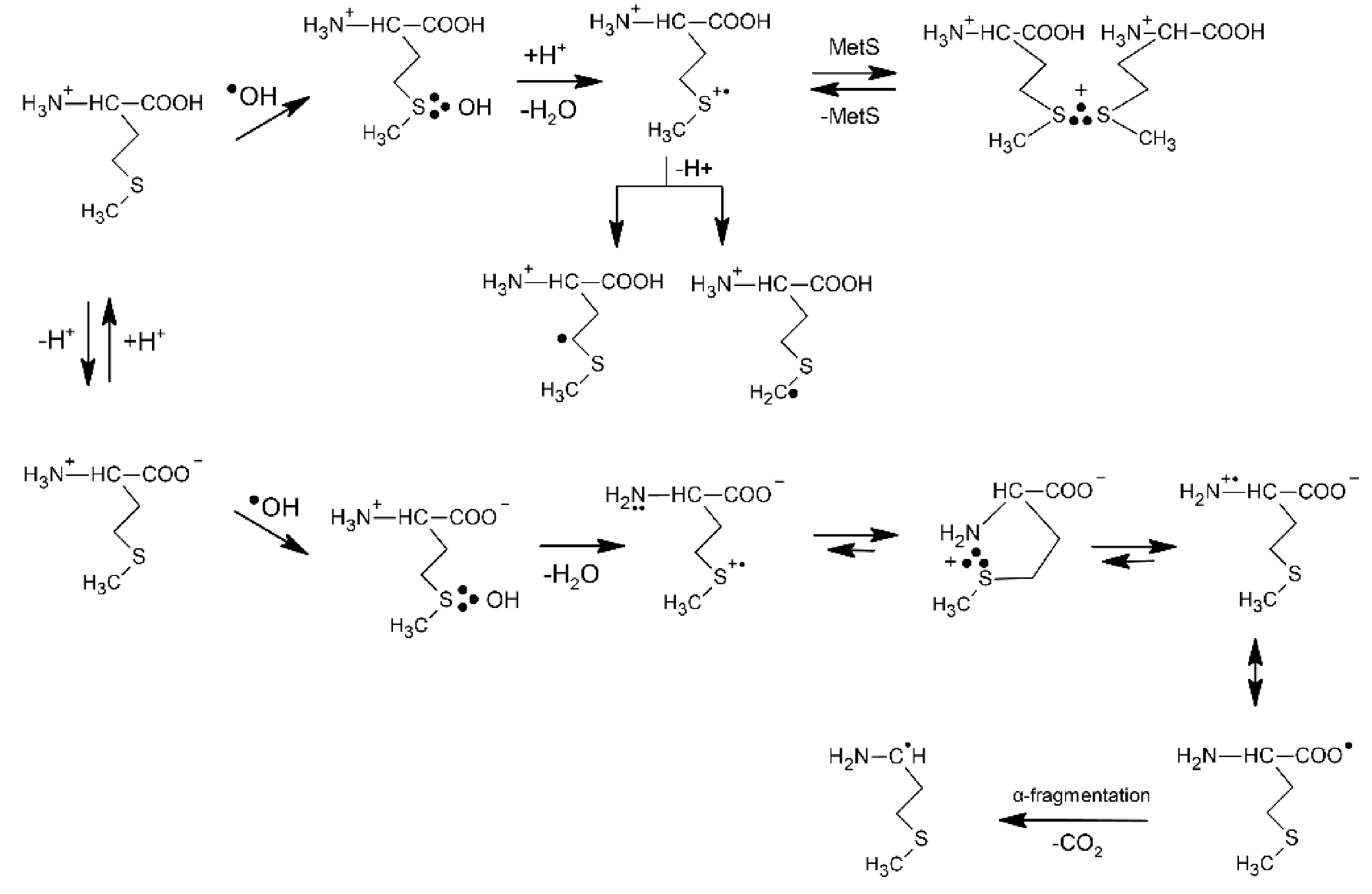
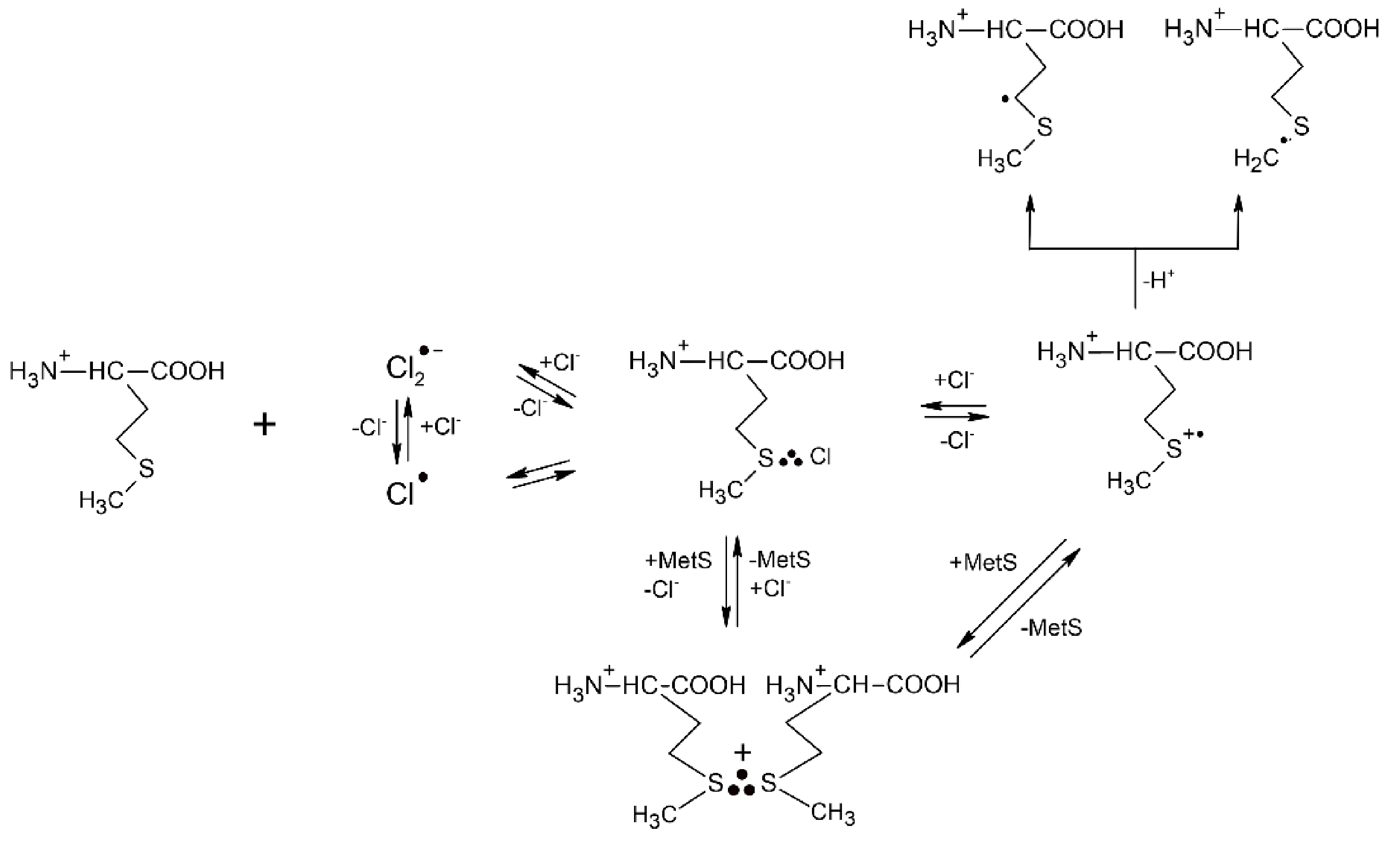
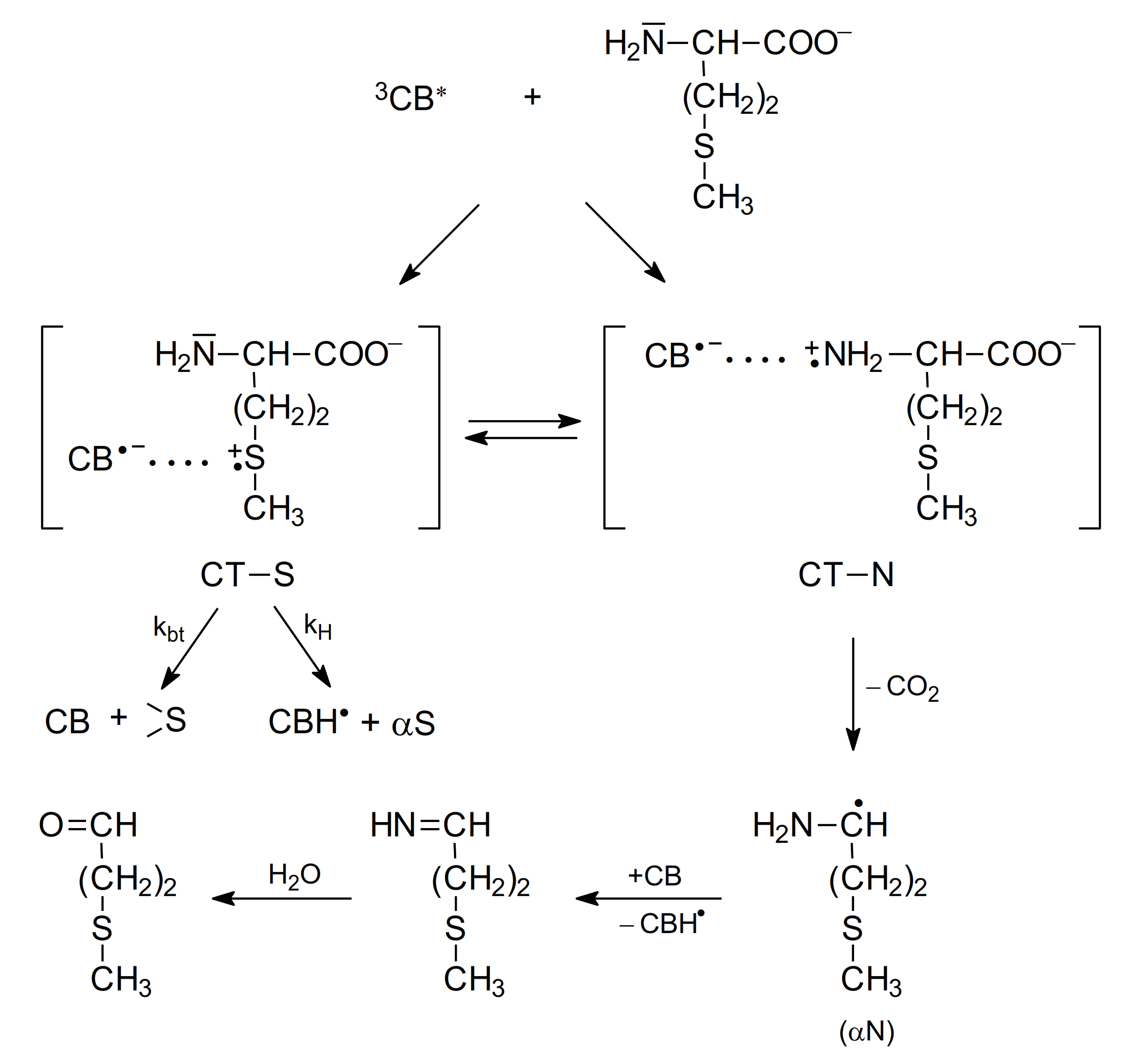
2.1.1. Methionine


| Oxidant | Met(NH3+,COOH) | Met(NH3+,COO−) | Met(NH2, COO−) | E0 (a) | Lit |
|---|---|---|---|---|---|
| ●OH | 2.3 × 1010 (b) | +2.72 | [7] | ||
| CCl3O2● | 2.9 × 107 (b,c) | +1.60 | [45] | ||
| CF3 CHClO2● | 1.4 × 106 (b,c) | +1.15 | [45] | ||
| CO3●− | 3.6 × 107 (b) | +1.57 | [50] | ||
| Cl2●− | 3.9 × 109 (b) | +2.13 | [52] | ||
| Br2●− | 2.5 × 109 (b) | 1.7 × 109 (b) | 2.0 × 109 (b,c) | +1.63 | [52] |
| Tl2+ | 2.5 × 109 (b) | +2.23 | [43] | ||
| Ag2+ | 3.3 × 108 (b) | +1.98 | [43] |
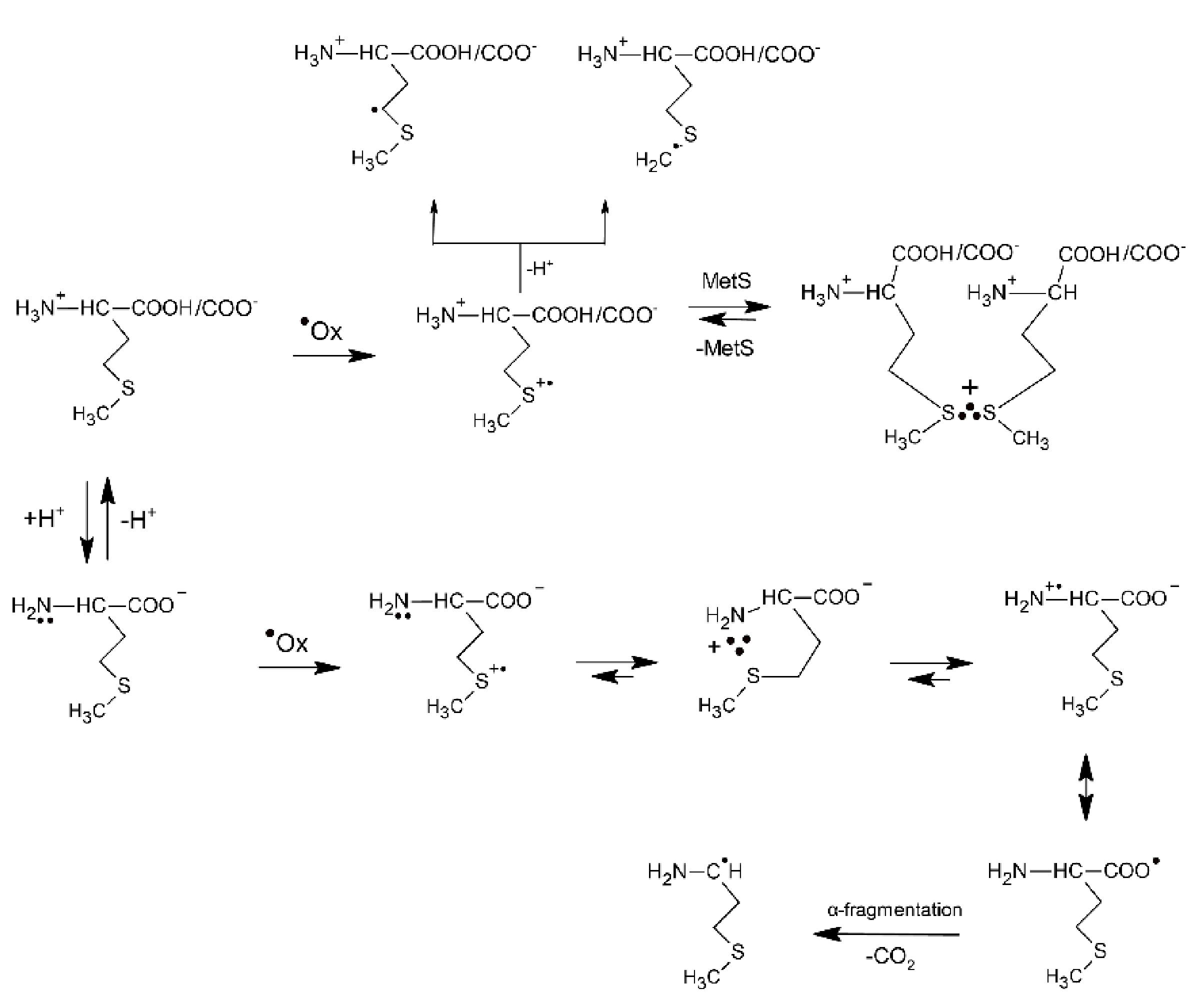
2.1.2. Methionine Derivatives
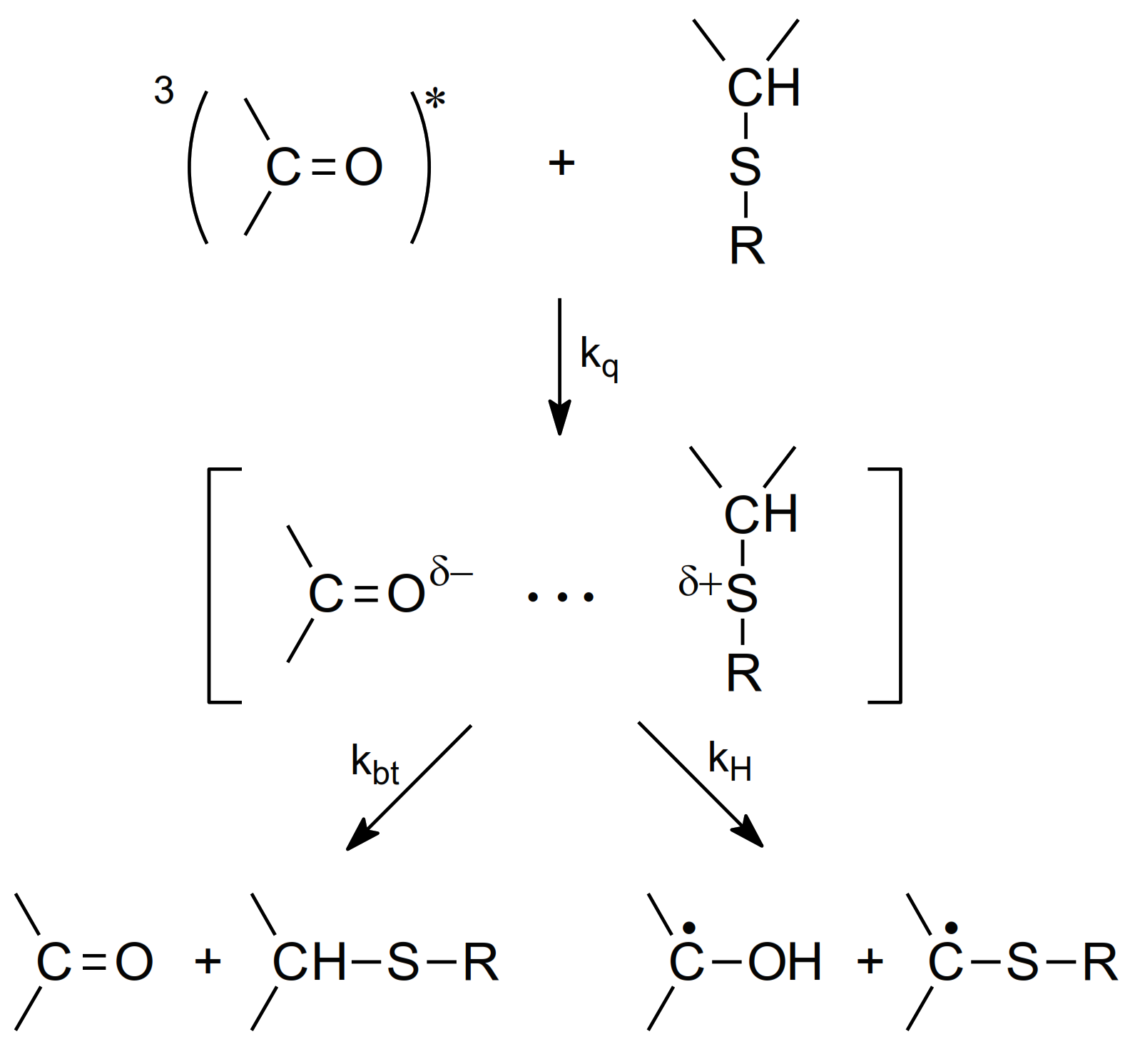
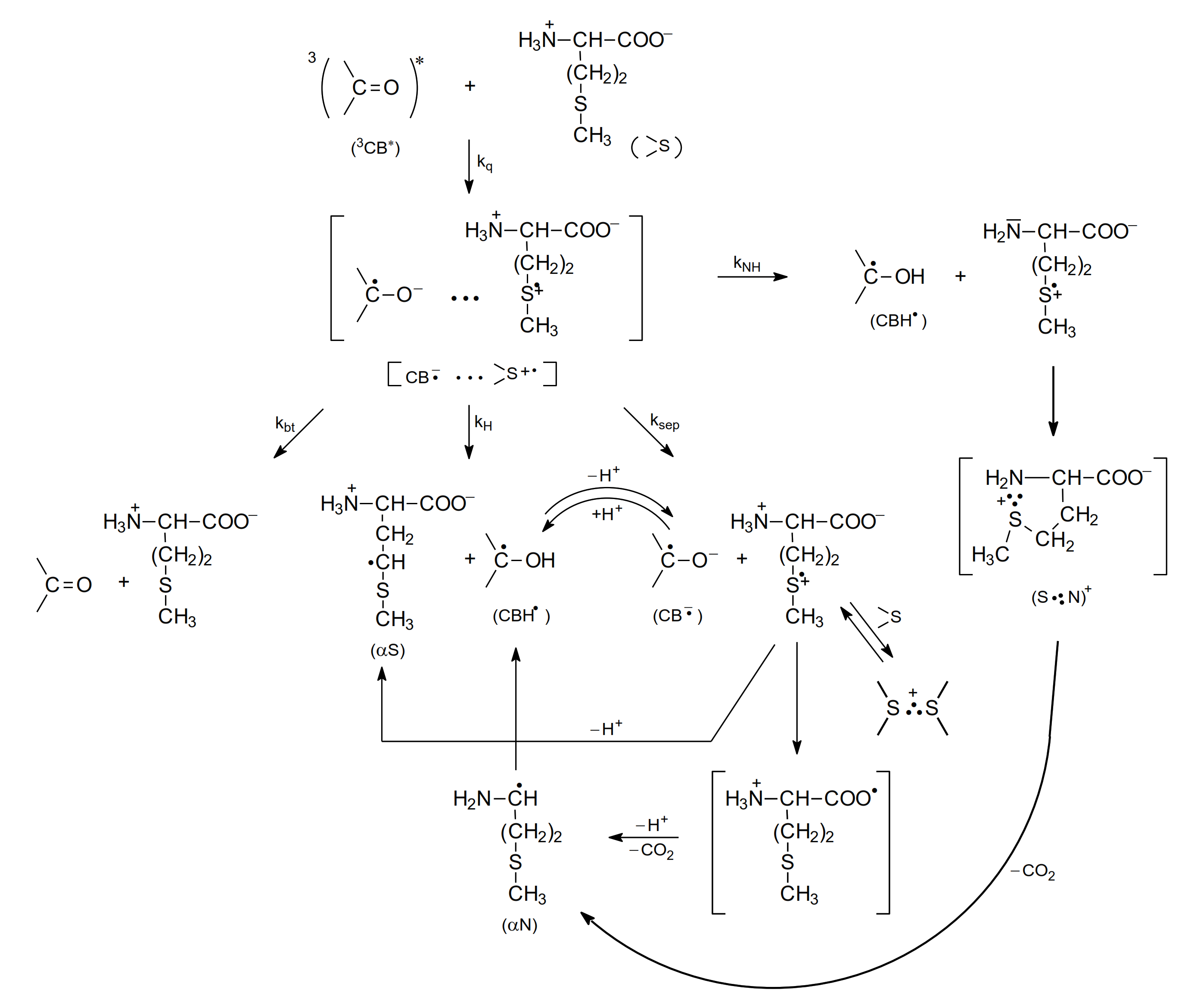
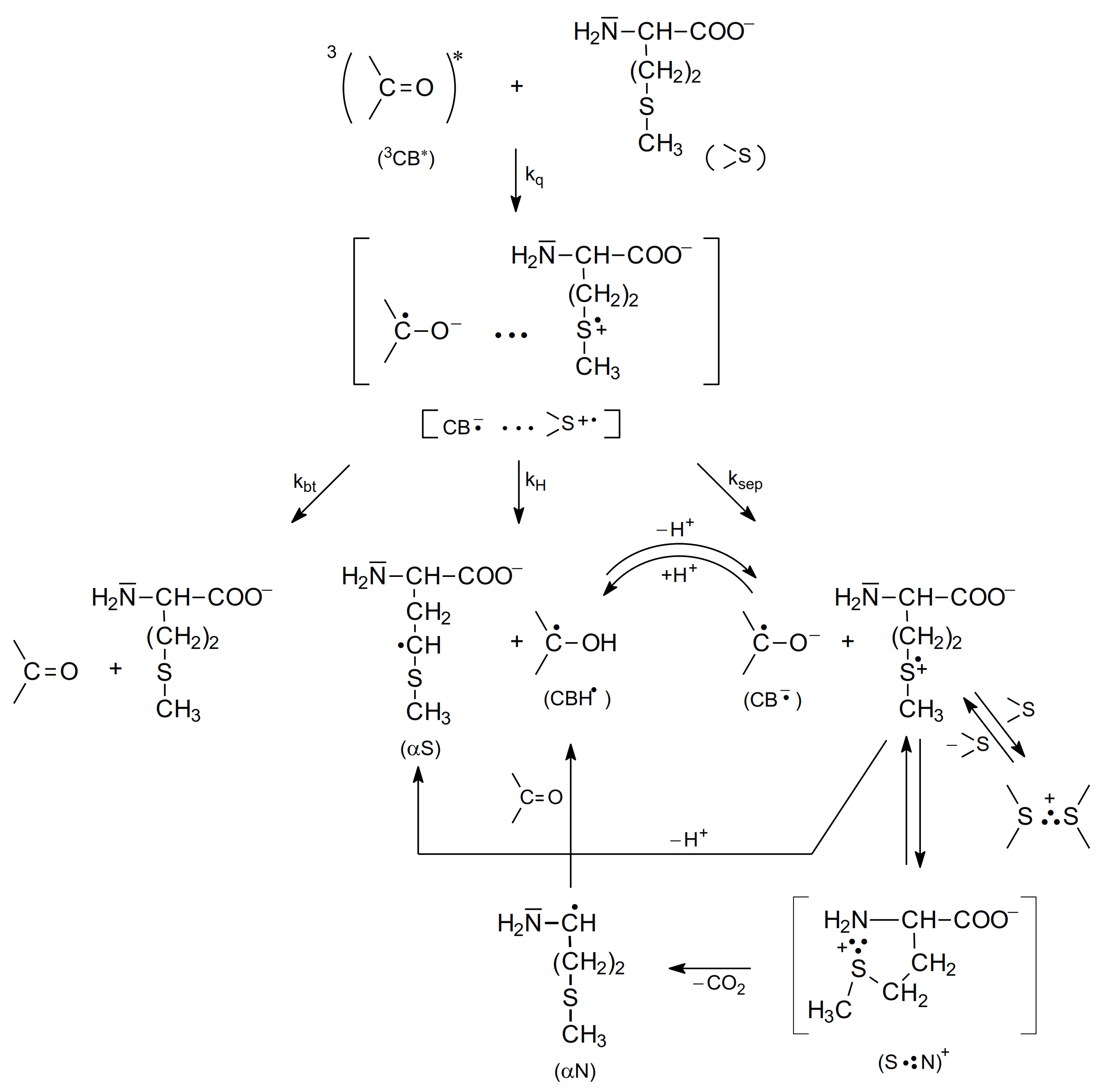
2.2. Photo-Induced Oxidation
- as shown in Table 2, the quenching rate constants were found to be in the range of 109 M−1 s−1 (diffusion controlled limit) for methionine and Met-containing compounds and were, respectively, three orders or one order of magnitude lower for amino acids without a sulfur atom (e.g., alanine) at pH 7 and pH 10,
- direct observation of radical-ion products in the transient absorption spectra (electron transfer intermediates, among them various two-center three-electron (2c-3e) bonded species) were products of oxidation of Met and CB radical anions and CB ketyl radicals were products of CB reduction,
- indirectly by the Rehm-Weller correlations of kq vs. ΔGel (free energy change for electron transfer).
| Amino Acid or Peptide | pH 7 | pH 10/11 |
|---|---|---|
| Methionine | 2.5 | 2.3 |
| N-Ac-Met | 1.9 | 1.6 |
| Met-OCH3 | 3.0 | 2.9 |
| Met-Gly | 2.1 | 2.3 |
| Gly-Met | 2.0 | 2.0 |
| Met-Lys | 1.8 | 1.3 |
| Lys-Met | 2.5 | 1.8 |
| Met-Gly-Gly | 2.3 | 2.2 |
| Gly-Gly-Met | 1.8 | 1.9 |
| Met-Met | 2.9 | 1.8 |
| Met-Enkephalin | 1.9 | 1.8 |
| Alanine | <0.0005 | 0.18 |
2.2.1. Methionine
2.2.2. Methionine Derivatives
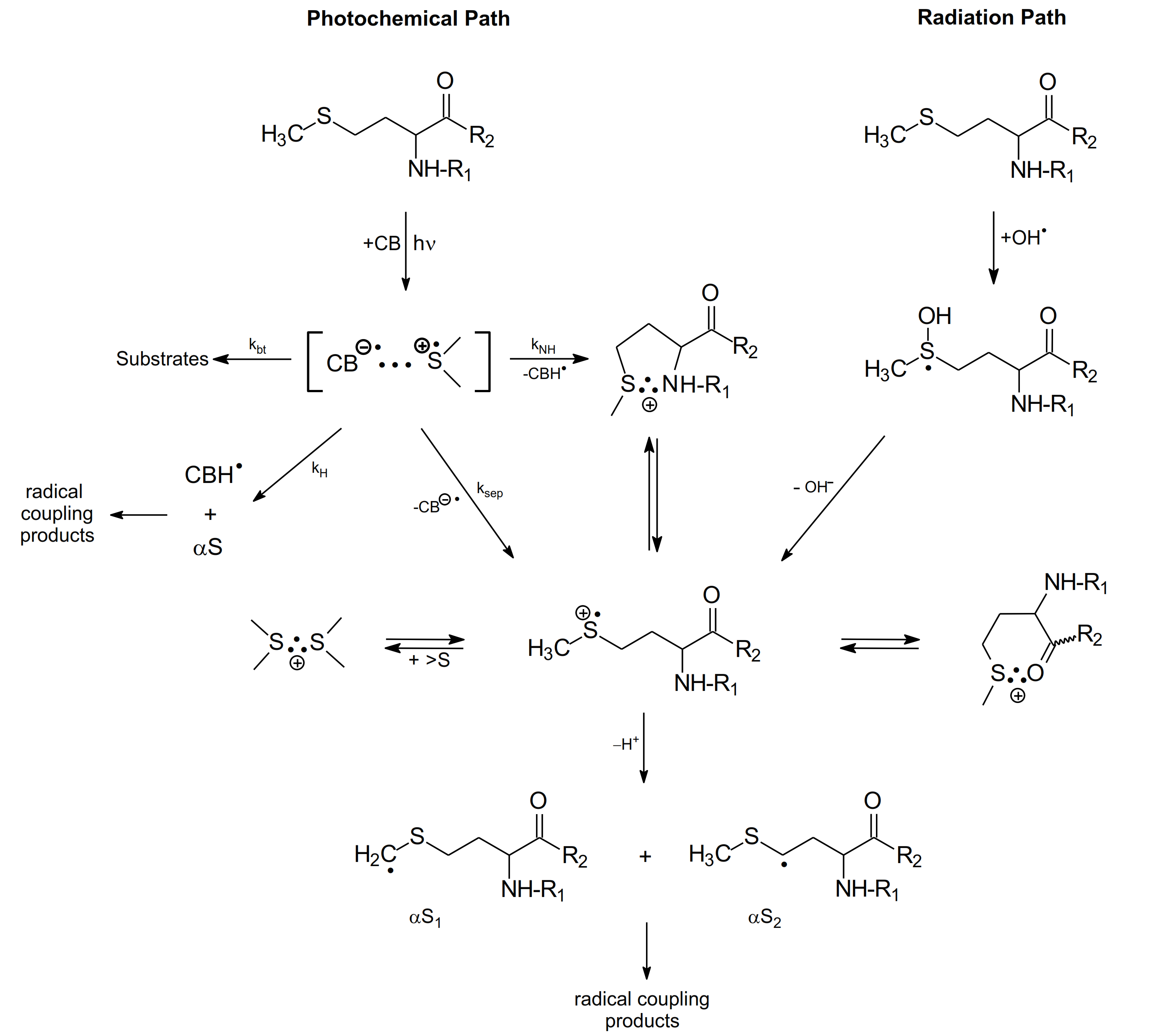
3. Methionine in Linear Peptides
3.1. Radiation-Induced Oxidation
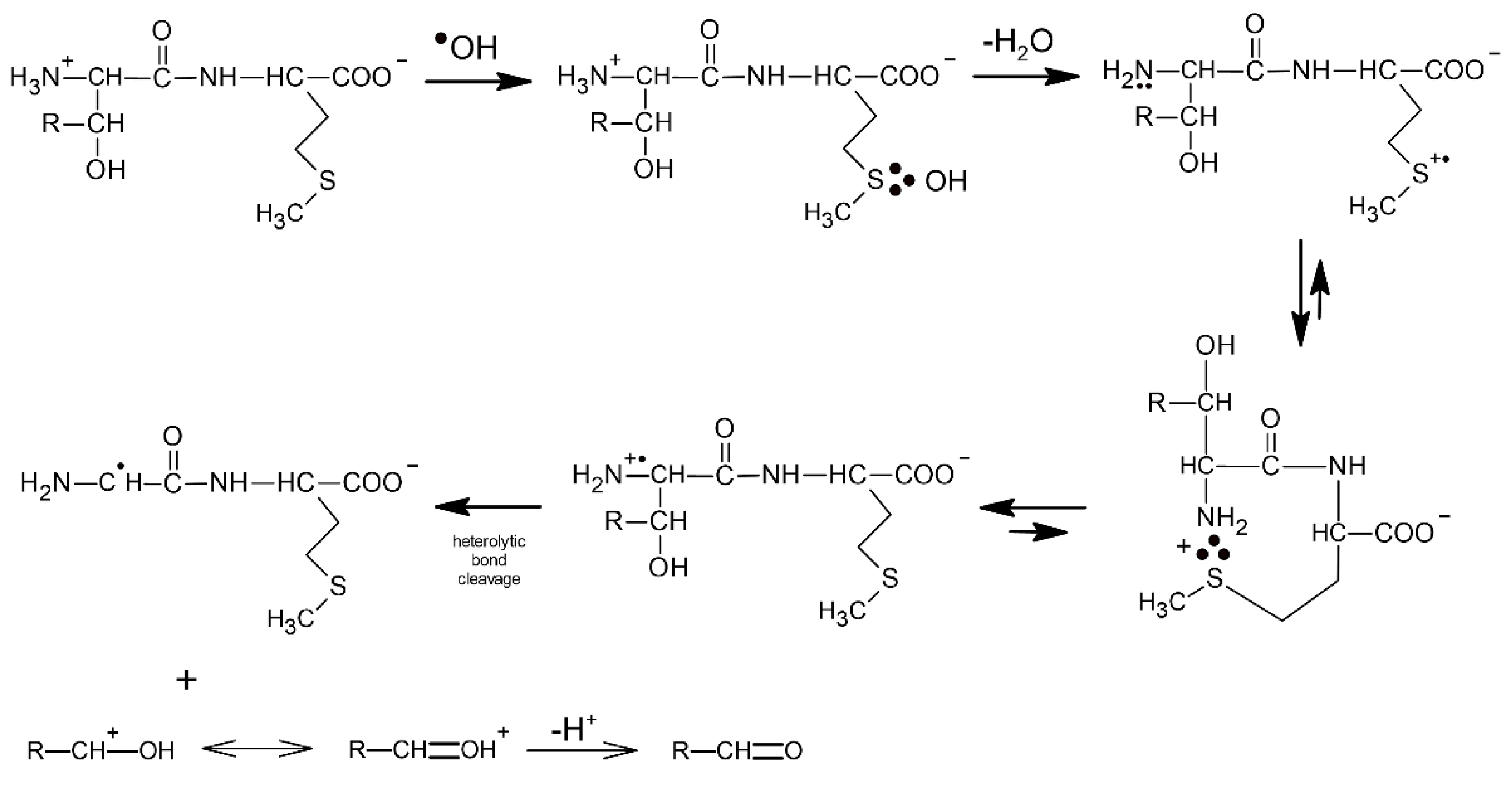
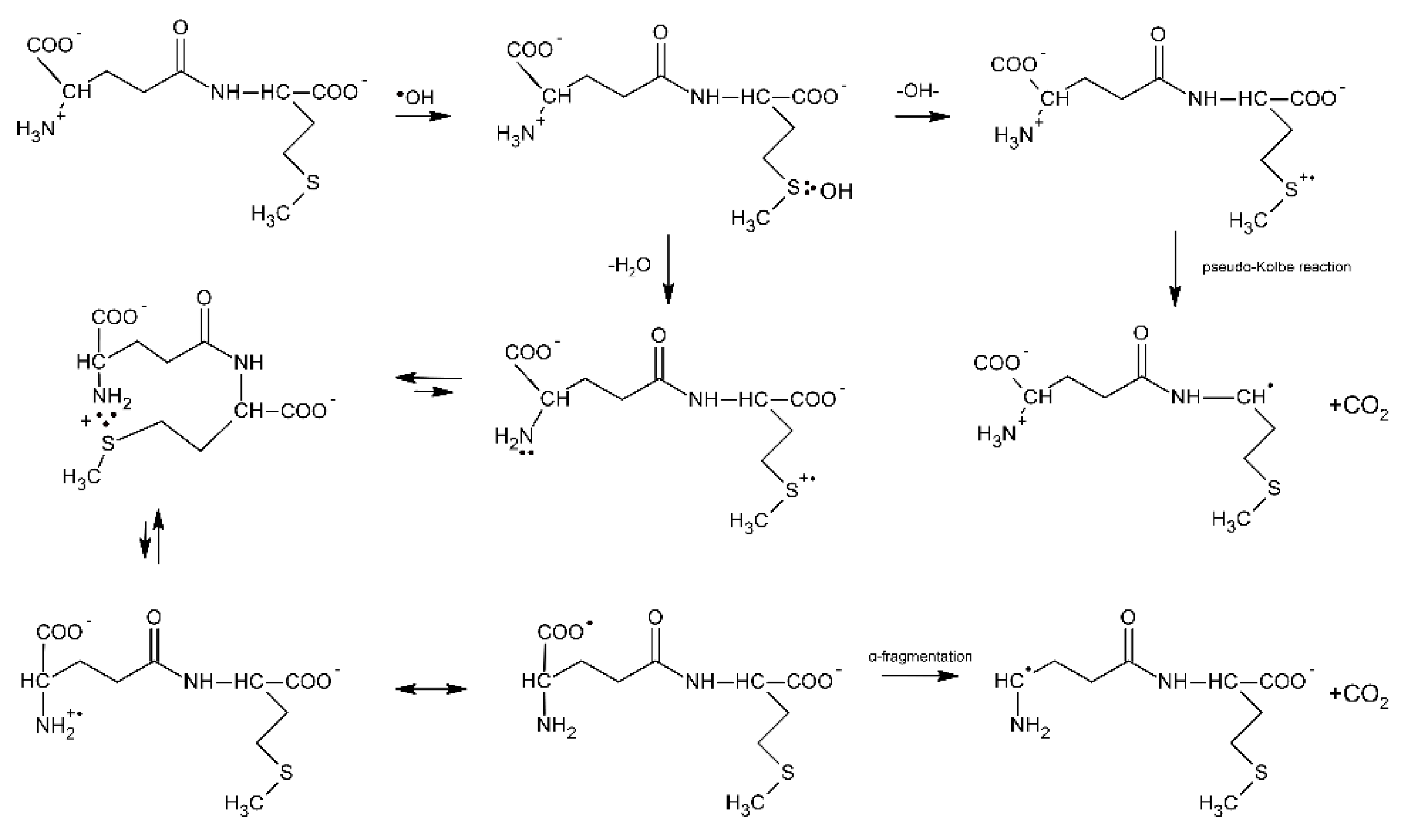
3.1.1. Methionine as the N/C-Terminal Amino Acid Residue


3.1.2. Methionine as the Internal Amino Acid Residue

3.1.3. Linear Peptides with Two Methionine Residues
3.2. Photo-Induced Oxidation
3.2.1. Methionine as the N/C-Terminal and the Internal Amino Acid Residue
3.2.2. Linear Peptides with Two Methionine Residues
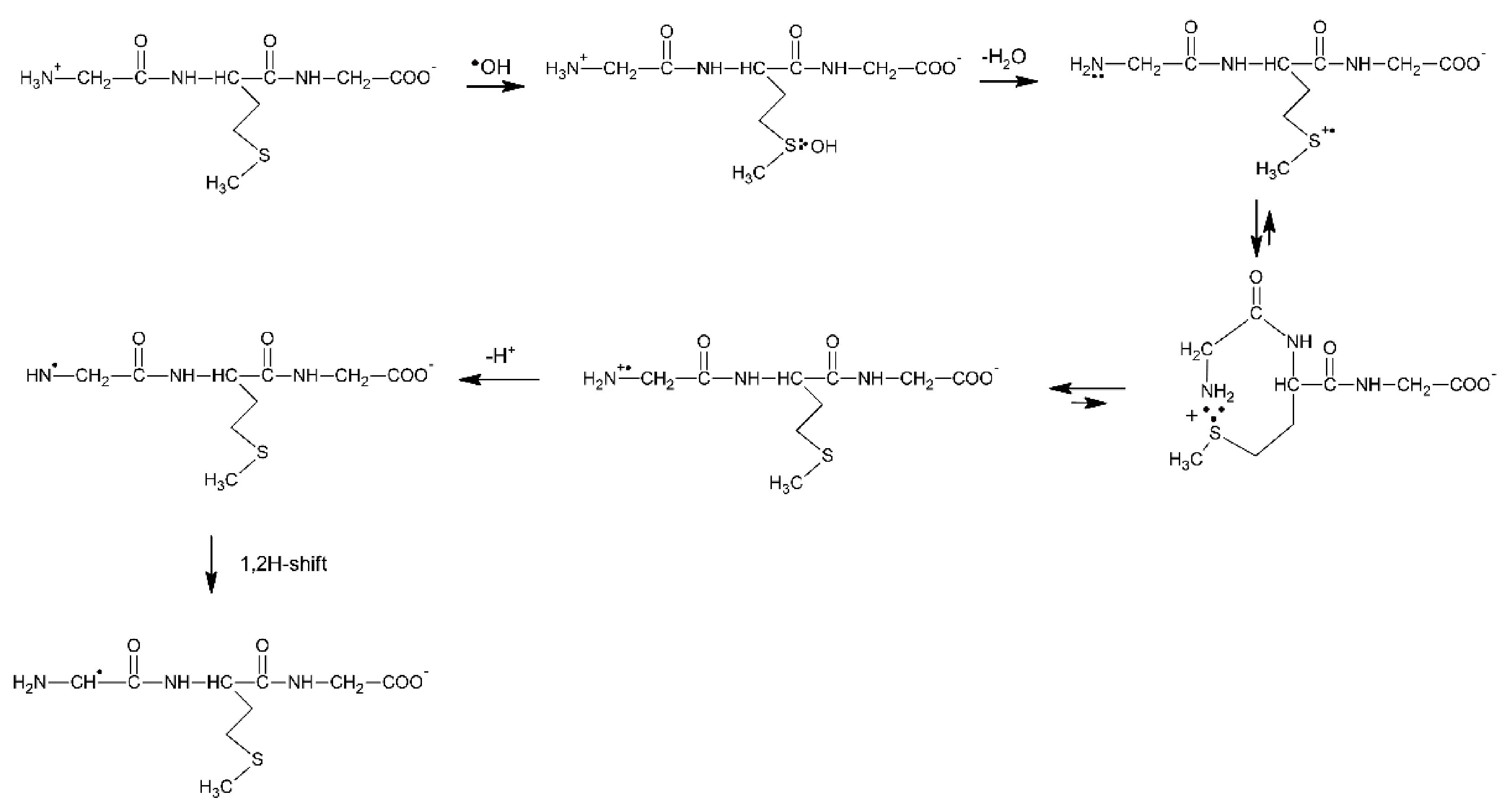
4. Methionine in Cyclic Peptides
4.1. Radiation-Induced Oxidation
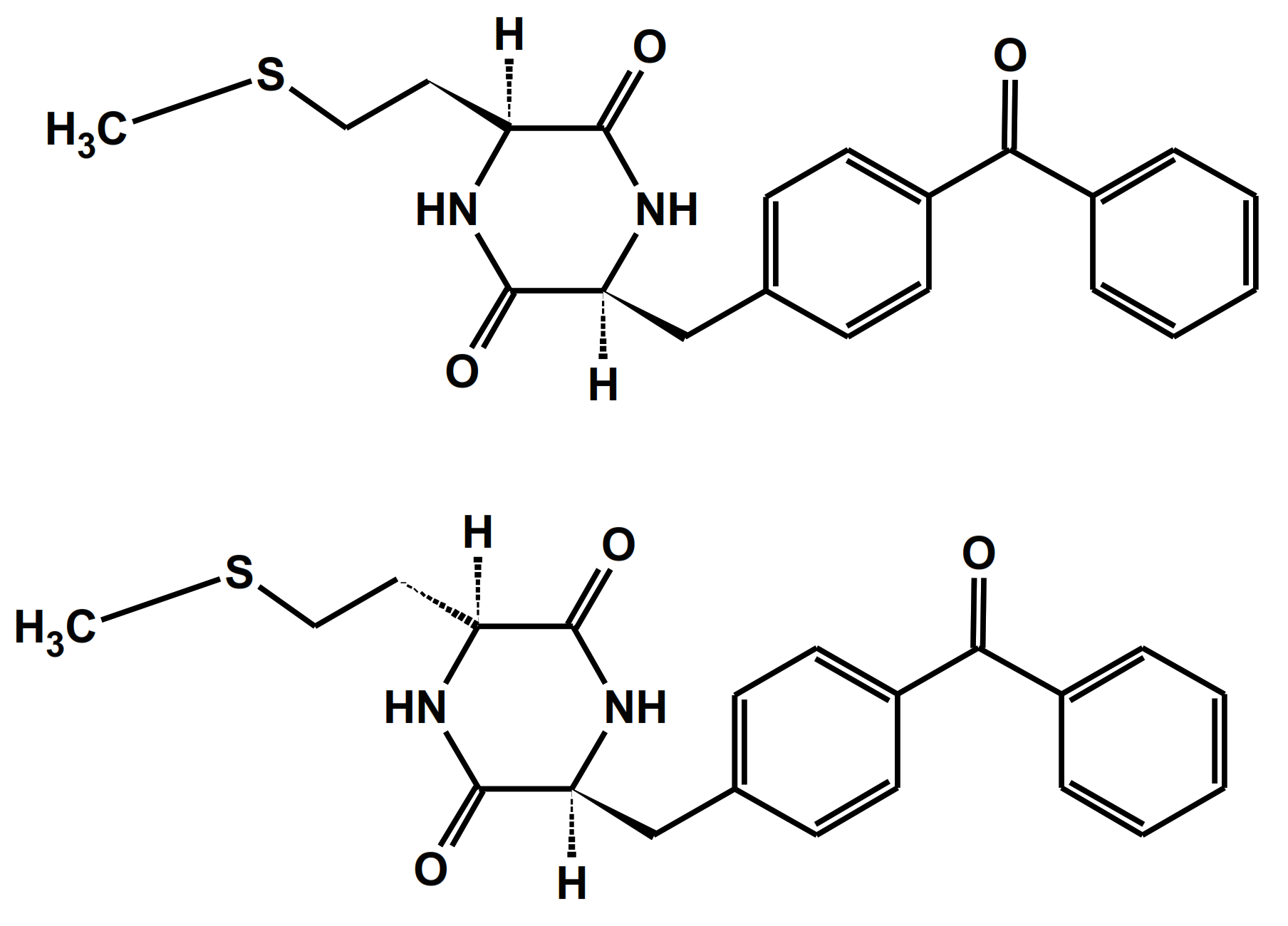
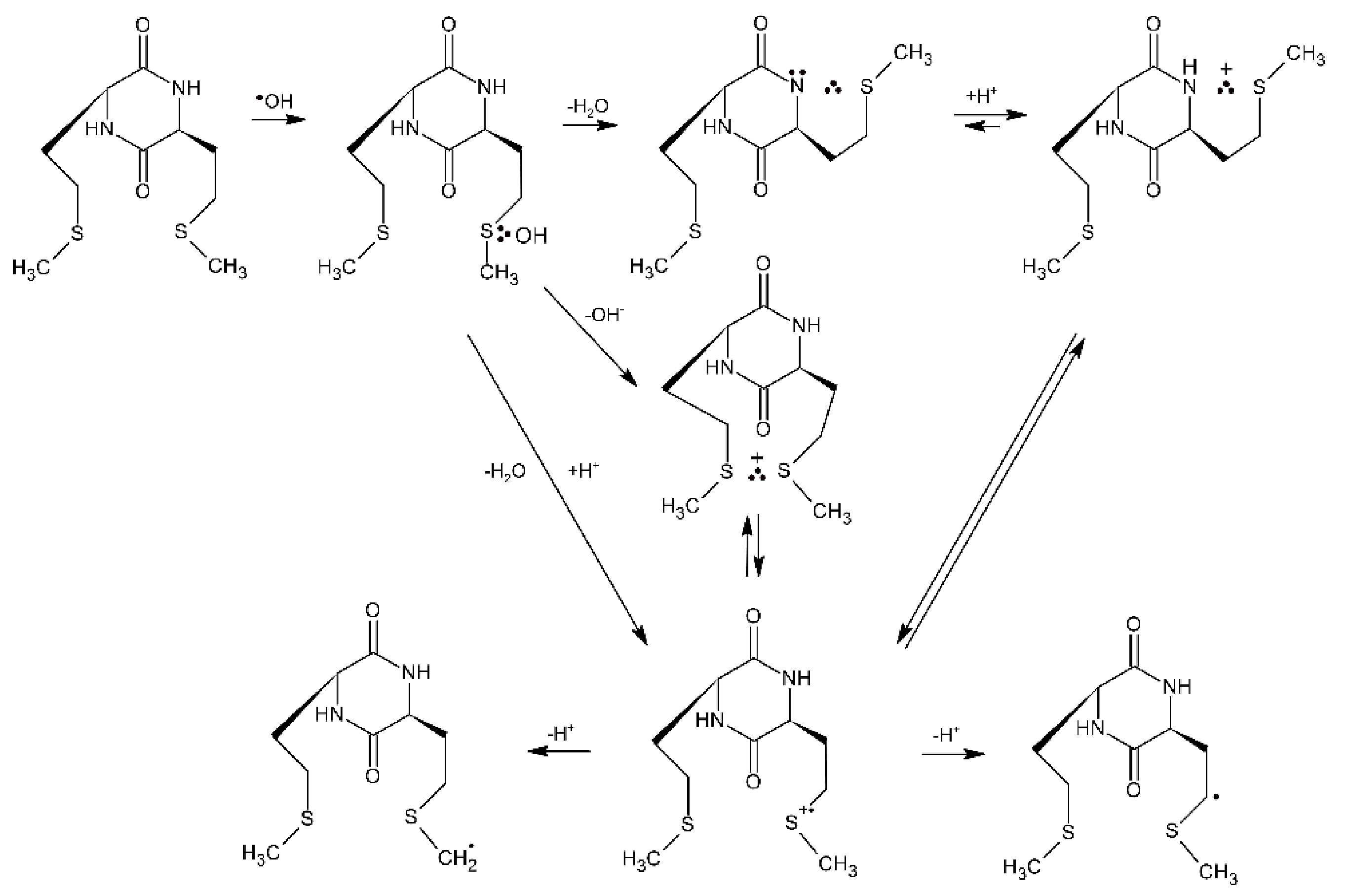
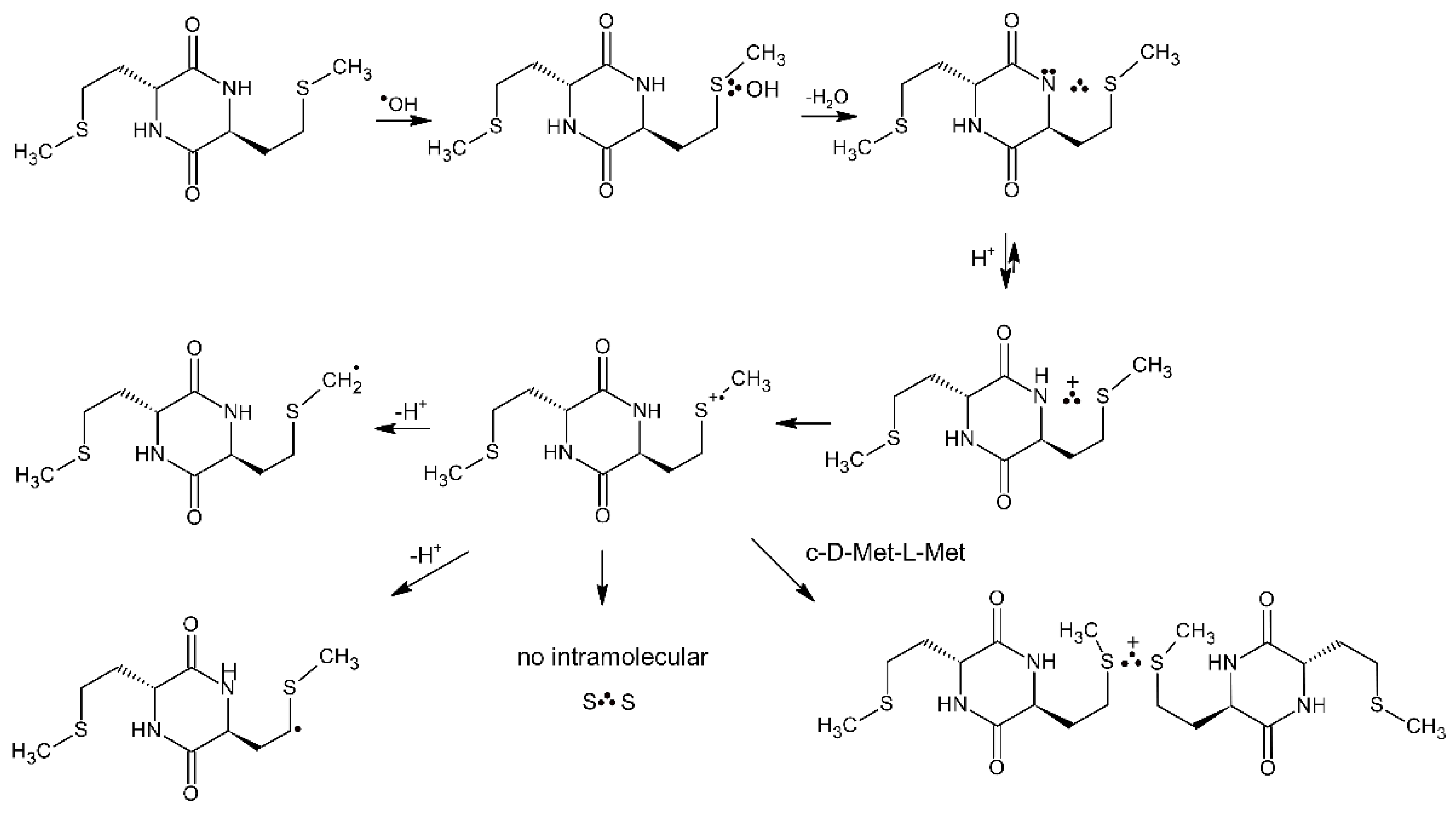
4.1.1. Cyclic Dipeptides with Single Met Residue
4.1.2. Cyclic Dipeptides with Two Met Residues
| Type of the Radical | pH 4 | pH 4.3 | pH 4.9 | pH 5.3 |
|---|---|---|---|---|
| Intramolecular Met(S∴N) | 0.26 (a) | 0.28 (a) | 0.25 (a) | 0.27 (a) |
| 0 | 0 | 0 | 0 | |
| Intramolecular Met(S∴S)+ | 0.22 (a) | 0.23 (a) | 0.22 (a) | 0.23 (a) |
| 0.45 (a) | 0.45 (a) | 0.32 (a) | 0.34 (a) |
4.2. Photo-Induced Oxidation
4.2.1. Cyclic Dipeptides with Single Met Residue
4.2.2. Cyclic Dipeptides with Two Met Residue
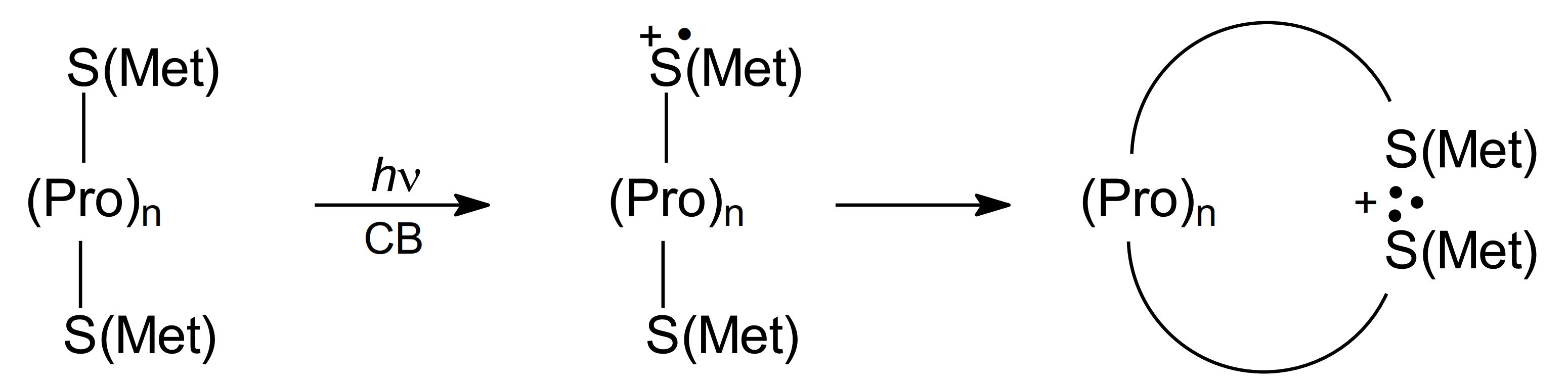
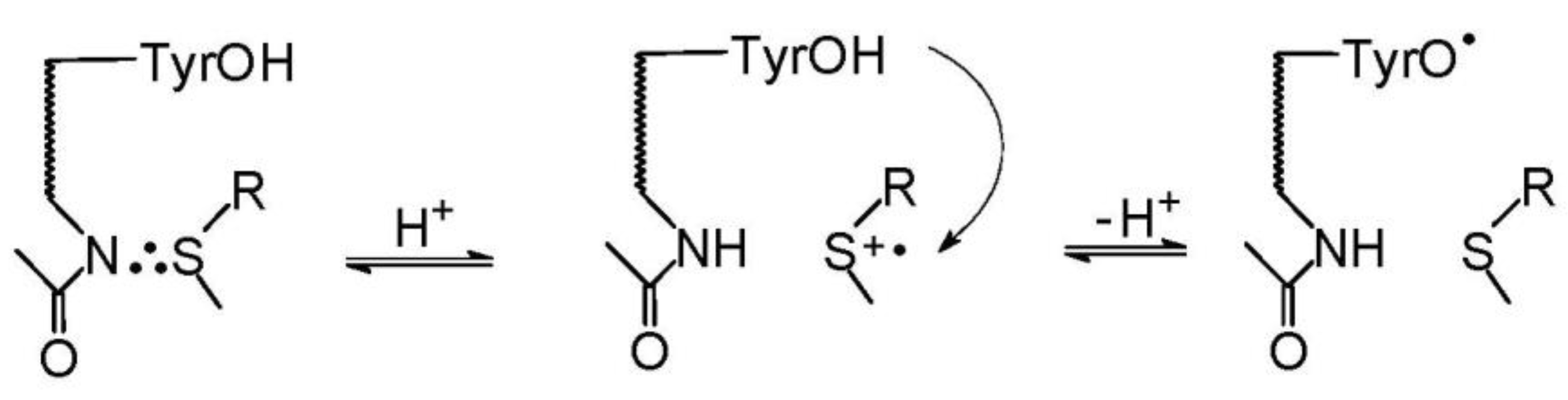
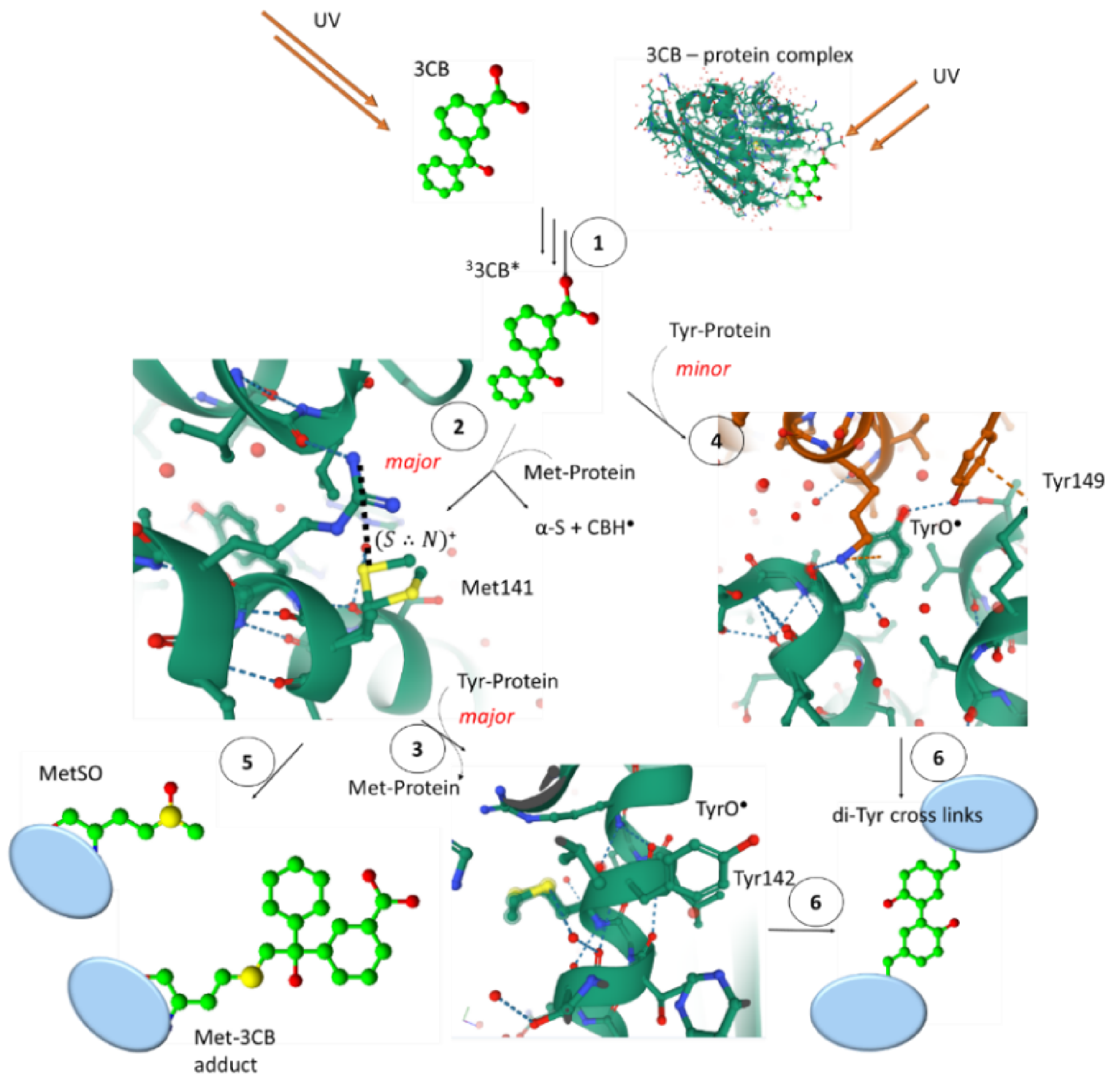
5. Methionine in Proteins
5.1. Radiation-Induced Oxidation

5.2. Photo-Induced Oxidation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levine, R.L.; Mosoni, L.; Berlett, B.S.; Stadtman, E.R. Methionine residues as endogenous antioxidants in proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 15036–15040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, W. Oxidation of Methionyl Residues in Proteins: Tools, Targets, and Reversal. Free Radic. Biol. Med. 1995, 18, 93–105. [Google Scholar] [CrossRef]
- Davies, M. The oxidative environment and protein damage. Biochim. Biophys. Acta 2005, 1703, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R.; Van Remmen, H.; Richardson, A.; Wehr, N.B.; Levine, R.L. Methionine oxidation and aging. Biochim. Biophys. Acta 2005, 1703, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Houée-Levin, C.; Bobrowski, K. The use of methods of radiolysis to explore the mechanisms of free radical modifications in proteins. J. Proteom. 2013, 92, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Ignasiak, M.T.; Marciniak, B.; Houée-Levin, C. A Long Story of Sensitized One-Electron Photo-oxidation of Methionine. Isr. J. Chem. 2014, 54, 248–253. [Google Scholar] [CrossRef]
- Hiller, K.-O.; Masloch, B.; Göbl, M.; Asmus, K.-D. Mechanism of the OH∙ radical induced oxidation of methionine in aqueous solution. J. Am. Chem. Soc. 1981, 103, 2734–2743. [Google Scholar] [CrossRef]
- Bobrowski, K.; Holcman, J. Formation of three-electron bonds in one-electron oxidized methionine dipeptides: A pulse radiolytic study. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1987, 52, 139–144. [Google Scholar] [CrossRef]
- Bobrowski, K.; Holcman, J. Formation and stability of intramolecular three-electron S∴N, S∴S, and S∴O bonds in one-electron-oxidized simple methionine peptides. Pulse radiolysis study. J. Phys. Chem. 1989, 93, 6381–6387. [Google Scholar] [CrossRef]
- Schöneich, C.; Pogocki, D.; Wisniowski, P.; Hug, G.L.; Bobrowski, K. Intramolecular Sulfur-Oxygen Bond Formation in Radical Cations of N-Acetylmethionine Amide. J. Am. Chem. Soc. 2000, 122, 10224–10225. [Google Scholar] [CrossRef]
- Schöneich, C.; Pogocki, D.; Hug, G.L.; Bobrowski, K. Free radical reactions of methionine in peptides: Mechanisms relevant to β-amyloid oxidation and Alzheimer’s disease. J. Am. Chem. Soc. 2003, 125, 13700–13713. [Google Scholar] [CrossRef]
- Bobrowski, K.; Hug, G.L.; Pogocki, D.; Marciniak, B.; Schöneich, C. Stabilization of sulfide radical cations through complexation with the peptide bond: Mechanisms relevant to oxidation of proteins containing multiple methionine residues. J. Phys. Chem. B 2007, 111, 9608–9620. [Google Scholar] [CrossRef]
- Hug, G.L.; Bobrowski, K.; Pogocki, D.; Hörner, G.; Marciniak, B. Conformational influence on the type of stabilization of sulfur radical cations in cyclic peptides. ChemPhysChem 2007, 8, 2202–2210. [Google Scholar] [CrossRef]
- Schöneich, C.; Zhao, F.; Madden, K.P.; Bobrowski, K. Side chain fragmentation of N-terminal threonine or serine residue induced through intramolecular proton transfer to hydroxy sulfuranyl radical formed at neighboring methionine in dipeptides. J. Am. Chem. Soc. 1994, 116, 4641–4652. [Google Scholar] [CrossRef]
- Bobrowski, K.; Marciniak, B.; Hug, G.L. 4-Carboxybenzophenone-sensitized photooxidation of sulfur-containing amino acids. Nanosecond laser flash photolysis and pulse radiolysis studies. J. Am. Chem. Soc. 1992, 114, 10279–10288. [Google Scholar] [CrossRef]
- Marciniak, B.; Hug, G.L.; Bobrowski, K.; Kozubek, H. Mechanism of 4-carboxybenzophenone-sensitized photooxidation of methionine-containing dipeptides and tripeptides in aqueous solution. J. Phys. Chem. 1995, 99, 13560–13568. [Google Scholar] [CrossRef]
- Hug, G.L.; Marciniak, B.; Bobrowski, K. Acid-base equilibria involved in secondary reactions following the 4-carboxybenzophenone sensitized photooxidation of methionyl-glycine in aqueous solution. Spectral and time resolution of the decaying (S∴N)+ radical cation. J. Phys. Chem. 1996, 100, 14914–14921. [Google Scholar] [CrossRef]
- Hug, G.L.; Marciniak, B.; Bobrowski, K. Sensitized photo-oxidation of sulfur-containing amino acids and peptides in aqueous solution. J. Photochem. Photobiol. A 1996, 95, 81–88. [Google Scholar] [CrossRef]
- Hug, G.L.; Bobrowski, K.; Kozubek, H.; Marciniak, B. Photooxidation of Methionine Derivatives by the 4-Carboxybenzophenone Triplet State in Aqueous Solution. Intramolecular Proton Transfer involving the Amino Group. Photochem. Photobiol. 1998, 68, 785–796. [Google Scholar] [CrossRef]
- Ignasiak, M.T.; Pedzinski, T.; Rusconi, F.; Filipiak, P.; Bobrowski, K.; Houée-Levin, C.; Marciniak, B. Photosensitized Oxidation of Methionine-Containing Dipeptides. From the Transients to the Final Products. J. Phys. Chem. B 2014, 118, 8549–8558. [Google Scholar] [CrossRef]
- Pedzinski, T.; Markiewicz, A.; Marciniak, B. Photosensitized oxidation of methionine derivatives. Laser flash photolysis studies. Res. Chem. Intermed. 2009, 35, 497–506. [Google Scholar] [CrossRef]
- Pedzinski, T.; Grzyb, K.; Kaźmierczak, F.; Frański, R.; Filipiak, P.; Marciniak, B. Early Events of Photosensitized Oxidation of Sulfur-Containing Amino Acids Studied by Laser Flash Photolysis and Mass Spectrometry. J. Phys. Chem. B 2020, 124, 7564–7573. [Google Scholar] [CrossRef] [PubMed]
- Goez, M.; Rozwadowski, J.; Marciniak, B. CIDNP spectroscopic observation by magnetic resonance of SN+ radical cations with two-center-three-electron-bond during the photooxidation of methionine. Angew. Chem. Int. Ed. 1998, 37, 785–796. [Google Scholar] [CrossRef]
- Morozova, O.B.; Korchak, S.E.; Vieth, H.-M.; Yurkovskaya, A.V. Photo-CIDNP Study of Transient Radicals of Met-Gly and Gly-Met Peptides in Aqueous Solution at Variable pH. J. Phys. Chem. B 2009, 113, 7398–7406. [Google Scholar] [CrossRef]
- Köchling, T.; Morozova, O.B.; Yurkovskaya, A.V.; Vieth, H.-M. Magnetic Resonance Characterization of One-Electron Oxidized Cyclic Dipeptides with Thioether Groups. J. Phys. Chem. B 2016, 120, 9277–9286. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, H.; White, R.C.; Yurkovskaya, A.V.; Forbes, M.D.E. Methionine Radical Cation: Structural Studies as a Function of pH Using X- and Q-Band Time-Resolved Electron Paramagnetic Resonance Spectroscopy. J. Phys. Chem. A 2005, 109, 5855–5864. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski, K.; Houée-Levin, C.; Marciniak, B. Stabilization and Reactions of Sulfur Radical Cations: Relevance to One-Electron Oxidation of Methionine in Peptides and Proteins. Chimia 2008, 62, 728–734. [Google Scholar] [CrossRef]
- Schöneich, C. Redox processes of methionine relevant to β-amyloid oxidation and Alzheimer’s disease. Arch. Biochem. Biophys. 2002, 397, 370–376. [Google Scholar] [CrossRef]
- Schöneich, C. Methionine oxidation by reactive oxygen species: Reaction mechanisms and relevance to Alzheimer’s disease. Biochim. Biophys. Acta 2005, 1703, 111–119. [Google Scholar] [CrossRef]
- Glass, R.S. Sulfur Radicals and Their Application. Top. Curr. Chem. 2018, 376, 22. [Google Scholar] [CrossRef]
- Schöneich, C.; Zhao, F.; Yang, J.; Miller, B. Mechanisms of Methionine Oxidation in Peptides. In Therapeutic Protein and Peptide Formulation and Delivery; Shahrokh, Z., Sluzky, V., Cleland, J.L., Shire, S.J., Randolph, T.W., Eds.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1997; Volume 675, pp. 79–89. [Google Scholar]
- Bobrowski, K. Chemistry of Sulfur-centered Radicals. In Recent Trends in Radiation Chemistry; Wishart, J.F., Rao, B.S.M., Eds.; World Scientific: Singapore, 2010. [Google Scholar]
- Bobrowski, K. Radiation-induced radicals and radical ions in amino acids and peptides. In Selectivity, Control, and Fine Tuning in High Energy Chemistry; Staas, D.V., Feldman, V.I., Eds.; Research Signpost: Trivandrum, India, 2011. [Google Scholar]
- Houée-Levin, C. One-electron redox processes in proteins. In Selectivity, Control, and Fine Tuning in High-Energy Chemistry; Stass, D.V., Feldman, V.I., Eds.; Research Signpost: Trivandrum, India, 2011; p. 59. [Google Scholar]
- Schöneich, C. Radical-Based Damage of Sulfur-Containing Amino Acid Residues. In Encyclopedia of Radical in Chemistry, Biology and Materials; Chatgilialoglu, C., Studer, A., Eds.; Chemical Biology; John Wiley & Sons, Ltd.: Chichester, UK, 2012; Volume 3, pp. 1459–1474. [Google Scholar]
- Capon, B.; Mc Manus, S.P. Neighboring Group Participation; Plenum Press: New York, NY, USA, 1976; Volume 1. [Google Scholar]
- Glass, R.S.; Hug, G.L.; Schöneich, C.; Wilson, G.S.; Kuznetsova, L.; Lee, T.; Ammam, M.; Lorance, E.; Nauser, T.; Nichol, G.S.; et al. Neighboring Amide Participation in Thioether Oxidation: Relevance to Biological Oxidation. J. Am. Chem. Soc. 2009, 131, 13791–13805. [Google Scholar] [CrossRef]
- Fourre, I.; Silvi, B. What Can We Learn from Two-Center Three-Electron Bonding with the Topological Analysis of ELF? Heteroat. Chem. 2007, 18, 135–160. [Google Scholar] [CrossRef]
- Fourre, I.; Bergès, J.; Houée-Levin, C. Structural and Topological Studies of Methionine Radical Cations in Dipeptides: Electron Sharing in Two-Center Three-Electron Bonds. J. Phys. Chem. A 2010, 114, 7359–7368. [Google Scholar] [CrossRef]
- Brunelle, P.; Rauk, A. One-electron oxidation of methionine in peptide environments: The effect of three-electron bonding on the reduction potential of the radical cation. J. Phys. Chem. A 2004, 108, 11032–11041. [Google Scholar] [CrossRef]
- Asmus, K.-D. Sulfur-centered three-electron bonded radical species. In Sulfur-Centered Reactive Intermediates in Chemistry and Biology; Chatgilialoglu, C., Asmus, K.-D., Eds.; NATO ASI Series, Series A: Life Sciences; Plenum Press: New York, NY, USA, 1990; Volume 197, pp. 155–172. [Google Scholar]
- Asmus, K.-D. Sulfur-centered Free Radicals. In Methods in Enzymology; Packer, L., Ed.; Academic Press: Orlando, FL, USA, 1990; Volume 186, pp. 168–180. [Google Scholar]
- Hiller, K.-O.; Asmus, K.-D. Tl2+ and Ag2+ metal-ion-induced oxidation of methionine in aqueous solution. A pulse radiolysis study. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1981, 40, 597–604. [Google Scholar] [CrossRef]
- Hiller, K.-O.; Asmus, K.-D. Formation and reduction reactions of α-amino radicals derived from methionine and its derivatives in aqueous solutions. J. Phys. Chem. 1983, 87, 3682–3688. [Google Scholar] [CrossRef]
- Mönig, J.; Göbl, M.; Asmus, K.-D. Free Radical One-electron versus Hydrogen Radical-induced Oxidation. Reaction of Trichloromethyl Peroxyl Radicals with Simple and Substituted Aliphatic Sulphides in Aqueous Solution. J. Chem. Soc. Perkin Trans. 1985, 647. [Google Scholar] [CrossRef]
- Asmus, K.-D.; Göbl, M.; Hiller, K.-O.; Mahling, S.; Mönig, J. S∴N and S∴O three-electron-bonded radicals and radical cations in aqueous solutions. J. Chem. Soc. Perkin Trans. 2 1985, 641–646. [Google Scholar] [CrossRef]
- Armstrong, D.A.; Huie, R.E.; Koppenol, W.H.; Lymar, S.V.; Merenyi, G.; Neta, P.; Ruscic, B.; Stanbury, D.M.; Steenken, S.; Wardman, P. Standard electrode potentials involving radicals in aqueois solutions: Inorganic radicals (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1139–1150. [Google Scholar] [CrossRef]
- Rauk, A.; Armstrong, D.A.; Fairlie, D.P. Is oxidative damage by β-amyloid and prion peptides mediated by hydrogen atom transfer from glycyne α-carbon to methionine sulfur within β-sheets? J. Am. Chem. Soc. 2000, 122, 9761–9767. [Google Scholar] [CrossRef]
- Merényi, G.; Lind, J.; Engman, L. One- and Two-electron reduction Potentials of Peroxyl Radicals and Related Species. J. Chem. Soc. Perkin Trans. 2 1994, 2551–2553. [Google Scholar] [CrossRef]
- Chen, S.N.; Hoffman, M.Z. Rate constants for the reaction of the carbonate radical with compounds of biochemical interest in neutral aqueous solution. Radiat. Res. 1973, 56, 40–47. [Google Scholar] [CrossRef]
- Wojnárovits, L.; Takács, E. Rate constants of dichloride radical anion reactions with molecules of environmental interest in aqueous solution: A review. Environ. Sci. Pollut. Res. 2021, 28, 41552–41575. [Google Scholar] [CrossRef]
- Hiller, K.-O.; Asmus, K.-D. Oxidation of methionine by X2∙− in aqueous solution and characterization of some >SX three-electron bonded intermediates. A pulse radiolysis study. Int. J. Radiat. Biol. 1981, 40, 583–595. [Google Scholar]
- Bonifačić, M.; Asmus, K.-D. Stabilization of oxidized sulfur centres by halide ions. Formation and properties of R2SX radicals in aqueous solutions. J. Chem. Soc. Perkin Trans. 2 1980, 758–762. [Google Scholar] [CrossRef]
- Pogocki, D.M. Investigation of Radical Processes Induced by Hydroxyl Radical in Amino Acids and Peptides Containing Thioether Group. Ph.D. Thesis, Institute of Nuclear Chemistry and Technology, Warsaw, Poland, 1996. [Google Scholar]
- Mishra, B.; Priyadarsini, K.I.; Mohan, H. Pulse radiolysis studies on reaction of OH radical with N-acetyl methionine in aqueous solutions. Res. Chem. Interm. 2005, 31, 625–632. [Google Scholar] [CrossRef]
- Shirdhonkar, M.; Maity, D.K.; Mohan, H.; Rao, B.S.M. Oxidation of methionine methyl ester in aqueous solution: A combined pulse radiolysis and quantum chemical study. Chem. Phys. Lett. 2006, 417, 116–123. [Google Scholar] [CrossRef]
- Bobrowski, K.; Schöneich, C.; Holcman, J.; Asmus, K.-D. •OH radical induced decarboxylation of methionine-containing peptides. Influence of peptide sequence and net charge. J. Chem. Soc., Perkin Trans. 2 1991, 353–362. [Google Scholar] [CrossRef]
- Steffen, L.K.; Glass, R.S.; Sabahi, M.; Wilson, G.S.; Schöneich, C.; Mahling, S.; Asmus, K.-D. ∙OH radical induced decarboxylation of amino acids. Decarboxylation vs bond formation in radical intermediates. J. Am. Chem. Soc. 1991, 113, 2141–2145. [Google Scholar] [CrossRef]
- Pedzinski, T.; Grzyb, K.; Skotnicki, K.; Filipiak, P.; Bobrowski, K.; Chatgilialoglu, C.; Marciniak, B. Radiation- and Photo-Induced Oxidation Pathways of Methionine in Model Peptide Backbone under Anoxic Conditions. Int. J. Mol. Sci. 2021, 22, 4773. [Google Scholar] [CrossRef]
- Guttenplan, J.B.; Cohen, S.G. Quenching and reduction of photoexcited benzophenone by thioethers and mercaptans. J. Org. Chem. 1973, 38, 2001–2007. [Google Scholar] [CrossRef]
- Wojcik, A.; Lukaszewicz, A.; Brede, O.; Marciniak, B. Competitive photosensitized oxidation of tyrosine and methionine residues in enkephalins and their model peptides. J. Photochem. Photobiol. A Chem. 2008, 198, 111–118. [Google Scholar] [CrossRef]
- Marciniak, B.; Bobrowski, K.; Hug, G.L. Quenching of triplet states of aromatic ketones by sulfur-containing amino acids in solution. Evidence for electron transfer. J. Phys. Chem. 1993, 97, 11937–11943. [Google Scholar] [CrossRef]
- Hug, G.L.; Bobrowski, K.; Kozubek, H.; Marciniak, B. Photo-oxidation of Methionine-containing Peptides by the 4-Carboxy-benzophenone Triplet State in Aqueous Solution. Competition between Intramolecular Two-centered Three-electron Bonded (S∴S)+ and (S∴N)+ Formation. Photochem. Photobiol. 2000, 72, 1–9. [Google Scholar] [CrossRef]
- Cohen, S.G.; Ojanpera, S. Photooxidation of methionine and related compounds. J. Am. Chem. Soc. 1975, 97, 5633–5634. [Google Scholar] [CrossRef]
- Encinas, M.V.; Lissi, E.A.; Olea, A.F. Quenching of triplet benzophenone by vitamins E and C and by sulfur containing aminoacids and peptides. Photochem. Photobiol. 1985, 42, 347–352. [Google Scholar] [CrossRef]
- Encinas, M.V.; Lissi, E.A.; Vasquez, M.; Olea, A.F.; Silva, E. Photointeraction of benzophenone triplet with lysozyme. Photochem. Photobiol. 1989, 49, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski, K.; Hug, G.L.; Marciniak, B.; Kozubek, H. 4-Carboxybenzophenone-sensitized photooxidation of sulfur-containing amino acids in alkaline aqueous solutions. Secondary photoreactions kinetics. J. Phys. Chem. 1994, 98, 537–544. [Google Scholar] [CrossRef]
- Filipiak, P.; Bobrowski, K.; Hug, G.L.; Pogocki, D.; Schöneich, C.; Marciniak, B. New Insights into the Reaction Paths of 4-Carboxybenzophenone Triplet with Oligopeptides Containing N- and C-Terminal Methionine Residues. J. Phys. Chem. B 2017, 121, 5247–5258. [Google Scholar] [CrossRef]
- Goez, M.; Rozwadowski, J. Reversible Pair Substitution in CIDNP: The Radical Cation of Methionine. J. Phys. Chem. A 1998, 102, 7945–7953. [Google Scholar] [CrossRef]
- Bonifačić, M.; Stefanic, I.; Hug, G.L.; Armstrong, D.A.; Asmus, K.-D. Glycine decarboxylation: The free radical mechanism. J. Am. Chem. Soc. 1998, 120, 9930–9940. [Google Scholar] [CrossRef]
- Marciniak, B.; Hug, G.L.; Rozwadowski, J.; Bobrowski, K. Excited Triplet State of N-(9-methylpurin-6-yl)pyridinium Cation as as Efficient Photosensitizer in the Oxidation of Sulfur-containing Amino Acids. Laser Flash and Steady-state Photolysis Studies. J. Am. Chem. Soc. 1995, 117, 127–134. [Google Scholar] [CrossRef]
- Bobrowski, K.; Hug, G.L.; Pogocki, D.; Marciniak, B.; Schöneich, C. Sulfur radical cation peptide bond complex in the one-electron oxidation of S-methylglutathione. J. Am. Chem. Soc. 2007, 129, 9236–9245. [Google Scholar] [CrossRef] [PubMed]
- Filipiak, P.; Hug, G.L.; Bobrowski, K.; Pedzinski, T.; Kozubek, H.; Marciniak, B. Sensitized Photooxidation of S-Methylglutathione in Aqueous Solution: Intramolecular (S∴O) and (S∴N) Bonded Species. J. Phys. Chem. B 2013, 117, 2359–2368. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Ferreri, C.; Zhang, T.; Permentier, H.; Bischoff, R.; Bobrowski, K.; Chatgilialoglu, C. Radiation chemical studies of Gly-Met-Gly in aqueous solution. Free Rad. Res. 2016, 50, S24–S39. [Google Scholar] [CrossRef] [Green Version]
- Hug, G.L.; Bobrowski, K.; Kozubek, H.; Marciniak, B. pH effects on the photooxidation of methionine derivatives by the 4-carboxybenzophenone triplet state. Nukleonika 2000, 45, 63–71. [Google Scholar]
- Lewandowska-Andralojć, A.; Kazmierczak, F.; Hug, G.L.; Hörner, G.; Marciniak, B. Photoinduced CC-coupling Reactions of Rigid Diastereomeric Benzophenone-Methionine Dyads. Photochem. Photobiol. 2013, 89, 14–23. [Google Scholar] [CrossRef]
- Ignasiak, M.T. Study of the Mechanism of Radiation- and Photo-Induced Oxidation of Methionine Containing Peptides. Ph.D. Thesis, Adam Mickiewicz University, Poznań, Poland, 2014. [Google Scholar]
- Bonifačić, M.; Hug, G.L.; Schöneich, C. Kinetics of the reactions between sulfide radical cation complexes, [S∴S]+ and [S∴N]+, and superoxide or carbon dioxide radical anions. J. Phys. Chem. A 2000, 104, 1240–1245. [Google Scholar] [CrossRef]
- Klug, D.; Rabani, J.; Fridovich, I. A direct demonstration of the catalytic action of superoxide dismutase through the use of pulse radiolysis. J. Biol. Chem. 1972, 247, 4839–4842. [Google Scholar] [CrossRef]
- Bobrowski, K.; Schöneich, C.; Holcman, J.; Asmus, K.-D. •OH radical induced decarboxylation of γ-glutamylmethionine and S-alkylglutathione derivatives: Evidence for two different pathways involving C- and N-terminal decarboxylation. J. Chem. Soc. Perkin Trans. 2 1991, 975–980. [Google Scholar] [CrossRef]
- Bobrowski, K.; Schöneich, C. Decarboxylation mechanism of the N-terminal glutamyl moiety in γ-glutamic acid and methionine containing peptides. Radiat. Phys. Chem. 1996, 47, 507–510. [Google Scholar] [CrossRef]
- Filipiak, P.; Bobrowski, K.; Hug, G.L.; Schöneich, C.; Marciniak, B. N-Terminal Decarboxylation as a Probe for Intramolecular Contact Formation in γ-Glu-(Pro)n-Met Peptides. J. Phys. Chem. B 2020, 124, 8082–8098. [Google Scholar] [CrossRef]
- Harriman, A. Further comment on the redox potentials of tryptophan and tyrosine. J. Phys. Chem. 1987, 91, 6102–6104. [Google Scholar] [CrossRef]
- Folkes, L.K.; Trujillo, M.; Bartesaghi, S.; Radi, R.; Wardman, P. Kinetics of reduction of tyrosine phenoxyl radicals by glutathione. Arch. Biochem. Biophys. 2011, 506, 242–249. [Google Scholar] [CrossRef]
- Merenyi, G.; Lind, J.; Engman, L. The Dimethylhydroxysulfuranyl Radical. J. Phys. Chem. 1996, 100, 8875–8881. [Google Scholar] [CrossRef]
- Stryer, L. Biochemistry; W. H. Freeman and Company: New York, NY, USA, 1988. [Google Scholar]
- Mozziconacci, O.; Mirkowski, J.; Rusconi, F.; Kciuk, G.; Wisniowski, P.; Bobrowski, K.; Houée-Levin, C. Methionine residue acts as a prooxidant in the OH-induced oxidation of enkephalins. J. Phys. Chem B 2012, 116, 9352–9362. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (∙OH/∙O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef] [Green Version]
- Bobrowski, K.; Wierzchowski, K.L.; Holcman, J.; Ciurak, M. Intramolecular electron transfer in peptides containing methionine, tryptophan, and tyrosine: A pulse radiolysis study. Int. J. Radiat. Biol. 1990, 57, 919–932. [Google Scholar] [CrossRef]
- Bobrowski, K.; Wierzchowski, K.L.; Holcman, J.; Ciurak, M. Pulse radiolysis, of intramolecular electron transfer in model peptides. IV. Met/S∴Br -> Tyr/O∙ radical transformation in aqueous solution of H-Tyr-(Pro)n-Met-OH peptides. Int. J. Radiat. Biol. 1992, 62, 507–516. [Google Scholar] [CrossRef]
- Bergès, J.; Trouillas, P.; Houée-Levin, C. Oxidation of protein tyrosine, or methionine residues: From the amino acid to the peptide. J. Phys. Conf. Ser. 2011, 261, 012003. [Google Scholar] [CrossRef]
- Kadlčik, V.; Sicard-Roselli, C.; Mattioli, T.A.; Kodiček, M.; Houée-Levin, C. One-electron oxidation of β−amyloid peptide: Sequence modulation of reactivity. Free Radic. Biol. Med. 2004, 37, 881–891. [Google Scholar] [CrossRef] [PubMed]
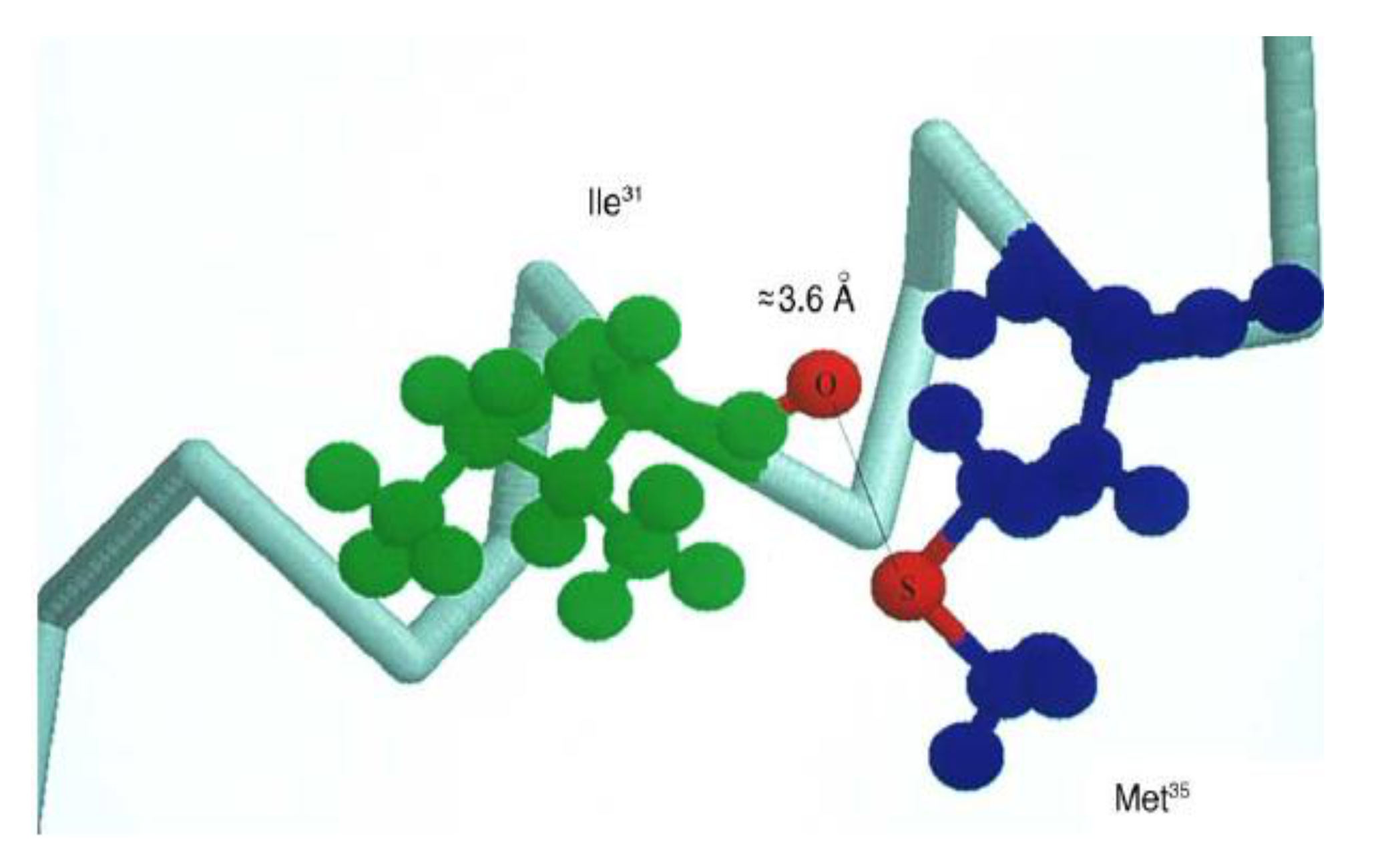
- Pogocki, D.; Schöneich, C. Redox properties of Met35 in neurotoxic β-amyloid peptide. A molecular modeling study. Chem. Res. Toxicol. 2002, 15, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Bush, A.I. Alzheimer’s amyloid β-peptide (1-42): Involvement of methionine residue 35 in the oxidative stress and neurotoxicity properties of this peptide. Neurobiol. Aging 2004, 25, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Kanski, J.; Varadarajan, S.; Aksenova, M.; Butterfield, D.A. Role of glycine-33 and methionine-35 in Alzheimer’s amyloid beta peptide 1–42-associated oxidative stress and neurotoxicity. Biochim. Biophys. Acta 2001, 1586, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Varadarajan, S.; Yatin, S.; Kanski, J.; Jahanshahi, F.; Butterfield, D.A. Methionine residue 35 is important in amyloid beta peptide-associated free radical oxidative stress. Brain Res. Bull. 1999, 50, 133–141. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Kimball-Boyd, D. The critical role of methionine 35 in Alzheimer’s amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochim. Biophys. Acta 2005, 1703, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Asmus, K.-D. Stabilization of oxidized sulfur centers in organic sulfides. Radical cations and odd-electron sulfur-sulfur bonds. Acc. Chem. Res. 1979, 12, 436–442. [Google Scholar] [CrossRef]
- Asmus, K.-D.; Bahnemann, D.; Fischer, C.-H.; Veltwisch, D. Structure and stability of radical cations from cyclic and open-chain dithia compounds in aqueous solutions. J. Am. Chem. Soc. 1979, 101, 5322–5329. [Google Scholar] [CrossRef]
- Filipiak, P.; Bobrowski, K.; Hug, G.L.; Pogocki, D.; Schöneich, C.; Marciniak, B. Formation of a Three-Electron Sulfur−Sulfur Bond as a Probe for Interaction between Side Chains of Methionine Residues. J. Phys. Chem. B 2016, 120, 9732–9744. [Google Scholar] [CrossRef]
- Zahn, R.; Liu, A.; Luhrs, T.; Riek, R.; Von Schroetter, C.; Lopez Garcia, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wüthrich, K. NMR solution structure of the human prion protein. Proc. Nat. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Glasser, C.B.; Yamin, G.; Uversky, V.N.; Fink, A.L. Methionine oxidation, α−synuclein and Parkinson’s disease. Biochim. Biophys. Acta 2005, 1703, 157–169. [Google Scholar] [CrossRef]
- Kobayashi, K. Pulse Radiolysis Studies for Mechanism in Biochemical Redox Reactions. Chem. Rev. 2019, 119, 4413–4462. [Google Scholar] [CrossRef]
- Bobrowski, K.; Holcman, J.; Poznanski, J.; Wierzchowski, K.L. Pulse radiolysis of intramolecular electron transfer in model peptides and proteins. 7. Trp → TyrO radical transformation in hen-egg white lysozyme. Effects of pH, temperature, Trp62 oxidation and inhibitor binding. Biophys. Chem. 1997, 63, 153–166. [Google Scholar] [CrossRef]
- Houée-Levin, C.; Bobrowski, K. Pulse radiolysis studies of free radical processes in peptides and proteins. In Radiation Chemistry: From Basics to Applications in Material and Life Sciences; Spotheim-Maurizot, M., Mostafavi, M., Douki, T., Belloni, J., Eds.; EDP Sciences: Bonchamp-Les Laval, France, 2008; pp. 233–247. [Google Scholar]
- Butler, J.; Land, E.J.; Prütz, W.A.; Swallow, A.J. Charge transfer between tryptophan and tyrosine in proteins. Biochim. Biophys. Acta 1982, 705, 150–162. [Google Scholar] [CrossRef]
- Audette-Stuart, M.; Blouquit, Y.; Faraggi, M.; Sicard-Roselli, C.; Houee-Levin, C.; Jolles, P. Re-evaluation of intramolecular long-range electron transfer between tyrosine and tryptophan in lysozymes. Eur. J. Biochem. 2003, 270, 3565–3571. [Google Scholar] [CrossRef]
- Sicard-Roselli, C.; Lemaire, S.; Jacquot, J.-P.; Favaudon, V.; Marchand, C.; Houee-Levin, C. Thioredoxin Ch1 of Chlamydomonas reinhardtii displays an unusual resistance toward one-electron oxidation. Eur. J. Biochem. 2004, 271, 3481–3487. [Google Scholar] [CrossRef]
- Nauser, T.; Jacoby, M.; Koppenol, W.H.; Squier, T.C.; Schöneich, C. Calmodulin methionine residues are targets for one-electron oxidation by hydroxyl radicals: Formation of S∴N three-electron bonded radical complexes. Chem. Commun. 2005, 587–589. [Google Scholar] [CrossRef]
- Ignasiak, M.; Nowicka-Bauer, K.; Grzechowiak, M.; Sikorski, M.; Shashikadze, B.; Jaskolski, M.; Marciniak, B. Sensitized photo-oxidation of plant cytokinin-specific binding protein—Does the environment of the thioether group influence the oxidation reaction? From primary intermediates to stable products. Free Radic. Biol. Med. 2021, 165, 411–420. [Google Scholar] [CrossRef]



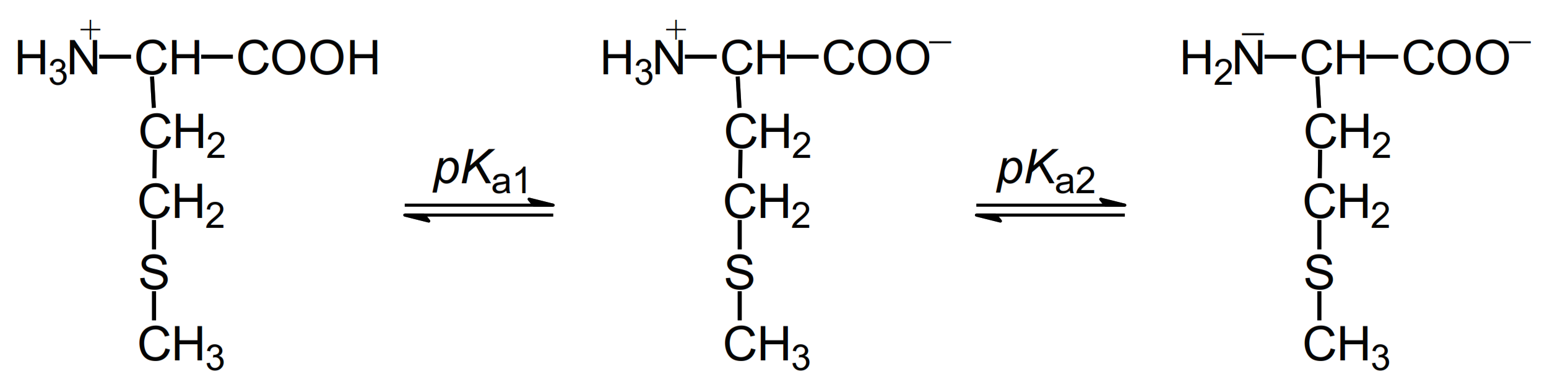{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













| pH | ΦCBH∙ | ΦCB∙− | ΦCBH∙ + ΦCB∙− | Φ(S∴N)+ | Φ(S∴S)+ | Φ’’ CBH∙ | ΦCO2- | Lit | |
|---|---|---|---|---|---|---|---|---|---|
| Methionine | 7 | 0.11 | 0.27 | 0.38 | 0 | 0.23 | 0.25 | 0.28 | [18] |
| Methionine | 11 | 0.05 | 0.66 | 0.71 | 0.38 (a) | 0.10 | 0.66 (b) | 0.55 | [18,67] |
| Met Derivatives | pH | ΦCBH∙ | ΦCB∙− | ΦCBH∙ + ΦCB∙− | Φ(S∴N)+ | Φ(S∴S)+ | ΦCO2− | Lit |
|---|---|---|---|---|---|---|---|---|
| Met-OCH3 | 6 | 0.39 | 0 | 0.39 | 0.20 | 0.04 | 0 | [19,75] |
| Met-OCH3 | 10 | 0 | 0.43 | 0.43 | 0.28 | 0.04 | 0 | [19,75] |
| Met-NH2 | 5 | 0.39 | 0 | 0.39 | 0.28 | 0.10 | 0 | [19,75] |
| Met-NH2 | 10 | 0 | 0.48 | 0.48 | 0.12 | 0.07 | 0 | [19] |
| N-Ac-Met | 7 | 0.12 | 0.29 | 0.41 | 0 | 0.28 | 0.05 | [18,19] |
| N-Ac-Met | 10 | 0.13 | 0.29 | 0.42 | 0 | 0.27 | − | [19,75] |
| N-Ac-Met-OCH3 | 6 | 0.22 | 0.04 | 0.26 | <0.05 | 0.07 | 0 | [19] |
| N-Ac-Met-NH2 | 6 | 0.19 | 0.07 | 0.26 | 0 | 0.09 | 0 | [19] |
| N-Ac-Met-NHCH3 | 7 | 0.32 | <0.02 | 0.34 | 0 | 0 | 0 | [22] |
| Time | α-N | α-S | α-C | Met(S∴N)+ | Met(S∴S)+ |
|---|---|---|---|---|---|
| 1.4 μs | 0.34 (57.1%) | 0.14 (23.6%) | 0.052 (8.6%) | 0.035 (5.9%) | 0.029 (4.8%) |
| 3.5 μs | 0.35 (58.7%) | 0.13 (21.8%) | 0.061 (10.2%) | 0.029 (4.8%) | 0.027 (4.5%) |
| Time | MetS∴OH | α-S | α-C | Met(S∴N)+ | Met(S∴S)+ |
|---|---|---|---|---|---|
| 1.4 μs | 0.18 (30.6%) | 0.17 (29.9%) | 0.05 (8.1%) | 0.10 (17.7%) | 0.08 (13.6%) |
| 3.5 μs | 0.07 (11.8%) | 0.29 (49.0%) | 0.02 (3.8%) | 0.11 (19.2%) | 0.10 (16.3%) |
| Met-Containing Peptides (a) | pH | ΦCBH∙ (b) | ΦCB∙− (b) | ΦCBH∙ + ΦCB∙− (b) | Φ(S∴N)+ (b) | Φ(S∴S)+ (b) | ΦCO2- (c) | Lit |
|---|---|---|---|---|---|---|---|---|
| Met-Gly | 6 | 0.25 | 0.13 | 0.38 | 0.18 | ~0.04 | 0.01 | [16,18] |
| Met-Gly | 11 | 0.06 | 0.55 | 0.61 | 0.51 | ~0.04 | 0.01 | [16,18,75] |
| Gly-Met | 6 | 0.15 | 0.22 | 0.37 | 0 | 0.22 | 0.02 | [18] |
| Gly-Met | 11 | 0.10 | 0.32 | 0.42 | 0 | 0.24 | 0.02 | [18] |
| Met-Gly-Gly | 6 | ~0.29 | ~0.06 | ~0.35 | 0.14 | ~0.08 | 0.01 | [16] |
| Met-Gly-Gly | 11 | 0.09 | 0.49 | 0.58 | 0.30 | <0.03 | − | [16] |
| Gly-Gly-Met | 6 | 0.19 | 0.25 | 0.44 | 0 | 0.24 | 0.02 | [16] |
| Gly-Gly-Met | 11 | 0.13 | 0.30 | 0.43 | 0 | 0.29 | − | [16] |
| Gly-Met-Gly | 6 | 0.24 | 0.05 | 0.29 | 0 | 0.08 | 0.02 | [16] |
| Gly-Met-Gly | 11 | 0.10 | 0.21 | 0.31 | 0 | 0.07 | − | [16] |
| Met-Lys | 6 | 0.55 | ~0.03 | 0.58 | 0.33 | 0 | 0 | [20] |
| Lys-Met | 6 | 0.63 | 0 | 0.63 | 0 | 0.19 | 0 | [20] |
| Met-Enkephalin | 7 | − | − | 0.82 | 0 | 0 | 0 | [61] |
| Met-Containing Peptides (a) | pH | ΦCBH∙ (b) | ΦCB∙− (b) | ΦCBH∙ + ΦCB∙− (b) | Φ(S∴N)+ (b) | Φ(S∴S)+ (b) | ΦCO2- (c) | Lit |
|---|---|---|---|---|---|---|---|---|
| L-Met-L-Met | 6 | 0.23 | 0.15 | 0.38 | 0.19 | 0.07 (d) | <0.01 | [63] |
| L-Met-L-Met | 10 | 0.13 | 0.41 | 0.54 | 0.43 | 0.01 (d) | <0.01 | [63] |
| D-Met-D-Met | 6 | 0.24 | 0.18 | 0.42 | 0.27 | 0.06 (d) | - | [63] |
| D-Met-D-Met | 10 | 0.09 | 0.40 | 0.49 | 0.41 | ~0.02 (d) | - | [63] |
| L-Met-D-Met | 6 | 0.17 | 0.23 | 0.40 | 0.18 | 0.15 (d) | <0.01 | [63] |
| L-Met-D-Met | 10 | 0.08 | 0.37 | 0.45 | 0.34 | 0.06 (d) | <0.01 | [63] |
| D-Met-L-Met | 6 | 0.15 | 0.25 | 0.40 | 0.15 | 0.16 (d) | <0.01 | [63] |
| D-Met-L-Met | 10 | 0.10 | 0.38 | 0.48 | 0.36 | 0.06 (d) | <0.01 | [63] |
| c-(L-Met-L-Met) | 7 | 0.17 | 0.19 | 0.36 | 0 | 0.14 (d) | - | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marciniak, B.; Bobrowski, K. Photo- and Radiation-Induced One-Electron Oxidation of Methionine in Various Structural Environments Studied by Time-Resolved Techniques. Molecules 2022, 27, 1028. https://doi.org/10.3390/molecules27031028
Marciniak B, Bobrowski K. Photo- and Radiation-Induced One-Electron Oxidation of Methionine in Various Structural Environments Studied by Time-Resolved Techniques. Molecules. 2022; 27(3):1028. https://doi.org/10.3390/molecules27031028
Chicago/Turabian StyleMarciniak, Bronislaw, and Krzysztof Bobrowski. 2022. "Photo- and Radiation-Induced One-Electron Oxidation of Methionine in Various Structural Environments Studied by Time-Resolved Techniques" Molecules 27, no. 3: 1028. https://doi.org/10.3390/molecules27031028
APA StyleMarciniak, B., & Bobrowski, K. (2022). Photo- and Radiation-Induced One-Electron Oxidation of Methionine in Various Structural Environments Studied by Time-Resolved Techniques. Molecules, 27(3), 1028. https://doi.org/10.3390/molecules27031028