
The Mechanism of a Retro-Diels–Alder Fragmentation of Luteolin: Theoretical Studies Supported by Electrospray Ionization Tandem Mass Spectrometry Results



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Equipment
2.3. Extraction of Dyes from Weld
2.4. LC-MS Analysis
2.5. Theoretical Calculations
3. Results
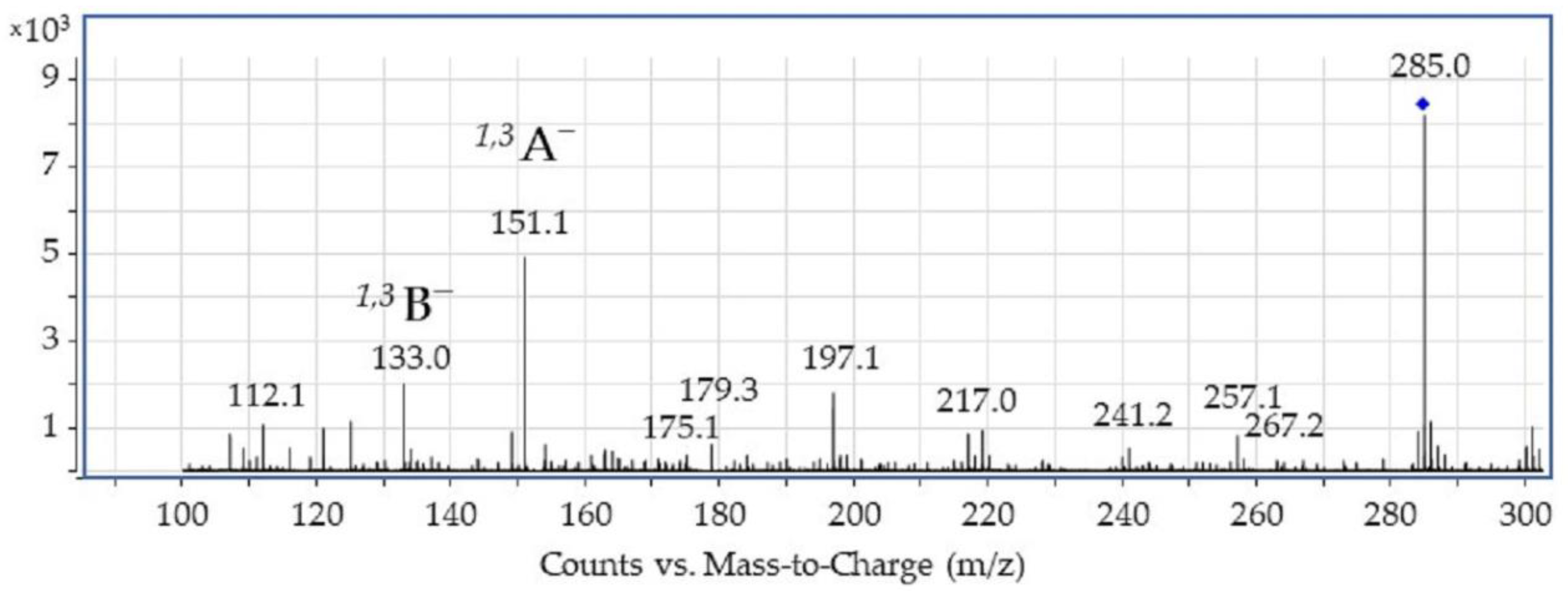
3.1. HPLC-ESI(-)-MS Study
3.2. Isomeric Structures Resulting from Deprotonation of Neutral Luteolin
3.3. Fragmentation Paths for LA1− Isomer (Leading to 1,3A− Product at m/z 151)
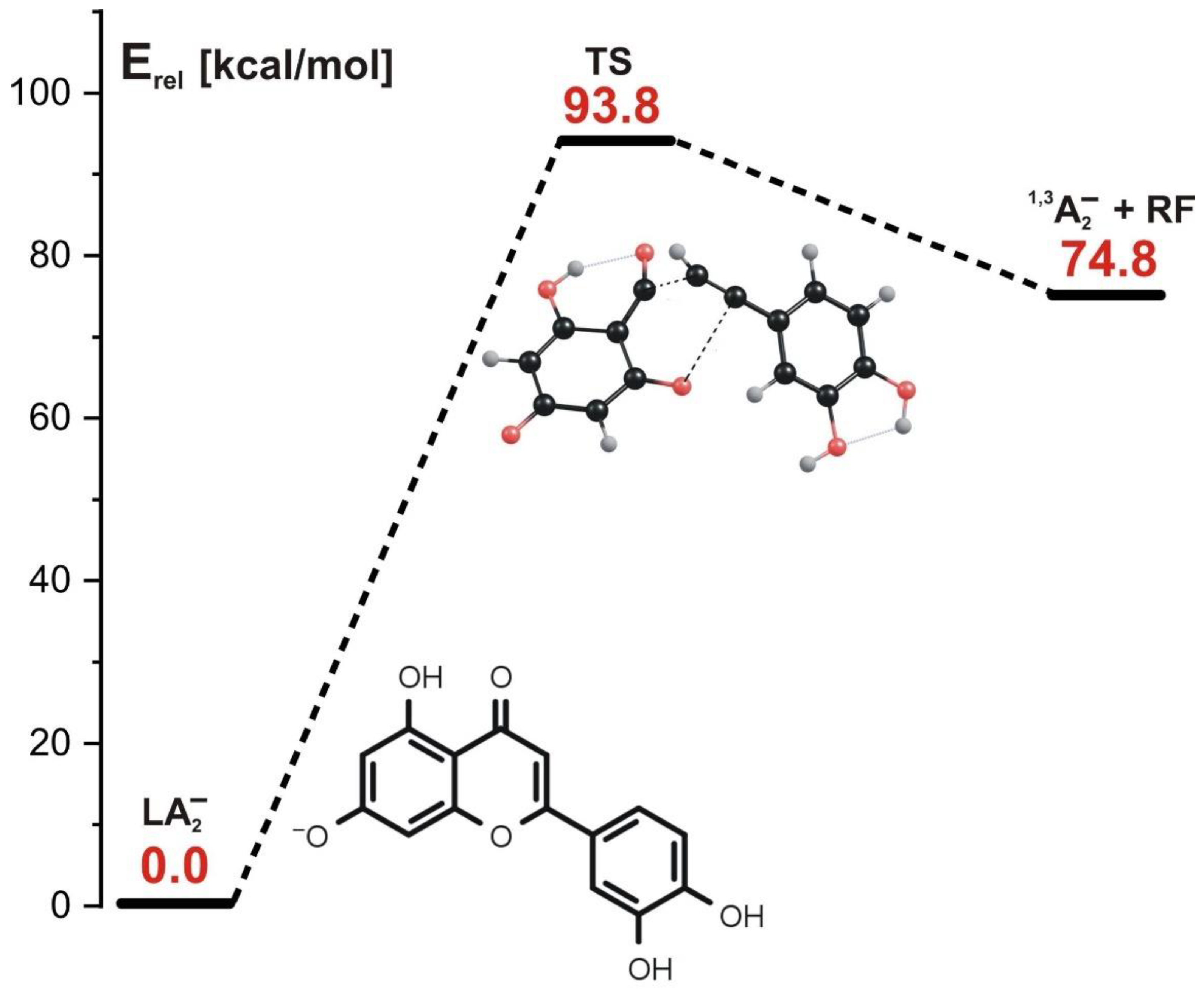
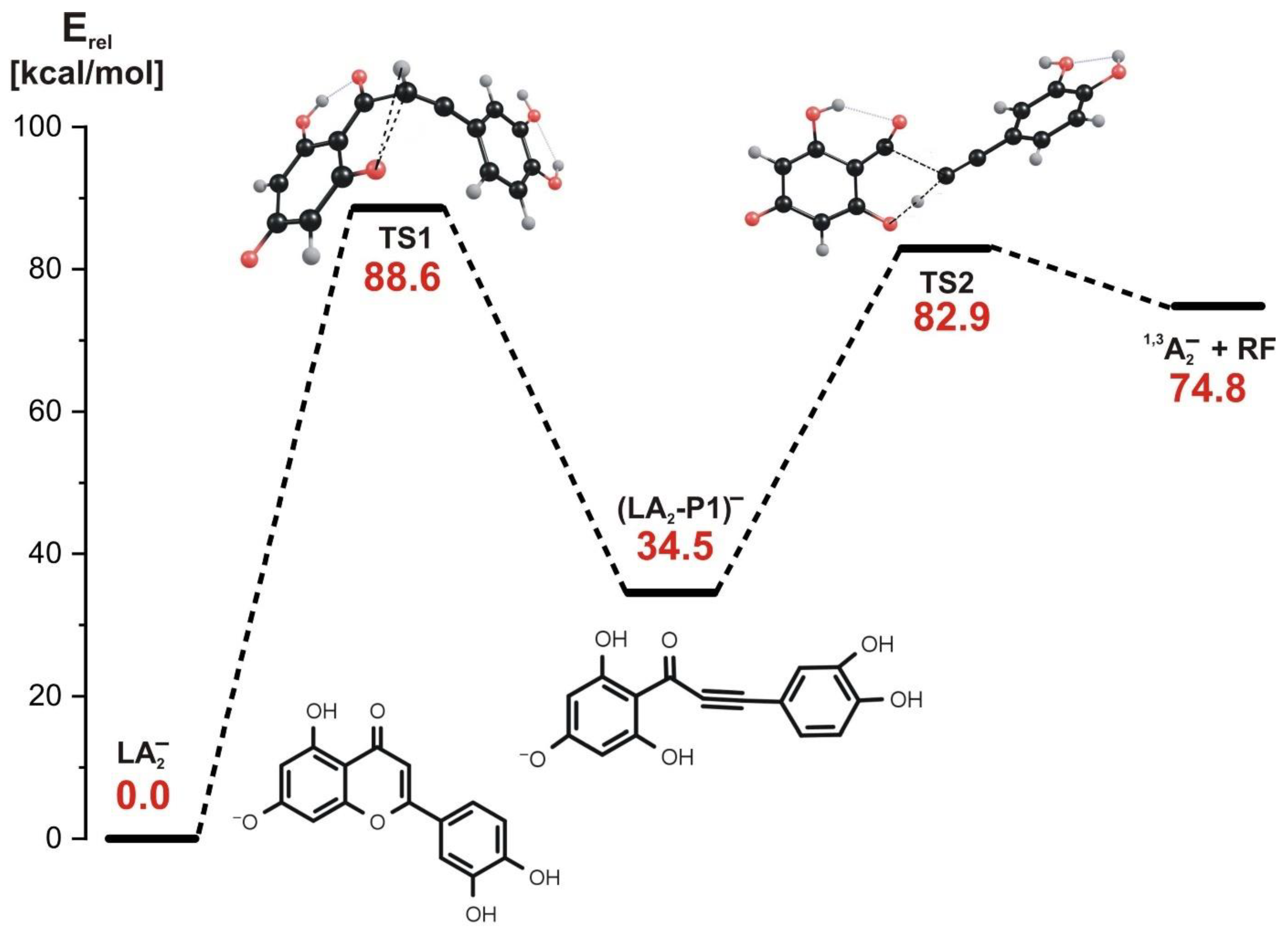
3.4. Fragmentation Paths for LA2− Isomer (Leading to 1,3A− Product at m/z 151)
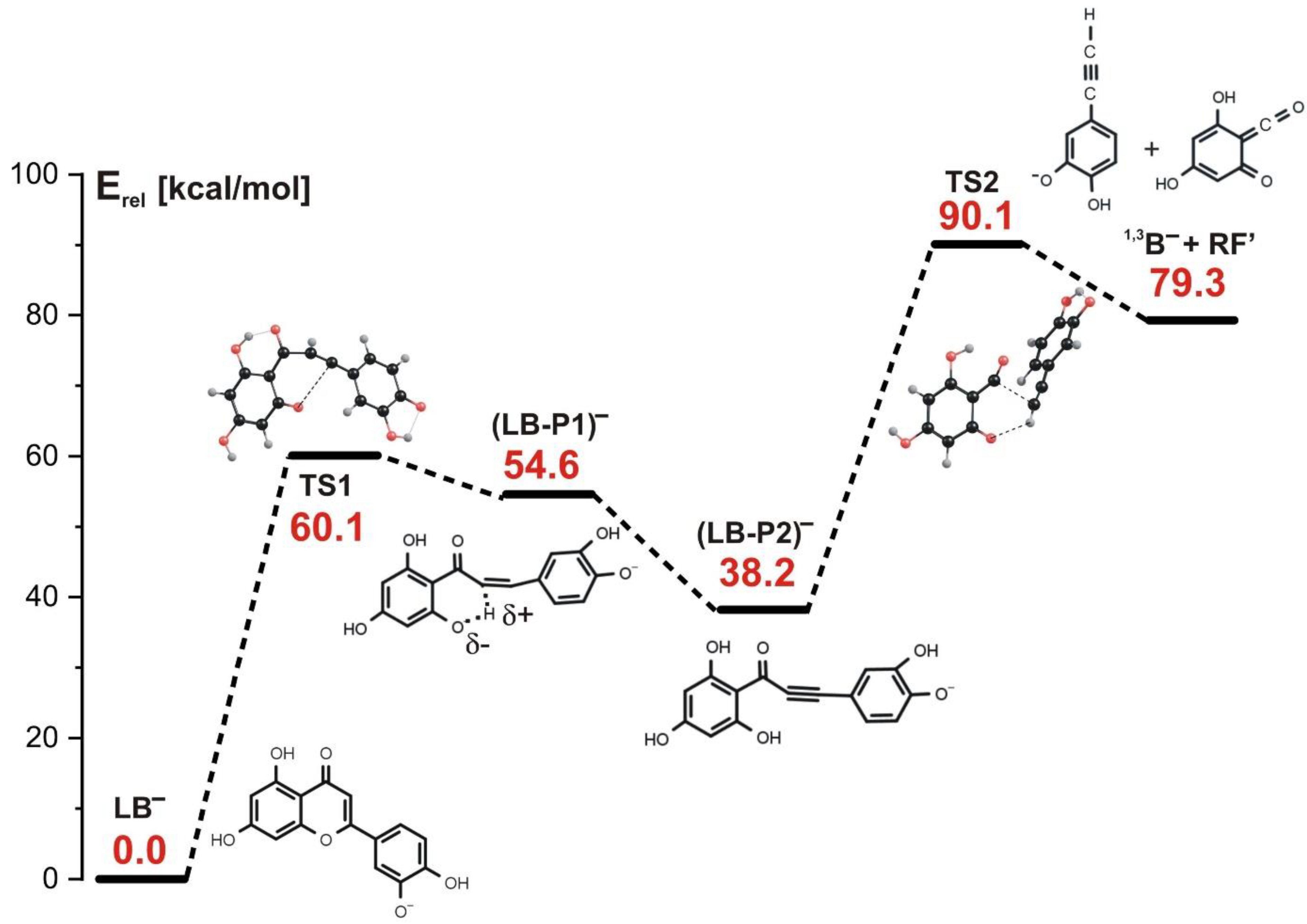
3.5. Fragmentation Paths for LB− Isomer (Leading to 1,3B− Product at m/z 133)
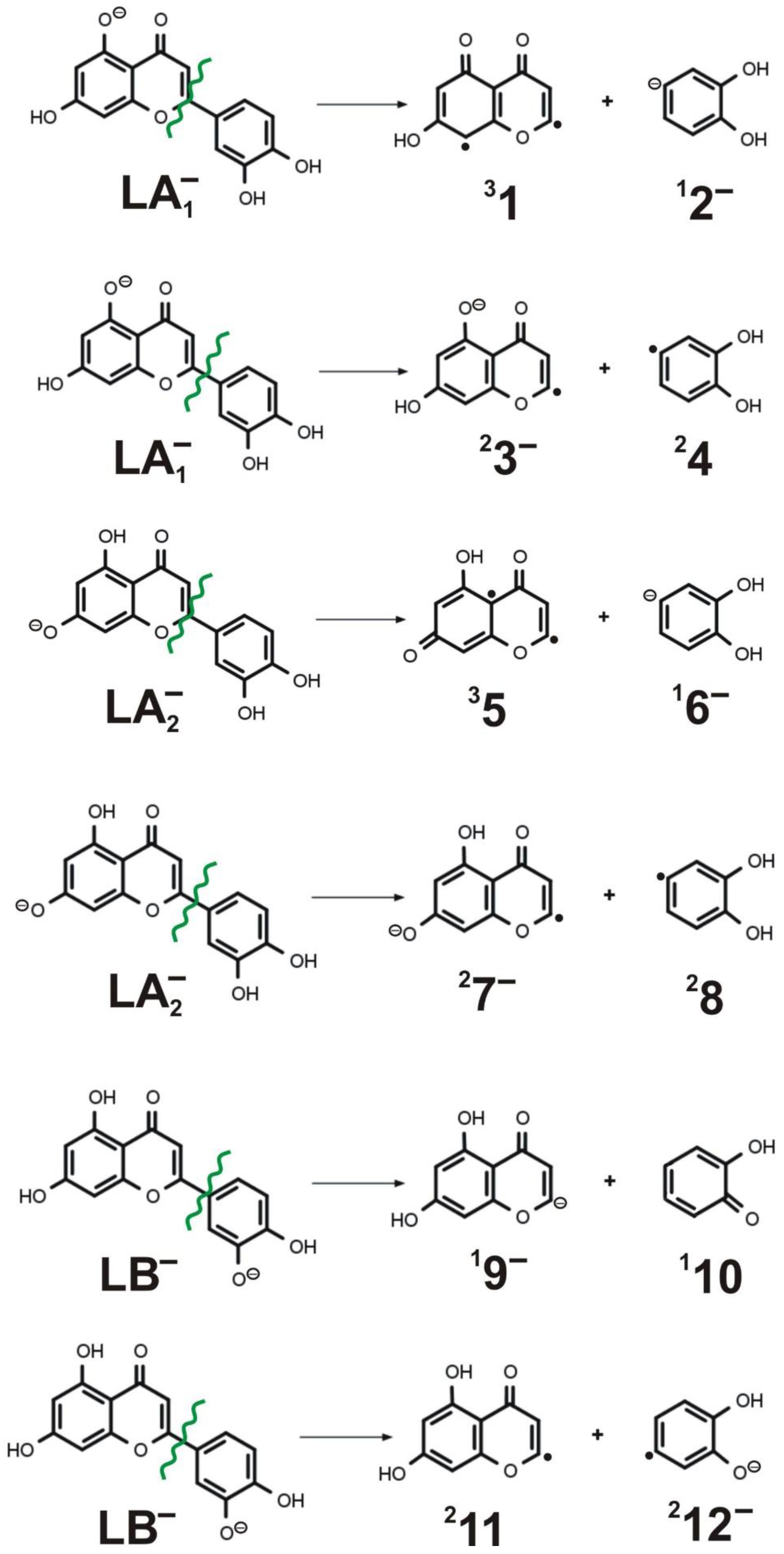
3.6. Alternative Fragmentation Paths for LA1−, LA2−, and LB− Isomers
4. Summary and Conclusions
- (i)
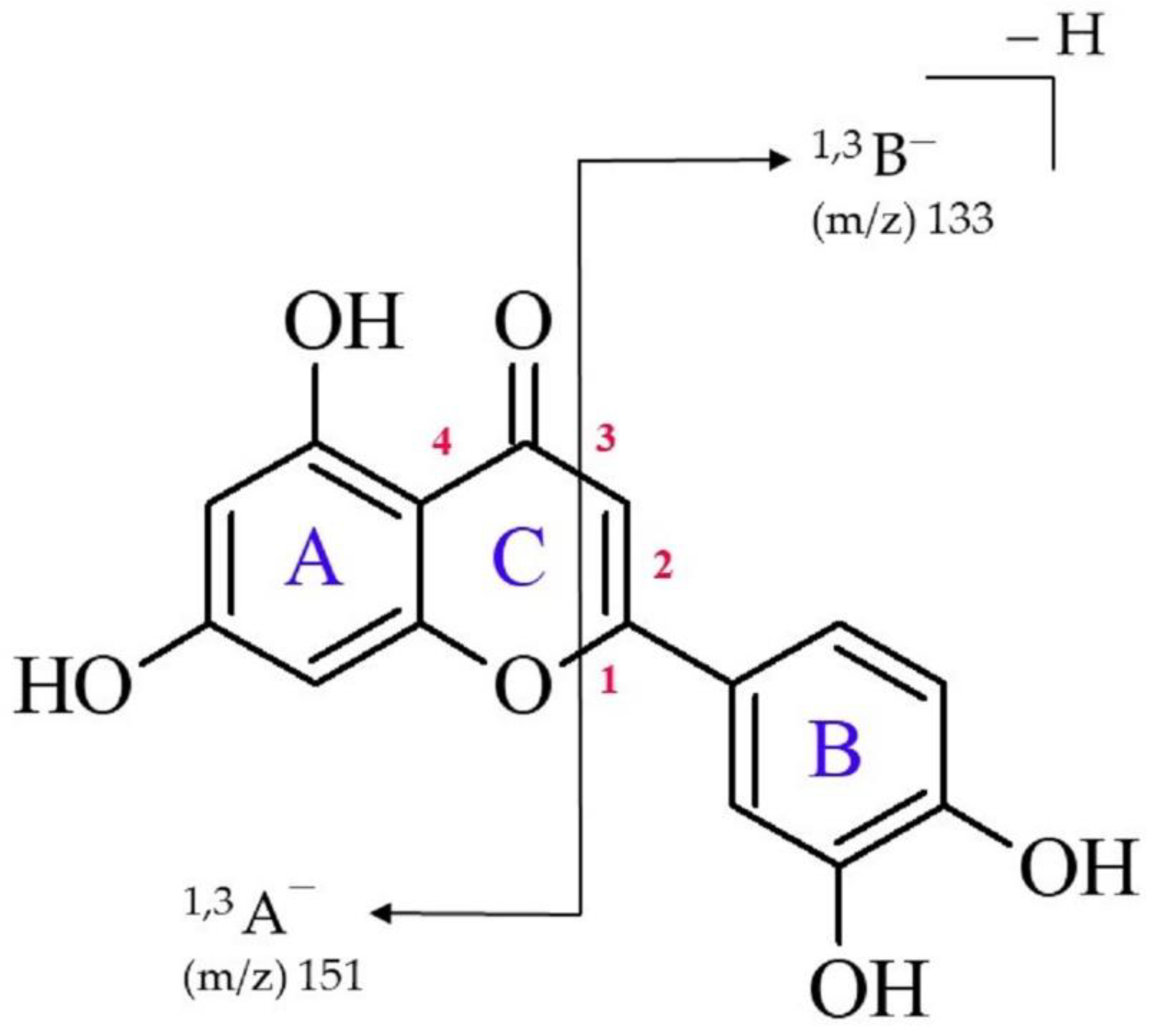
- Two of the most intensive spectral features observed in the ESI-MS spectrum of luteolin (acquired in negative ion mode) correspond to the 1,3A− ion at m/z 151 and the 1,3B− ion at m/z 133 (with the former being the main fragment ion observed).
- (ii)
- The presence of two fragment ions (i.e., 1,3A− and 1,3B−) in the ESI-MS spectrum and the absence of the signal corresponding to the [M-B]− fragment ion (which would appear if the bond connecting luteolin’s B and C rings were ruptured) indicates that the luteolin molecule undergoes the 1,3-retrocyclization process, whereas a direct cleavage of a single C-C bond representing the linkage between the aromatic rings (observed for some flavonols) does not occur in this case.
- (iii)
- Theoretical studies indicate that the 1,3-retrocyclization of deprotonated luteolin may proceed according to either the concerted or stepwise mechanism (when one of the hydroxyl groups connected to the A ring is initially deprotonated) or according to the stepwise mechanism only (when the initial deprotonation is related to one of the OH groups connected to the B ring).
- (iv)
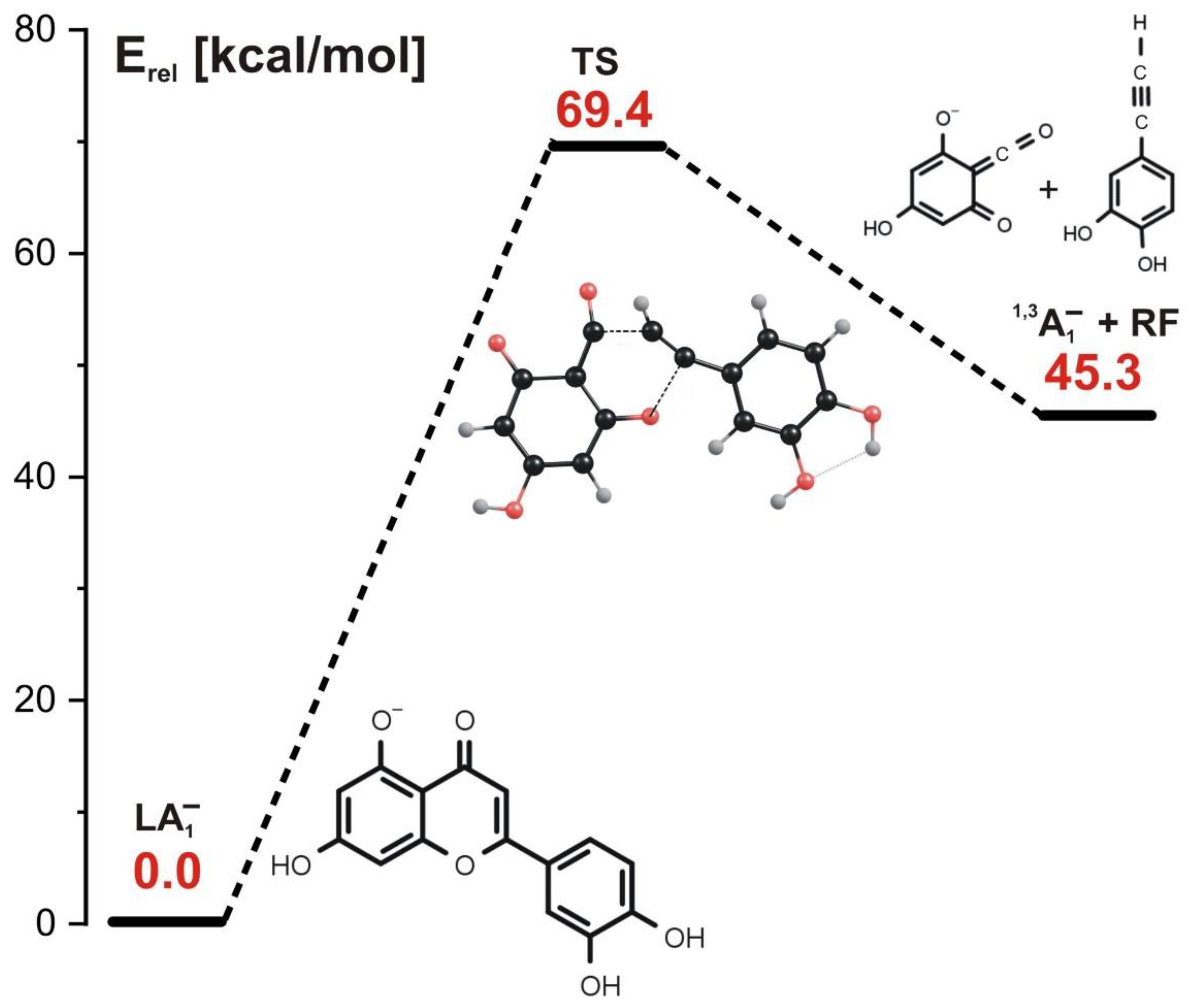
- (In the concerted mechanism involving LA1− or LA2− ionized luteolin isomer as a starting structure, the simultaneous cleavage of two bonds (C-O and C-C) in the C ring occurs. Such a process requires a single kinetic barrier whose height is equal to 69 kcal/mol for LA1− and 94 kcal/mol for LA2− to be overcome and results in the formation of 1,3A1− or 1,3A2− anions which are experimentally indistinguishable (as they both appear as the fragment ion at m/z 151 in ESI-MS spectrum).
- (v)
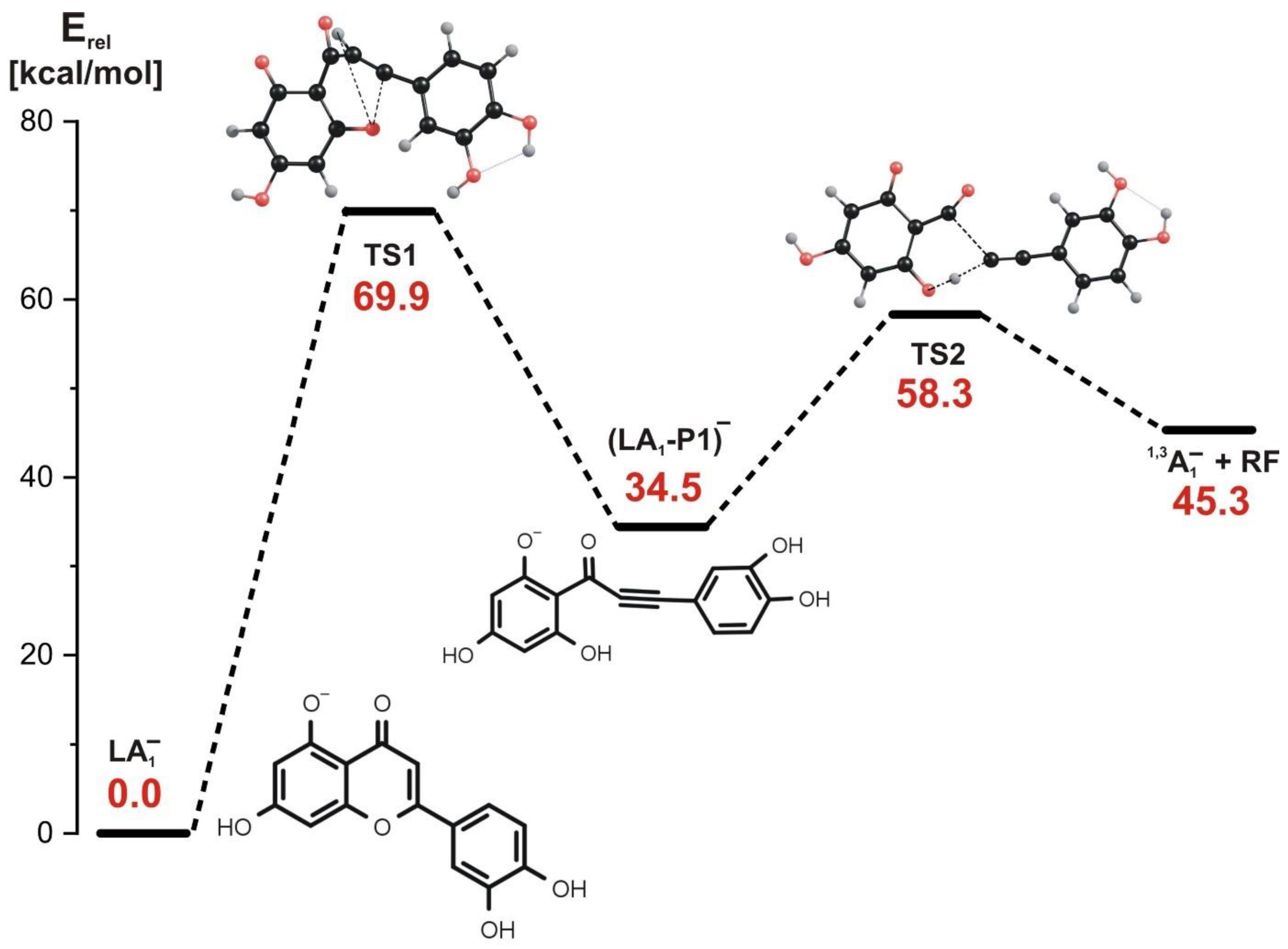
- In the stepwise mechanism involving LA1− or LA2− as a starting structure, two kinetic barriers have to be surmounted: the first barrier (70–89 kcal/mol, depending on the isomer) is related to the reaction step involving the rupture of the C-O bond in the C ring and a simultaneous H transfer from C to O, and the second barrier (24–48 kcal/mol, depending on the isomer) is related to the next step involving the rupture of the C-C bond and a simultaneous H transfer from the O atom back to the C atom. Regardless of the starting structure considered (LA1− or LA2−), the stepwise fragmentation path leads to the formation of the final product appearing as 1,3A− ion at m/z 151 in ESI-MS spectrum.
- (vi)
- The fragmentation involving the LB− isomer as a starting reagent proceeds according to the stepwise mechanism (which is the only operative mechanism for this isomer) and involves three steps: the first step related to the C-O bond cleavage (with the barrier of 60 kcal/mol), the barrierless proton transfer from C to O as the second step, and the final step (requiring the activation energy of 52 kcal/mol) related to both the C-C bond rupture and the proton transfer from O back to C. This reaction path leads to the formation of the 1,3B− anion observed in the ESI-MS experiment at m/z 133.
- (vii)
- The absence of the spectral feature corresponding to the [M-B]− fragment in the ESI-MS spectrum is likely caused by the fact that such a fragment ion formation would require much higher energy barriers (131–166 kcal/mol) related to the rupture of the C-C bond connecting luteolin’s B and C rings to be surmounted.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harborne, J.B.; Mabry, T.J.; Mabry, H. The Flavonoids; Science + Business Media Dordrecht, Chapman and Hall Ltd.: Dordrecht, The Netherlands, 1975; ISBN 978-1-4899-2909-9. [Google Scholar]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant Flavonoids: Chemical Characteristics and Biological Activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef] [PubMed]
- Hofenk de Graaff, J.H.; Reolofs, W.G.T.; Van Bommel, M.R. The Colourful Past: Origins, Chemistry and Identification of Natural Dyestuffs; Archetype Publications: London, UK, 2004. [Google Scholar]
- Cardon, D. Natural Dyes; Archetype Publications: London, UK, 2007. [Google Scholar]
- Grotewold, E. The Science of Flavonoids; Springer Science + Business Media, Inc.: Columbus, OH, USA, 2006. [Google Scholar]
- Rupasinghe, H.V. Flavonoids and Their Disease Prevention and Treatment Potential. Molecules 2020, 25, 4746. [Google Scholar] [CrossRef]
- Fernández, J.; Silván, B.; Entrialgo-Cadierno, R.; Villar, C.J.; Capasso, R.; Uranga, J.A.; Lombó, F.; Abalo, R. Antiproliferative and palliative activity of flavonoids in colorectal cancer. Biomed. Pharmacother. 2021, 143, 112241. [Google Scholar] [CrossRef]
- Rodriguez-Arce, E.; Saldias, M. Antioxidant properties of flavonoid metal complexes and their potential inclusion in the devel-opment of novel strategies for the treatment against neurodegenerative diseases. Biomed. Pharmacother. 2021, 143, 112236. [Google Scholar] [CrossRef] [PubMed]
- Sapian, S.; Taib, I.S.; Latip, J.; Katas, H.; Chin, K.-Y.; Mohd Nor, N.A.; Jubaidi, F.F.; Budin, S.B. Therapeutic Approach of Fla-vonoid in Ameliorating Diabetic Cardiomyopathy by Targeting Mitochondrial-Induced Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 11616. [Google Scholar] [CrossRef] [PubMed]
- Kaul, R.; Paul, P.; Kumar, S.; Büsselberg, D.; Dwivedi, V.D.; Chaari, A. Promising Antiviral Activities of Natural Flavonoids against SARS-CoV-2 Targets: Systematic Review. Int. J. Mol. Sci. 2021, 22, 11069. [Google Scholar] [CrossRef] [PubMed]
- Somerset, S.M.; Johannot, L. Dietary Flavonoid Sources in Australian Adults. Nutr. Cancer 2008, 60, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Rauf, A.; Abu-Izneid, T.; Nadeem, M.; Shariati, M.A.; Khan, I.A.; Imran, A.; Orhan, I.E.; Rizwan, M.; Atif, M.; et al. Luteolin, a flavonoid, as an anticancer agent: A review. Biomed. Pharmacother. 2019, 112, 108612. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lim, T.; Han, M.S.; Lee, S.H.; Baek, S.H.; Nan, H.Y.; Lee, C. Anticancer effect of luteolin is mediated by downregula-tion of TAM receptor tyrosine kinases, but not interleukin-8, in non-small cell lung cancer cells. Oncol. Rep. 2017, 37, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Wei, P.; Song, J.; Jia, X.; Zhang, Z. Enhanced anticancer activity in vitro and in vivo of luteolin incorporated into long-circulating micelles based on DSPE-PEG2000 and TPGS. J. Pharm. Pharmacol. 2016, 68, 1290–1298. [Google Scholar] [CrossRef]
- McNab, H.; Ferreira, E.S.B.; Hulmea, A.N.; Quye, A. Negative ion ESI–MS analysis of natural yellow dye flavonoids—An iso-topic labelling study. Int. J. Mass Spectrom. 2008, 284, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Sammani, M.S.; Clavijo, S.; Cerdà, V. Recent, advanced sample pretreatments and analytical methods for flavonoids determi-nation in different samples. Trends Analyt. Chem. 2021, 138, 116220. [Google Scholar] [CrossRef]
- Ma, Y.L.; Li, Q.M.; Van den Heuvel, H.; Claeys, M. Characterization of Flavone and Flavonol Aglycones by Collision-induced Dissociation Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 1997, 11, 1357–1364. [Google Scholar] [CrossRef]
- Wu, W.; Yan, C.; Li, L.; Liu, Z.; Liu, S. Studies on the flavones using liquid chromatography-electrospray ionization tandem mass spectrometry. J. Chromatogr. A 2004, 1047, 213–220. [Google Scholar] [CrossRef]
- Wojtanowski, K.K.; Mroczek, T. Detection, identification and structural elucidation of flavonoids using liquid chromatography coupled to mass spectrometry. Curr. Org. Chem. 2020, 24, 104–112. [Google Scholar] [CrossRef]
- Yang, M.; Li, J.; Zhao, C.; Xiao, H.; Fang, X.; Zheng, J. LC-Q-TOF-MS/MS detection of food flavonoids: Principle, methodology, and applications. Crit. Rev. Food Sci. Nutr. 2021, 21, 1–21. [Google Scholar] [CrossRef]
- Cuyckens, F.; Claeys, M. Mass spectrometry in the structural analysis of flavonoids. Biol. Mass Spectrom. 2004, 39, 1–15. [Google Scholar] [CrossRef]
- Fabre, N.; Rustan, I.; De Hoffmann, E.; Quetin-Leclercq, J. Determination of Flavone, Flavonol, and Flavanone Aglycones by Negative Ion Liquid Chromatography Electrospray Ion Trap Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2001, 12, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.L.; Vedernikova, I.; Van den Heuvel, H.; Clayes, M. Stable isotope incorporation triples the upper mass limit for determination of elemental composition by accurate mass measurement. J. Am. Soc. Mass Spectrom. 2000, 11, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Otłowska, O.; Ślebioda, M.; Wachowiak, M.; Śliwka-Kaszyńska, M. Identification and characterization of the Indian Yellow dyestuff and its degradation products in historical oil paint tube by liquid chromatography mass spectrometry. RSC Adv. 2015, 5, 48786–48792. [Google Scholar] [CrossRef]
- Otłowska, O.; Ślebioda, M.; Wachowiak, M.; Śliwka-Kaszyńska, M. A multi-analytical approach to the characterization of natural organic dyestuffs and inorganic substrates present in the 19th-century artistic oil paints manufactured by a French art materials supplier Richard Ainès. Anal. Methods 2017, 9, 94–102. [Google Scholar] [CrossRef]
- Otłowska, O.; Ślebioda, M.; Kot-Wasik, A.; Karczewski, J.; Śliwka-Kaszyńska, M. Chromatographic and spectroscopic identifi-cation and recognition of natural dyes, uncommon dyestuff components and mordants in 16th century carpet. Molecules 2018, 23, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Śliwka-Kaszyńska, M.; Ślebioda, M.; Brillowska-Dąbrowska, A.; Mroczyńska, M.; Karczewski, J.; Marzec, A.; Rybiński, P.; Drążkowska, A. Multi-Technique Investigation of Grave Robes from 17th and 18th Century Crypts Using Combined Spectroscopic, Spectrometric Techniques, and New-Generation Sequencing. Materials 2021, 14, 3535. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Coskun, D.; Jerome, S.V.; Friesner, R.A. Evaluation of the Performance of the B3LYP, PBE0, and M06 DFT Functionals, and DBLOC-Corrected Versions, in the Calculation of Redox Potentials and Spin Splittings for Transition Metal Containing Systems. J. Chem. Theory Comput. 2016, 12, 1121–1128. [Google Scholar] [CrossRef]
- López-López, J.A.; Ayala, R. Assessment of the performance of commonly used DFT functionals vs. MP2 in the study of IL-Water, IL-Ethanol and IL-(H2O)3 clusters. J. Mol. Liq. 2016, 220, 970–982. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Goel, N.; Yadav, T.C.; Pruthi, V. Quantum chemical, ADMET and molecular docking studies of ferulic acid amide derivatives with a novel anticancer drug target. Med. Chem. Res. 2017, 26, 1822–1834. [Google Scholar] [CrossRef]
- Lai, W.-J.; Lu, J.-H.; Jiang, L.-H.; Lei, F.-H.; Shen, L.-Q.; Wu, A.-Q.; Yang, J.; Qi, W.-L. Structural stabilities and transformation mechanism of rhynchophylline and isorhynchophylline in aqueous and methanol solution based on high-performance liquid chromatography and density functional theory. J. Mol. Struct. 2021, 1236, 130300. [Google Scholar] [CrossRef]
- Bauschlicher, C.W., Jr. A comparison of the accuracy of different functionals. Chem. Phys. Lett. 1995, 246, 40–44. [Google Scholar] [CrossRef]
- Besler, B.H.; Merz, K.M., Jr.; Kollman, P.A. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MS (m/z) | Fragment Ions |
|---|---|
| 285 | [M-H]− |
| 267 | [M-H-H2O]− |
| 257 | [M-H-CO]− |
| 241 | [M-H-CO2]− |
| 217 | [M-H-C3O2]− |
| 197 | [M-H-2CO2]− |
| 175 | [M-H-C3O2-C2H2O]− |
| 151 | 1,3A− |
| 133 | 1,3B− |
| Process No. | Fragmentation Reaction | Cleavage Type | Reaction Energy |
|---|---|---|---|
| (1) | LA1− → 31 + 12− | heterolytic | 156.5 |
| (2) | LA1− → 23− + 24 | homolytic | 131.3 |
| (3) | LA2− → 35 + 16− | heterolytic | 165.6 |
| (4) | LA2− → 27− + 28 | homolytic | 134.2 |
| (5) | LB− → 19− + 110 | heterolytic | 136.7 |
| (6) | LB− → 211 + 212− | homolytic | 134.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Śliwka-Kaszyńska, M.; Anusiewicz, I.; Skurski, P. The Mechanism of a Retro-Diels–Alder Fragmentation of Luteolin: Theoretical Studies Supported by Electrospray Ionization Tandem Mass Spectrometry Results. Molecules 2022, 27, 1032. https://doi.org/10.3390/molecules27031032
Śliwka-Kaszyńska M, Anusiewicz I, Skurski P. The Mechanism of a Retro-Diels–Alder Fragmentation of Luteolin: Theoretical Studies Supported by Electrospray Ionization Tandem Mass Spectrometry Results. Molecules. 2022; 27(3):1032. https://doi.org/10.3390/molecules27031032
Chicago/Turabian StyleŚliwka-Kaszyńska, Magdalena, Iwona Anusiewicz, and Piotr Skurski. 2022. "The Mechanism of a Retro-Diels–Alder Fragmentation of Luteolin: Theoretical Studies Supported by Electrospray Ionization Tandem Mass Spectrometry Results" Molecules 27, no. 3: 1032. https://doi.org/10.3390/molecules27031032
APA StyleŚliwka-Kaszyńska, M., Anusiewicz, I., & Skurski, P. (2022). The Mechanism of a Retro-Diels–Alder Fragmentation of Luteolin: Theoretical Studies Supported by Electrospray Ionization Tandem Mass Spectrometry Results. Molecules, 27(3), 1032. https://doi.org/10.3390/molecules27031032