
HDAC Inhibitors: Innovative Strategies for Their Design and Applications




Abstract
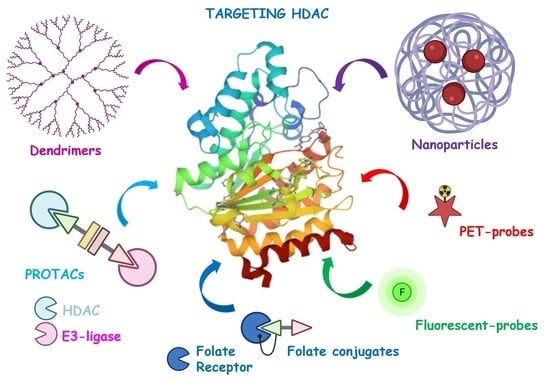
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. PROteolysis-TArgeting Chimeras (PROTACs)
3. Tumor-Targeted HDAC Inhibitors
3.1. Folate-Based Tumor-Targeted HDAC Inhibitors
3.2. Dendrimers-Based Tumor-Targeted HDAC Inhibitors
3.3. Nanoparticles-Based Tumor-Targeted HDAC Inhibitors
3.3.1. Lipid-Based NPs
3.3.2. Polymer-Based NPs
3.3.3. Sugar-Based NPs
4. Imaging Probes
4.1. PET Ligands
4.2. Fluorescent Probes
5. Conclusions
6. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tough, D.F.; Tak, P.P.; Tarkhovsky, A.; Prinjha, R.K. Epigenetic drug discovery: Breaking through the immune barrier. Nat. Rev. Drug Discov. 2016, 15, 835–853. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, H. HDAC and HDAC inhibitor: From cancer to cardiovascular diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bolden, J.; Peart, M.; Johnstone, R. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold. Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Marks, P.A.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesham, H.M.; Lasheen, D.S.; Abouzid, K.A.M. Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med. Res. Rev. 2018, 38, 2058–2109. [Google Scholar] [CrossRef] [PubMed]
- McClure, J.J.; Li, X.; Chou, J. Chapter Six–Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2016, 16, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Chi, K.R. Drug developers delve into the cell’s trash-disposal machinery. Nat. Rev. Drug Discov. 2016, 15, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, S.J.L.; Timmers, H.T.M. The family of ubiquitin-conjugating enzymes (E2s): Deciding between life and death of proteins. FASEB J. 2009, 24, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell. Chem. Biol. 2018, 25, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, K.G.; Crews, C.M. Proteolysis-Targeting Chimeras: Harnessing the ubiquitin-proteasome system to induce degradation of specific target proteins. Annu. Rev. Cancer Biol. 2018, 2, 41–58. [Google Scholar] [CrossRef]
- Chamberlain, P.P.; D’Agostino, L.A.; Ellis, J.M.; Hansen, J.D.; Matyskiela, M.E.; McDonald, J.J.; Riggs, J.R.; Hamann, L.G. Evolution of Cereblon-mediated protein degradation as a therapeutic modality. Med. Chem. Lett. 2019, 10, 1592–1602. [Google Scholar] [CrossRef]
- Scheepstra, M.; Hekking, K.F.W.; van Hijfte, L.; Folmer, R.H.A. Bivalent ligands for protein degradation in drug discovery. Comput. Struct. Biotechnol. J. 2019, 17, 160–176. [Google Scholar] [CrossRef]
- López-Cantudo, L.; Ramos, A.; Coderch, C.; de Pascual-Teresa, B. Proteasomal Degradation of Zn-Dependent Hdacs: The E3-Ligases Implicated and the Designed Protacs That Enable Degradation. Molecules 2021, 26, 5606. [Google Scholar] [CrossRef]
- Vogelmann, A.; Robaa, D.; Sippl, W.; Jung, M. Proteolysis targeting chimeras (PROTACs) for epigenetics research. Curr. Opin. Chem. Biol. 2020, 57, 8–16. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, X. Recent advances in small molecular modulators targeting histone deacetylase 6. Future Drug. Discov. 2020, 2, FDD53. [Google Scholar] [CrossRef]
- Zhou, X.; Dong, R.; Zhang, J.Y.; Zheng, X.; Sun, L.P. PROTAC: A promising technology for cancer treatment. Eur. J. Med. Chem. 2020, 203, 112539. [Google Scholar] [CrossRef] [PubMed]
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer therapy with HDAC inhibitors: Mechanism-based combination strategies and future perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically induced degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting-Chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Biorg. Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Qin, C.; Bai, L.; Wang, S. Small-molecule PROTAC degraders of the Bromodomain and Extra Terminal (BET) proteins—A review. Drug Dicov. Today Technol. 2018, 25, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective small molecule induced degradation of the BET bromodomain protein BRD 4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. 2019, 55, 14848–14851. [Google Scholar] [CrossRef]
- Wu, H.; Yang, K.; Zhang, Z.; Leisten, E.D.; Li, Z.; Xie, H.; Liu, J.; Smith, K.A.; Novakova, Z.; Barinka, C.; et al. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J. Med. Chem. 2019, 62, 7042–7057. [Google Scholar] [CrossRef]
- Yang, K.; Zhao, Y.; Nie, X.; Wu, H.; Wang, B.; Almodovar-Rivera, C.M.; Xie, H.; Tang, W. A cell-based target engagement assay for identification of cereblon E3 ubiquitin ligase ligands and their application in HDAC6 degraders. Cell Chem. Biol. 2020, 27, 866–876. [Google Scholar] [CrossRef]
- Yang, K.; Wu, H.; Zhang, Z.; Leisten, E.D.; Nie, X.; Liu, B.; Wen, Z.; Zhang, J.; Cunningham, M.D.; Tang, W. Development of selective histone deacetylase 6 (HDAC6) degraders recruiting von Hippel–Lindau (VHL) E3 ubiquitin ligase. ACS Med. Chem. Lett. 2020, 11, 575–581. [Google Scholar] [CrossRef]
- Smalley, J.P.; Adams, G.E.; Millard, C.J.; Song, Y.; Norris, J.K.S.; Schwabe, J.W.R.; Cowley, S.M.; Hodkinson, J.T. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem. Commun. 2020, 56, 4476–4479. [Google Scholar] [CrossRef]
- Cao, F.; de Weerd, S.; Chen, D.; Zwinderman, M.R.H.; van der Wouden, P.E.; Dekker, F.J. Induced protein degradation of histone deacetylases 3 (HDAC3) by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem. 2020, 208, 112800. [Google Scholar] [CrossRef]
- Roatsch, M.; Vogelmann, A.; Herp, D.; Jung, M.; Olsen, C.A. Proteolysis-Targeting Chimeras (PROTACs) based on macrocyclic tetrapeptides selectively degrade class I histone deacetylases 1–3. ChemRxiv. Camb. Open Engag. 2020. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef] [PubMed]
- Sinatra, L.; Bandolik, J.J.; Roatsch, M.; Sönnichsen, M.; Schroeder, C.T.; Hamacher, A.; Schöler, A.; Borkhardt, A.; Meiler, J.; Bhatia, S.; et al. Hydroxamic acids immobilized on resins (HAIRs): Synthesis of dual-targeting HDAC inhibitors and HDAC degraders (PROTACs). Angew. Chem. Int. Ed. 2020, 59, 22494–22499. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhao, W.; Zhao, C.; Liu, Q.; Li, S.; Zhang, G.; Chou, C.J.; Zhang, Y. Development of a Bestatin-SAHA hybrid with dual inhibitory activity against APN and HDAC. Molecules 2020, 25, 4991. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.A.H.; Chen, L. Multi-targeted histone deacetylase inhibitors in cancer therapy. Curr. Med. Chem. 2012, 19, 475–487. [Google Scholar] [CrossRef]
- Vlahov, I.R.; Leamon, C.P. Engineering folate-drug conjugates to target cancer: From chemistry to clinic. Bioconjugate Chem. 2012, 23, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Clod Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, C.; Guan, X.; Ma, H.; Cong, H.; Zhang, W.; Miao, Z. Small molecule-drug conjugates: A novel strategy for cancer-targeted treatment. Eur. J. Med. Chem. 2019, 163, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Srinivasarao, M.; Low, P.S. Ligand-targeted drug delivery. Chem. Rev. 2017, 117, 12133–12164. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ke, J.; Zhou, X.E.; Yi, W.; Brunzelle, J.S.; Li, J.; Young, E.L.; Xu, H.E.; Melcher, K. Structural basis for molecular recognition of folic acid by folate receptors. Nature 2013, 500, 486–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Min, S.H.; Wang, Y.; Campanella, E.; Low, P.S.; Goldman, I.D. A role for the proton-coupled folate transporter (PCFT-SLC46A1) in folate receptor-mediated endocytosis. J. Biol. Chem. 2009, 284, 4267–4274. [Google Scholar] [CrossRef] [Green Version]
- Elnakat, H.; Ratnam, M. Folate receptor-targeted drugs for cancer and inflammatory diseases. Adv. Drug Deliv. Rev. 2004, 56, 1067–1084. [Google Scholar] [CrossRef]
- Saul, J.M.; Annapragada, A.; Natarajan, J.V.; Bellamkonda, R.V. Controlled targeting of liposomal doxorubicin via the folate receptor in vitro. J. Control. Release 2003, 92, 49–67. [Google Scholar] [CrossRef]
- Crane, L.M.A.; Arts, H.J.G.; van Oosten, M.; Low, P.S.; van der Zee, A.G.J.; van Dam, G.M.; Bart, J. The effect of chemotherapy on expression of folate receptor-alpha in ovarian cancer. Cell. Oncol. 2012, 35, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Srinivasarao, M.; Galliford, C.V.; Low, P.S. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. 2015, 14, 203–219. [Google Scholar] [CrossRef]
- Suzuki, T.; Hisakawa, S.; Itoh, Y.; Suzuki, N.; Takahashi, K.; Kawahata, M.; Yamaguchi, K.; Nakagawa, H.; Miyata, N. Design, synthesis, and biological activity of folate receptor-targeted prodrugs of thiolate histone deacetylase inhibitors. Bioorg. Med. Chem Lett. 2007, 17, 4208–4212. [Google Scholar] [CrossRef]
- Carrasco, M.P.; Enyedy, E.A.; Krupenko, N.I.; Krupenko, S.A.; Nuti, E.; Tuccinardi, T.; Santamaria, S.; Rossello, A.; Martinelli, A.; Santos, M.A. Novel folate-hydroxamate based antimetabolites: Synthesis and biological evaluation. Med. Chem. 2011, 7, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Sodji, Q.H.; Kornacki, J.R.; McDonald, J.F.; Mrksich, M.; Oyelere, A.K. Design and structure activity relationship of tumor-homing histone deacetylase inhibitors conjugated to folic and pteroic acid. Eur. J. Med. Chem. 2015, 96, 340–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Shah, D.; Selwa, K.; Tsuchida, R.E.; Rattan, R.; Mohan, J.; Stein, A.B.; Otis, J.B.; Goonewardena, S.N. Design and evaluation of tumor-specific dendrimer epigenetic therapeutics. ChemistryOpen 2015, 4, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Hattori, Y.; Yamada, T.; Uesato, S.; Maitani, Y.; Nagaoka, Y. Histone deacetylase inhibitor prodrugs in nanoparticle vector enhanced gene expression in human cancer cells. Eur. J. Med. Chem. 2009, 44, 4603–4610. [Google Scholar] [CrossRef]
- Foglietta, F.; Serpe, L.; Canaparo, R.; Vivenza, N.; Riccio, G.; Imbalzano, E.; Gasco, P.; Zara, G.P. Modulation of butyrate anticancer activity by solid lipid nanoparticle delivery: An in vitro investigation of human breast cancer and leukemia cell lines. J. Pham. Pham. Sci. 2014, 17, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Nusbaum, O.; Chen, X.; Zhu, Y. Valeric acid suppresses liver cancer development by acting as a novel HDAC inhibitor. Mol. Ther. Oncolytics 2020, 19, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Torres-Hernández, B.A.; Del Valle-Mojica, L.M.; Ortíz, J.G. Valerenic acid and Valeriana officinalis extracts delay onset of Pentylenetetrazole (PTZ)-induced seizures in adult Danio rerio (Zebrafish). BMC Complement. Altern. Med. 2015, 15, 228. [Google Scholar] [CrossRef] [Green Version]
- Sankar, R.; Ravikumar, V. Biocompatibility and biodistribution of suberoylanilide hydroxamic acid loaded poly (DL-lactide-co-glycolide) nanoparticles for targeted drug delivery in cancer. Biomed. Pharmacother. 2014, 68, 865–871. [Google Scholar] [CrossRef]
- Wang, E.C.; Min, Y.; Palm, R.C.; Fiordalisi, J.J.; Wagner, K.T.; Hyder, N.; Cox, A.D.; Caster, J.; Tian, X.; Wang, A.Z. Nanoparticle formulation of histone deacetylase inhibitors for effective chemoradiotherapy in solid tumors. Biomaterials 2015, 51, 208–215. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Liang, Y.; Liu, X.; Zhou, S.; Liu, L.; Zhang, F.; Xie, C.; Cai, S.; Wei, J.; Zhu, Y.; et al. PLGA-PEG Nanoparticles coated with Anti-CD45RO and loaded with HDAC plus protease inhibitors activate latent HIV and inhibit viral spread. Nanoscale Res. Lett. 2015, 10, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wightman, F.; Ellenberg, P.; Churchill, M.; Lewin, S.R. HDAC inhibitors in HIV. Immunol. Cell Biol. 2012, 90, 47–54. [Google Scholar] [CrossRef]
- El Bahhaj, F.; Denis, I.; Pichavant, L.; Delatouche, R.; Collette, F.; Linot, C.; Pouliquen, D.; Grégoire, M.; Héeoguez, V.; Blanquart, C.; et al. Histone deacetylase inhibitors delivery using nanoparticles with intrinsic passive tumor targeting properties for tumor therapy. Theranostics 2016, 6, 795–807. [Google Scholar] [CrossRef]
- Hadden, M.J.; Advani, A. Histone deacetylase inhibitors and diabetic kidney disease. Int. J. Mol. Sci. 2018, 19, 2630. [Google Scholar] [CrossRef]
- Denis, I.; el Bahhaj, F.; Collette, F.; Delatouche, R.; Gueugnon, F.; Pouliquen, D.; Pichavant, L.; Héroguez, V.; Grégoire, M.; Bertrand, P.; et al. Histone deacetylase inhibitor-polymer conjugate nanoparticles for acid-responsive drug delivery. Eur. J. Med. Chem. 2015, 95, 369–376. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Y.; Zheng, Y.; Pan, C.; Yang, X.; Dou, T.; Wang, B.; Lu, W. Homing in on an intracellular target for delivery of loaded nanoparticles functionalized with a histone deacetylase inhibitor. Oncotarget 2017, 8, 68242–68251. [Google Scholar] [CrossRef] [Green Version]
- Thapa, R.K.; Nquyen, H.T.; Jeong, J.H.; Shin, B.S.; Ku, S.K.; Choi, H.G.; Yong, C.S.; Kim, J.O. Synergistic anticancer activity of combined histone deacetylase and proteasomal inhibitor-loaded zein nanoparticles in metastatic prostate cancers. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, L.; Ding, Y.; Wu, J.; Hu, Y.; Yuan, A. Synergy of hypoxia relief and chromatin remodeling to overcome tumor radiation resistance. Biomater. Sci. 2020, 8, 4739–4749. [Google Scholar] [CrossRef]
- Lee, S.Y.; Hong, E.H.; Jeong, J.Y.; Cho, J.; Seo, J.H.; Ko, H.J.; Cho, H.J. Esterase-sensitive cleavable histone deacetylase inhibitor-coupled hyaluronic acid nanoparticles for boosting anticancer activities against lung adenocarcinoma. Biomater. Sci. 2019, 7, 4624–4635. [Google Scholar] [CrossRef] [PubMed]
- Alp, E.; Damkaci, F.; Guven, E.; Tenniswood, M. Starch nanoparticles for delivery of the histone deacetylase inhibitor CG-1521 in breast cancer treatment. Int. J. Nanomed. 2019, 14, 1335–1346. [Google Scholar] [CrossRef] [Green Version]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMPO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, H.; Kühne, M.; Grune, C.; Warncke, P.; Hofmann, S.; Koschella, A.; Godmann, M.; Fischer, D.; Heinzel, T.; Heinze, T. Polysaccharide nanoparticles bearing HDAC inhibitor as nontoxic nanocarrier for drug delivery. Macromol. Biosci. 2020, 20, 2000039. [Google Scholar] [CrossRef] [Green Version]
- Kühne, M.; Lindemann, H.; Grune, C.; Schröder, D.; Cseresnyés, Z.; Godmann, M.; Koschella, A.; Figge, M.T.; Eggeling, C.; Fischer, D.; et al. Biocompatible sulfated valproic acid-coupled polysaccharide-based nanocarriers with HDAC inhibitory activity. J. Control. Release 2021, 329, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, S.; Folwer, M.J.; Baker, C.; Stopka, S.A.; Regan, M.S.; Sablatura, L.; Broughton, C.W.; Knight, B.E.; Stabenfeldt, S.E.; Agar, N.Y.R.; et al. β-Cyclodextrin-poly (β-Amino ester) nanoparticles are a generalizable strategy for high loading and sustained release of HDAC inhibitors. ACS Appl. Mater. Interfaces 2021, 13, 20960–20973. [Google Scholar] [CrossRef]
- Politis, M. Neuroimaging in Parkinson disease: From research setting to clinical practice. Nat. Rev. Neurol. 2014, 10, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, N.P.D.; Cancela, M.C.; Marins, L.F.L.; Souza, D.L.B. Spatial distribution of advanced stage diagnosis and mortality of breast cancer: Socioeconomic and health service offer inequalities in Brazil. PLoS ONE 2021, 16, e0246333. [Google Scholar] [CrossRef] [PubMed]
- Neal, R.D.; Tharmanathan, P.; France, B.; Din, N.U.; Cotton, S.; Fallon-Ferguson, J.; Hamilton, W.; Hendry, A.; Hendry, M.; Lewis, R.; et al. Is increased time to diagnosis and treatment in symptomatic cancer associated with poorer outcomes? Systemic review. Br. J. Cancer 2015, 112, S92–S107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, Y.J.; Kim, W.J.; Kim, K.; Kwon, I.C. Advances in the strategies for designing receptor-targeted molecular imaging probes for cancer research. J. Control. Release 2019, 305, 1–17. [Google Scholar] [CrossRef]
- Holland, J.P.; Liang, S.H.; Rostein, B.H.; Collier, T.L.; Stephenson, T.L.; Greguric, I.; Vasdev, N. Alternative approaches for PET radiotracer development in Alzheimer’s disease: Imaging beyond plaque. J. Label. Comp. Radiopharm. 2014, 57, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Tago, T.; Toyohara, J. Advances in the development of PET ligands targeting histone deacetylases for the assessment of neurodegenerative diseases. Molecules 2018, 23, 300. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, U.; Tong, W.P.; Gelovani, J.G.; Alauddin, M.M. Radiosynthesis of 6-([18F]fluoroacetamido)-1-hexanoicanilide ([18F]FAHA) for PET imaging of histone deacetylase (HDAC). J. Label. Compd. Radiopharm. 2006, 49, 997–1006. [Google Scholar] [CrossRef]
- Hooker, J.M.; Kim, S.W.; Alexoff, D.; Xu, Y.; Shea, C.; Reid, A.; Volkow, N.; Folwer, J.S. Histone deacetylase inhibitor, MS-275, exhibits poor brain penetration: PK studies of [11C]MS-275 using Positron Emission Tomography. ACS Chem. Neorosci. 2010, 1, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, J.A.; Keliher, E.J.; Marinelli, B.; Reiner, T.; Weissleder, R.; Mazitschek, R. In vivo PET imaging of histone deacetylase by 18F-suberoylanilide hydroxamic acid (18F-SAHA). J. Med. Chem. 2011, 54, 5576–5582. [Google Scholar] [CrossRef] [Green Version]
- Zeglis, B.M.; Pillarsetty, N.; Divilov, V.; Blasberg, R.A.; Lewis, J.S. The synthesis and evaluation of N1-(4-(2-[18F]-fluoroethyl)phenyl)-N8-hydroxyoctanediamide ([18F]-FESAHA), a PET radiotracer designed for delineation of histone deacetylase expression in cancer. Nucl. Med. Biol. 2011, 38, 683–696. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Hooker, J.M.; Otto, N.; Win, K.; Muench, L.; Shea, C.; Carter, P.; King, P.; Reid, A.E.; Volkow, N.D.; et al. Whole-body pharmacokinetics of HDAC inhibitor drugs, butyric acid, valproic acid and 4-phenylbutyric acid measured with carbon-11 labeled analogs by PET. Nucl. Med. Biol. 2013, 40, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Li, F.; Jiang, S.; Li, Z. Novel 64Cu-labeled CUDC-101 for in vivo PET imaging histone deacetylases. ACS Med. Chem. Lett. 2013, 4, 858–862. [Google Scholar] [CrossRef]
- Seo, Y.J.; Muench, L.; Reid, A.; Chen, J.; Kang, Y.; Hooker, J.M.; Volkow, N.D.; Fowler, J.S.; Kim, S.W. Radionuclide labeling and evaluation of candidate radioligands for PET imaging of histone deacetylase in the brain. Bioorganic Med. Chem. Lett. 2013, 23, 6700–6705. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Eessalu, T.E.; Barth, V.N.; Mitch, C.H.; Wagner, F.F.; Hong, Y.; Neelamegam, R.; Schroeder, F.A.; Holson, E.B.; Haggarty, S.J.; et al. Design, synthesis, and evaluation of hydroxamic acid-based molecular probes for in vivo imaging of histone deacetylase (HDAC) in brain. Am. J. Nucl. Med. Mol. 2014, 4, 29–38. [Google Scholar]
- Wang, C.; Schroeder, F.A.; Wey, H.Y.; Borra, R.; Wagner, F.F.; Reis, S.; Kim, S.W.; Holson, E.B.; Haggarty, S.J.; Hooker, J.M. In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J. Med. Chem. 2014, 57, 7999–8009. [Google Scholar] [CrossRef] [PubMed]
- Hooker, J.M.; Wang, C.; Schroeder, F.A. Imaging Histone Deacetylases with a Radiotracer Using Positron Emission Tomography. Patent WO 2015/058106 A1, 23 April 2015. [Google Scholar]
- Bonomi, R.; Mukhopadhyay, U.; Shavrin, A.; Yeh, H.H.; Majhi, A.; Dewage, S.W.; Najjar, A.; Lu, X.; Cisneros, G.A.; Tong, W.P.; et al. Novel histone deacetylase class IIa selective substrate radiotracers for PET imaging of epigenetic regulation in the brain. PLoS ONE 2015, 10, e0133512. [Google Scholar] [CrossRef] [Green Version]
- Strebl, M.G.; Wang, C.; Schroeder, F.A.; Placzek, M.S.; Wey, H.Y.; Van de Bittner, G.C.; Neelamegam, R.; Hooker, J.M. Development of a fluorinated Class-I HDAC radiotracer reveals key chemical determinants of brain penetrance. ACS Chem. Neurosci. 2016, 7, 528–533. [Google Scholar] [CrossRef]
- Strebl, M.G.; Campbell, A.J.; Zhao, W.N.; Schroeder, W.N.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 brain mapping with [18F]Bavarostat enabled by a Ru-mediated deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Hooker, J.; Wang, C.; Strebl-Bantillo, M.G. HDAC6 Inhibitors and Imaging Agents. Patent WO 2018/191360 A1, 2018. [Google Scholar]
- Lu, S.; Zhang, Y.; Kalin, J.H.; Cai, I.; Kozikowski, A.P.; Pike, V.W. Exploration of the labeling of [11C]tubastatin a at the hydroxamic acid site with [11C]carbon monoxide. J. Label. Compd. Radiopharm. 2016, 59, 9–13. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Zhang, L.; Kalin, J.; Liow, J.S.; Gladding, R.L.; Innis, R.B.; Kozikowski, A.P.; Pike, V.W. Synthesis and evaluation of [methyl-11C]KB631–A candidate radioligand for histone deacetylase isozyme 6 (HDAC6). J. Label. Comp. Radiopharm. 2013, 56, S319. [Google Scholar] [CrossRef]
- Bonomi, R.E. Development of Novel Radiotracers for PET Imaging of HDAC-Mediated Epigenetic Regulation. Ph.D. Dissertation, Wayne State University, Detroit, MI, USA, 2016. [Google Scholar]
- Wang, C.; Schroeder, F.A.; Hooker, J.M. Visualizing epigenetics: Current advances and advantages in HDAC PET imaging techniques. Neuroscience 2014, 264, 186–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kommidi, H.; Tosi, U.; Maahani, U.B.; Guo, H.; Marnell, C.S.; Law, B.; Souweidane, M.M.; Ting, R. 18F-radiolabeled Panobinostat allows for positron emission tomography guided delivery of a histone deacetylase inhibitor. ACS Med. Chem. Lett. 2018, 9, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Tosi, U.; Kommidi, H.; Adeuyan, O.; Guo, H.; Maachani, U.B.; Chen, N.; Su, T.; Zhang, G.; Pisapia, D.J.; Dahmane, N.; et al. PET, image-guided HDAC inhibition of pediatric diffuse midline glioma improves survival in murine models. Sci. Adv. 2020, 6, eabb4105. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.S.; Kim, H.S.; Kim, S.; Kwon, J.; Kim, E.M.; Hwang, H.; Oh, P.S.; Lim, S.L.; Sohn, M.H.; Kim, D.H.; et al. Synthesis and evaluation of 2-[18F]fluorethyltriazolesuberohydroxamine acid for histone deacetylase in a tumor model as a positron emission tomography radiotracer. Cancer Biother. Radiopharm. 2018, 33, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Shiue, C.Y.; Chang, H.C.; Feng, C.F. Compound and Analogues for Tracking Histone Acetylation Inhibitors PET Imaging for Diagnosis and Treatment of Tumors. U.S. Patent 2018/0099933 A1, 2018. [Google Scholar]
- Li, M.H.; Shiue, C.Y.; Chang, H.C.; Feng, C.F. Precursor of a Histone Deacetylase Inhibitor PET Imaging Compound for Tracking Cerebral Neurodegenerative and Tumor Diseases. U.S. Patent 2019/0076552 A1, 2019. [Google Scholar]
- Li, M.H.; Chang, H.C.; Feng, C.F.; Yu, H.W.; Shiue, C.Y. Synthesis and evaluation of 18F-INER-1577-3 as a central nervous system (CNS) histone deacetylase imaging agent. Curr. Med. Imaging 2020, 16, 979–990. [Google Scholar] [CrossRef]
- Tago, T.; Toyohara, J.; Ishii, K. Radiosynthesis and preliminary evaluation of an 18F-labeled tubastatin A analog for PET imaging of histone deacetylase 6. J. Label. Compd. Radiopharm. 2020, 63, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Tago, T.; Toyohara, J.; Ishii, K. Preclinical evaluation of an 18F-labeled SW-100 derivative for PET imaging of histone deacetylase 6 in the brain. ACS Chem. Neurosci. 2021, 12, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Turkman, N.; Liu, D.; Pirola, I. Novel late-stage radiosynthesis of 5-[18F]-trifluoromethyl-1,2,4-oxiadiazole (TFMO) containing molecules for PET imaging. Sci. Rep. 2021, 11, 10668. [Google Scholar] [CrossRef] [PubMed]
- Donovan, L.L.; Magnussen, J.H.; Dyssegaard, A.; Lehel, S.; Hooker, J.M.; Knudsen, G.M.; Hansen, H.D. Imaging HDACs in vivo: Cross-validation of the [11C]Martinostat radioligand in the pig brain. Mol. Imaging. Biol. 2020, 22, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, T.M.; Zürcher, N.R.; Wu, C.J.; Bhanot, A.; Hightower, B.G.; Kim, M.; Albrecht, D.S.; Wey, H.Y.; Schroeder, F.A.; Rodriguez-Thompson, A.; et al. PET neuroimaging reveals histone deacetylase dysregulation in schizophrenia. Clin. Investig. 2019, 129, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Laws, M.T.; Bonomi, R.E.; Kamal, S.; Gelovani, D.J.; Llaniguez, J.; Potukutchi, S.; Lu, X.; Mangner, T.; Gelovani, J.G. Molecular imaging HDACs class IIa expression-activity and pharmacologic inhibition in intracerebral glioma models in rats using PET/CT/(MRI) with [18F]TFAHA. Sci. Rep. 2019, 9, 3595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, K.; Ahmed, M.; Luyten, K.; Bormans, G. Evaluation of [11C]KB631 as a PET tracer for in vivo visualization of HDAC6 in B16.F10 melanoma. Nucl. Med. Biol. 2019, 74, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Celen, S.; Rokka, J.; Gilbert, T.M.; Koole, M.; Vermeulen, I.; Serdons, K.; Schroeder, F.A.; Wagner, F.F.; Bleeser, T.; Hightower, B.G.; et al. Translation of HDAC6 PET imaging using [18F]EKZ-001–cGMP production and measurement of HDAC6 target occupancy in NHPs. ACS Chem. Neurosci. 2020, 11, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Koole, M.; Van Weehaeghe, D.; Serdons, K.; Herbots, M.; Cawthorne, C.; Celen, S.; Schroeder, F.A.; Hooker, J.M.; Bormans, G.; de Hoon, J.; et al. Clinical validation of the novel HDAC6 radiotracer [18F]EKZ-001 in the human brain. Eur. J. Nucl. Med. 2021, 48, 596–611. [Google Scholar] [CrossRef]
- Terai, T.; Nagano, T. Small-molecule fluorophores and fluorescent probes for bioimaging. Pflugers Arch.-Eur. J. Psychol. 2013, 465, 347–359. [Google Scholar] [CrossRef]
- Hori, Y.; Kikuchi, K. Chemical tools with fluorescence switches for verifying epigenetic modifications. Acc. Chem. Res. 2019, 52, 2849–2857. [Google Scholar] [CrossRef]
- Mazitschek, R.; Patel, V.; Wirth, D.F.; Clardy, J. Development of a fluorescence polarization based assay for histone deacetylase ligand discovery. Bioorganic Med. Chem. Lett. 2008, 18, 2809–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.K.; Mandal, T.; Balasubramanian, N.; Cook, G.; Srivastava, D.K. Coumarin-suberoylanilide hydroxamic acid as a fluorescent probe for determining binding affinities and off-rates of histone deacetylase inhibitors. Anal. Biochem. 2011, 408, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Jung, M.; Wang, K.; Grindrod, S.; Velena, A.; Lee, S.A.; Dakshanamurthy, S.; Yang, Y.; Miessau, M.; Zheng, C.; et al. Histone deacetylase cytoplasmic trapping by a novel fluorescent HDAC inhibitor. Mol. Cancer Ther. 2011, 10, 1591–1599. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Liu, Z.; Li, F.; Ma, J.; Wang, H.; Huan, Y.; Li, Z. An HDAC-targeted imaging probe LBH589-Cy5.5 for tumor detection and therapy evaluation. Mol. Pharm. 2015, 12, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.S.; Lee, D.G.; Lee, J.H.; Jeong, H.J.; Seo, Y.J. Characterization and histone deacetylase inhibitory activity of three novel fluorescent benzamide derivatives. Bull. Korean Chem. Soc. 2015, 36, 553–558. [Google Scholar] [CrossRef]
- Fleming, C.A.; Ashton, T.D.; Nowell, C.; Devlin, M.; Natoli, A.; Schreuders, J.; Pfeffer, F.M. A fluorescent histone deacetylase (HDAC) inhibitor for cellular imaging. Chem. Commun. 2015, 51, 7827–7830. [Google Scholar] [CrossRef] [Green Version]
- Fleming, C.L.; Natoli, A.; Schreuders, J.; Devlin, M.; Yoganantharajah, P.; Gibert, Y.; Leslie, K.G.; New, E.J.; Ashton, T.D.; Pfeffer, F.M. Highly fluorescent and HDAC6 selective scriptaid analogues. Eur. J. Med. Chem. 2019, 162, 321–333. [Google Scholar] [CrossRef]
- Hearn, K.N.; Ashton, T.D.; Acharya, R.; Feng, Z.; Gueven, N.; Pfeffer, F.M. Direct amidation to access 3-amido-1,8-naphtalimides including fluorescent scriptaid analogues as HDAC inhibitors. Cells 2021, 10, 1505. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, J.; Yao, T.P. Discovery of a fluorescent probe with HDAC6 selective inhibition. Eur. J. Med. Chem. 2017, 141, 596–602. [Google Scholar] [CrossRef]
- Meyners, C.; Mertens, M.; Wessig, P.; Meyer-Almes, F.J. A fluorescence lifetime based binding assay for class IIa histone deacetylases. Chem. Eur. J. 2017, 23, 3107–3116. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.H.; Wang, K.J.; Hung, P.Y.; Cheng, Y.S.; Liu, J.R.; Fung, J.R.; Liang, P.H.; Chern, J.W.; Yu, C.W. A highly HDAC6-selective inhibitor acts as a fluorescent probe. Org. Biomol. Chem. 2018, 16, 7820–7832. [Google Scholar] [CrossRef] [PubMed]
- Raudszus, R.; Nowotny, R.; Gertzen, C.G.W.; Schöler, A.; Krizan, A.; Gockel, I.; Kalwa, H.; Gohlke, H.; Thieme, R.; Hansen, F.K. Fluorescent analogs of peptoid-based HDAC inhibitors: Synthesis, biological activity and cellular uptake kinetics. Bioorganic Med. Chem. 2019, 27, 115039. [Google Scholar] [CrossRef]
- Zhou, X.; Dong, G.; Song, T.; Wang, G.; Li, Z.; Qin, X.; Du, L.; Li, M. Environment-sensitive fluorescent inhibitors of histone deacetylase. Bioorganic Med. Chem. Lett. 2020, 30, 127128. [Google Scholar] [CrossRef]
- Tang, C.; Du, Y.; Liang, Q.; Cheng, Z.; Tian, J. Development of a novel histone deacetylase-targeted near-infrared probe for hepatocellular carcinoma imaging and fluorescence image-guided surgery. Mol. Imaging. Biol. 2020, 22, 476–485. [Google Scholar] [CrossRef]
- Huang, Y.; Ru, H.B.; Bao, B.; Yu, J.H.; Li, J.; Zang, Y.; Lu, W. The design of a novel near-infrared fluorescent HDAC inhibitor and image of tumor cells. Bioorganic Med. Chem. 2020, 28, 115639. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Huang, C.; Shu, Y.; Wen, H.; Shan, C.; Wang, X.; Liu, J.; Li, W. An HDAC8-selective fluorescent probe for imaging in living tumor cell lines and tissue slices. Org. Bioorganic Chem. 2021, 19, 8352–8366. [Google Scholar] [CrossRef]


































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daśko, M.; de Pascual-Teresa, B.; Ortín, I.; Ramos, A. HDAC Inhibitors: Innovative Strategies for Their Design and Applications. Molecules 2022, 27, 715. https://doi.org/10.3390/molecules27030715
Daśko M, de Pascual-Teresa B, Ortín I, Ramos A. HDAC Inhibitors: Innovative Strategies for Their Design and Applications. Molecules. 2022; 27(3):715. https://doi.org/10.3390/molecules27030715
Chicago/Turabian StyleDaśko, Mateusz, Beatriz de Pascual-Teresa, Irene Ortín, and Ana Ramos. 2022. "HDAC Inhibitors: Innovative Strategies for Their Design and Applications" Molecules 27, no. 3: 715. https://doi.org/10.3390/molecules27030715
APA StyleDaśko, M., de Pascual-Teresa, B., Ortín, I., & Ramos, A. (2022). HDAC Inhibitors: Innovative Strategies for Their Design and Applications. Molecules, 27(3), 715. https://doi.org/10.3390/molecules27030715