
A Molecular-Wide and Electron Density-Based Approach in Exploring Chemical Reactivity and Explicit Dimethyl Sulfoxide (DMSO) Solvent Molecule Effects in the Proline Catalyzed Aldol Reaction


Abstract
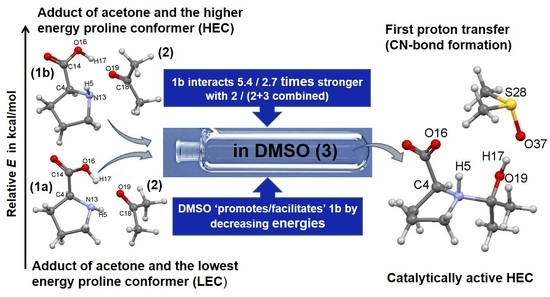
:
1. Introduction
2. Computational Details
3. Basic Concepts of REP-FAMSEC Method Applicable to This Work
- Formation of a poly-molecular complex from separate molecule—from this, one can learn how and why molecules arrange themselves relative to each other, and which atoms drive such arrangement.
- Inclusion of a solvent molecule to a poly-molecular complex—does this impact relative placement of molecules in the complex, what is the solvent molecule’s preferred site and why.
- Can molecules re-arrange themselves ‘freely’ within a complex and which atoms drive the molecules to attain their lowest, or global minimum structure.
- What drives molecules to better pre-organization required for subsequent bond formation or breaking, etc.
4. Results and Discussion
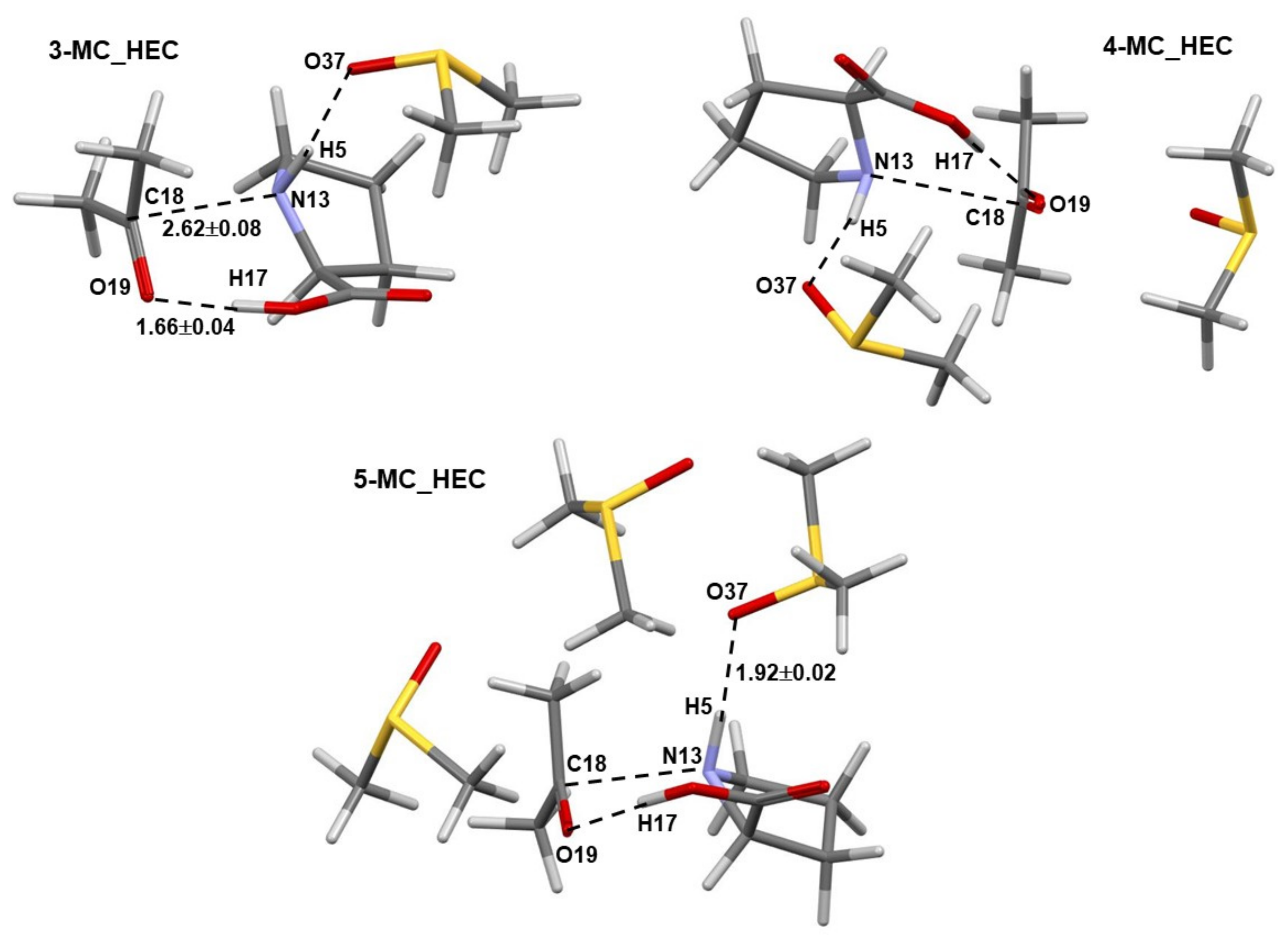
4.1. Exploring the Number of DMSO Molecules in an Explicit Solvation Model
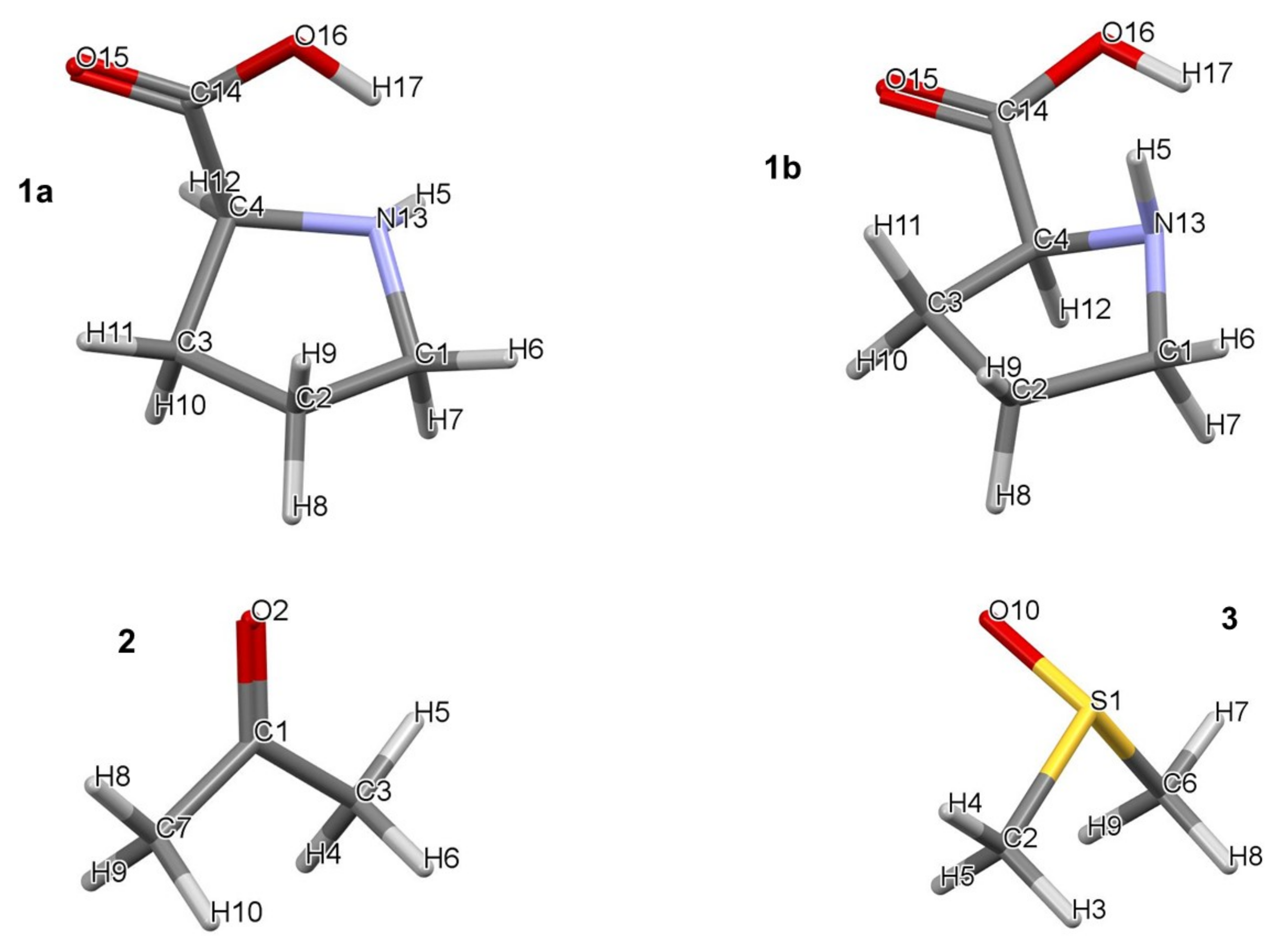
- The minimum number of explicit DMSO solvent molecules needed to strike a balance between the computational cost and insights derived knowing that the computational time and a number of intermolecular interactions increase exponentially with a number of atoms in a molecular system. We decided to limit the number of DMSO solvent molecules to three at most and use a smaller basis set in our preliminary studies, namely 6-31+G(d,p), rather than 6-311++G(d,p) employed in this work.
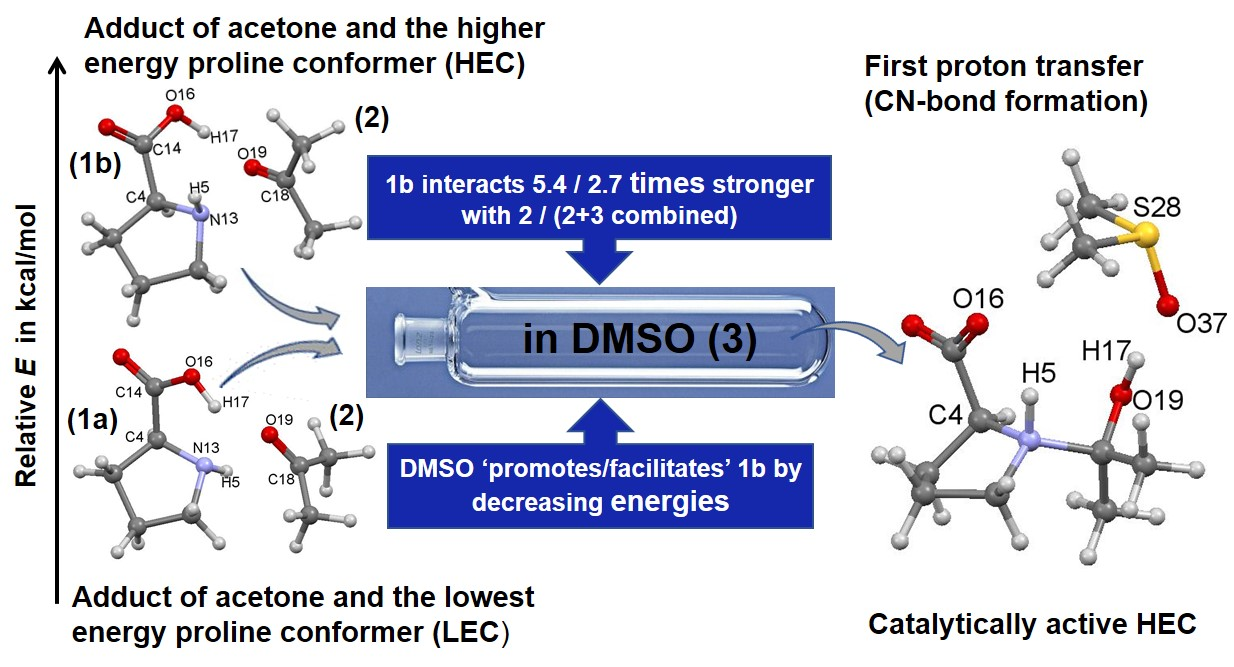
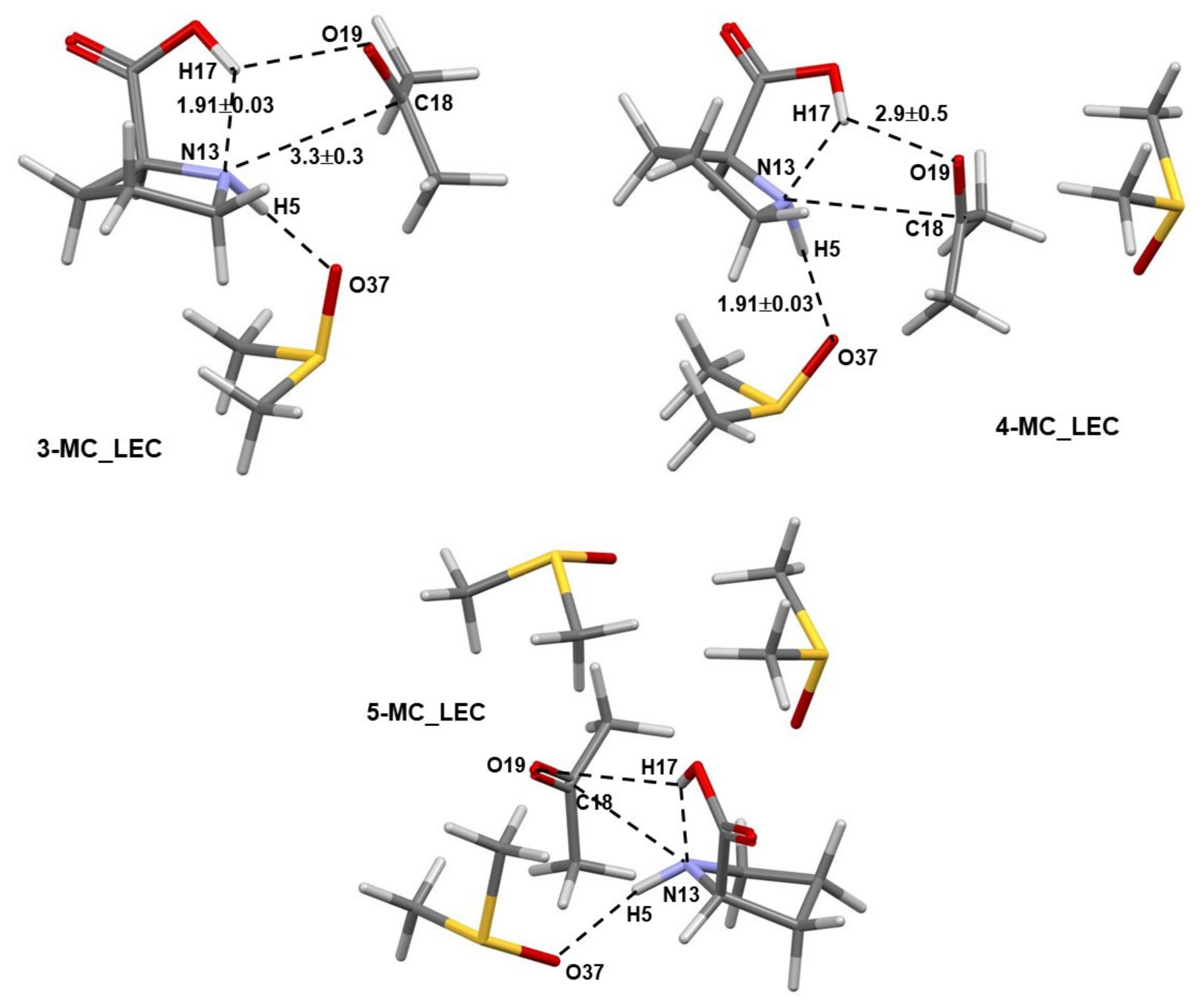
- Specific properties of our molecular system in terms of leading intermolecular interactions between proline 1 and acetone 2. The input structures for conformational searches in Spartan had a relative arrangement of 1 and 2 such that the intermolecular H-bonding O16–H17···O19 was preserved. This is because our recent findings [23] revealed that the transfer of H17 from proline 1 to O19 of acetone 2 must take place as it largely facilitates the CN-bond formation occurring between N13 (in 1) and C18 (in 2).
4.1.1. Geometrical Considerations
4.1.2. Leading Diatomic Interactions
4.1.3. The Energy Barrier Computed for the First Step of the Catalytic Process
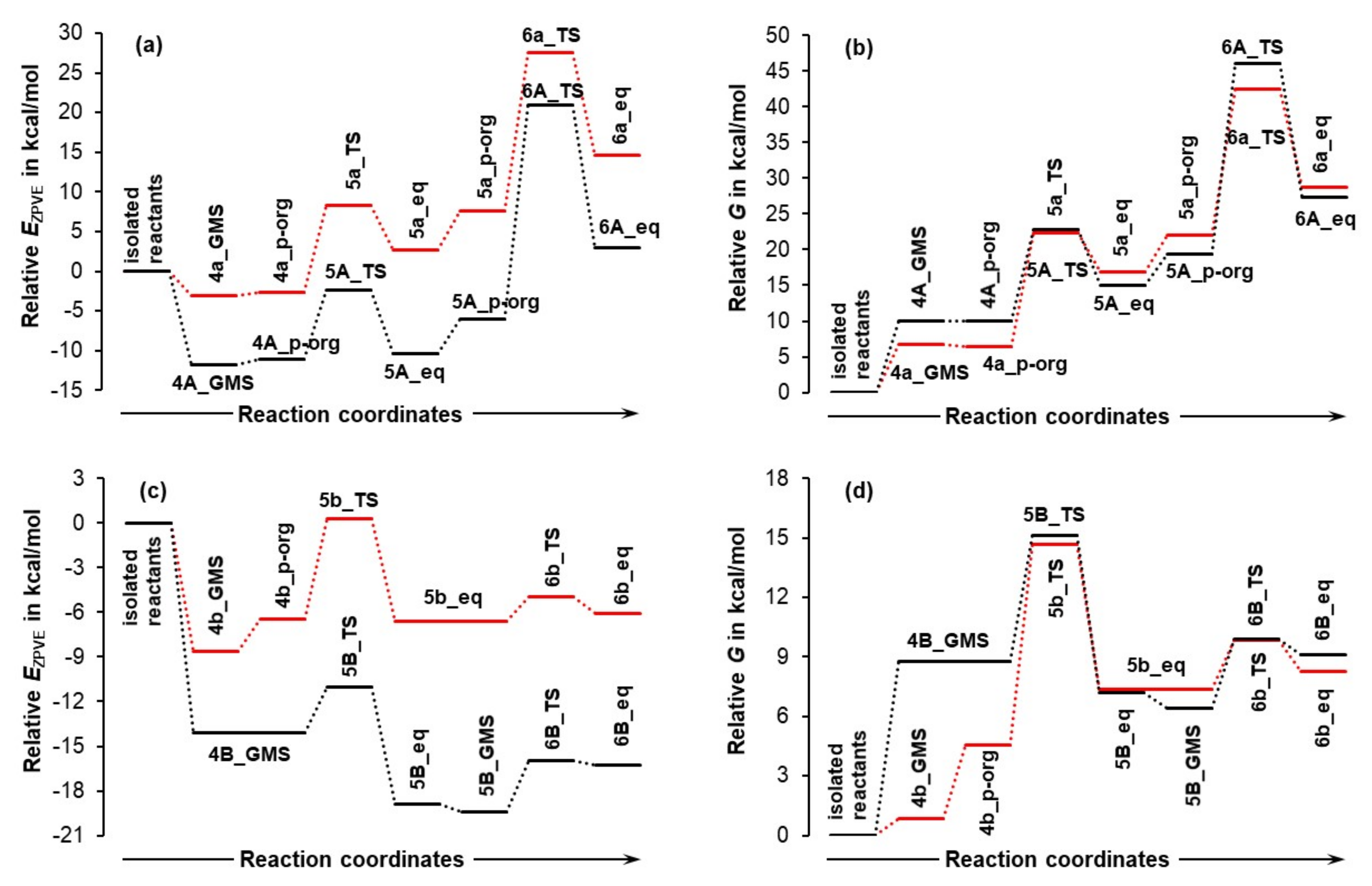
4.2. Impact of a DMSO Solvent Molecule on the Reaction Energy Profile
4.2.1. Concomitant First Proton Transfer and the C–N Bond Formation Step
- Proline plus acetone strengthened significantly as changed favorably (became more negative) by –55.3 and –75.7 kcal/mol for 5A_TS and 5B_TS, respectively. Hence, exactly the same set of diatomic intermolecular interactions, between 3 and 1 plus between 3 and 2, became stronger, by –20.4 kcal/mol, in the case of the 1b-containing MS.
- Acetone strengthened more, by –7.1 kcal/mol, in the case of the LEC-containing MS; the change in the term of –14.7 (at 5A_TS) and –7.6 (at 5B_TS) kcal/mol was obtained.
- Proline strengthened a lot and in favor of 1b-containing MS by –27.5 kcal/mol; the change in the term of –40.6 (5A_TS) and –68.1 (5B_TS) kcal/mol was obtained.
4.2.2. Second Proton Transfer
- The energy barriers ΔE‡ZPVE and ΔG‡ (from 5B_GMS to 6B_TS) are very low, just a few kcal/mol for both energy terms.
- The energy difference between a transition state 6B_TS and the product of the second proton transfer 6B_eq decreased slightly in the presence of DMSO.
4.3. Molecular Interactions Driving a Chemical Change
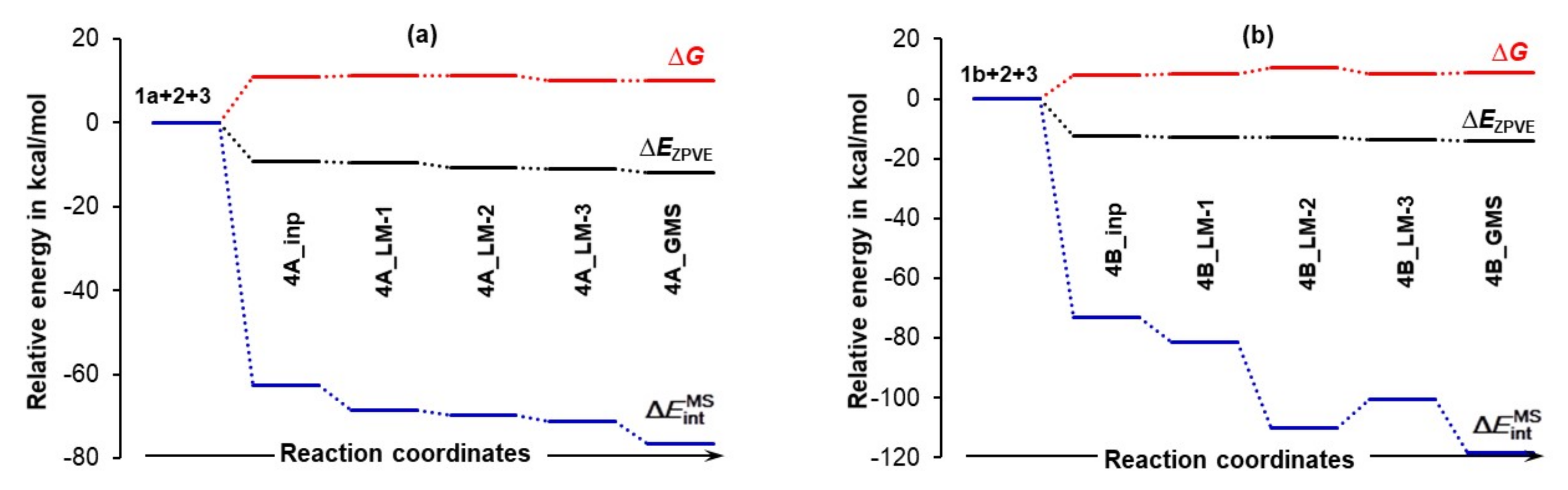
4.3.1. Changes in the Electronic Energy, Gibbs Free Energy, and the Total Molecular System Interaction Energy
- The interactions drive the 3-MCs formation in the first place.
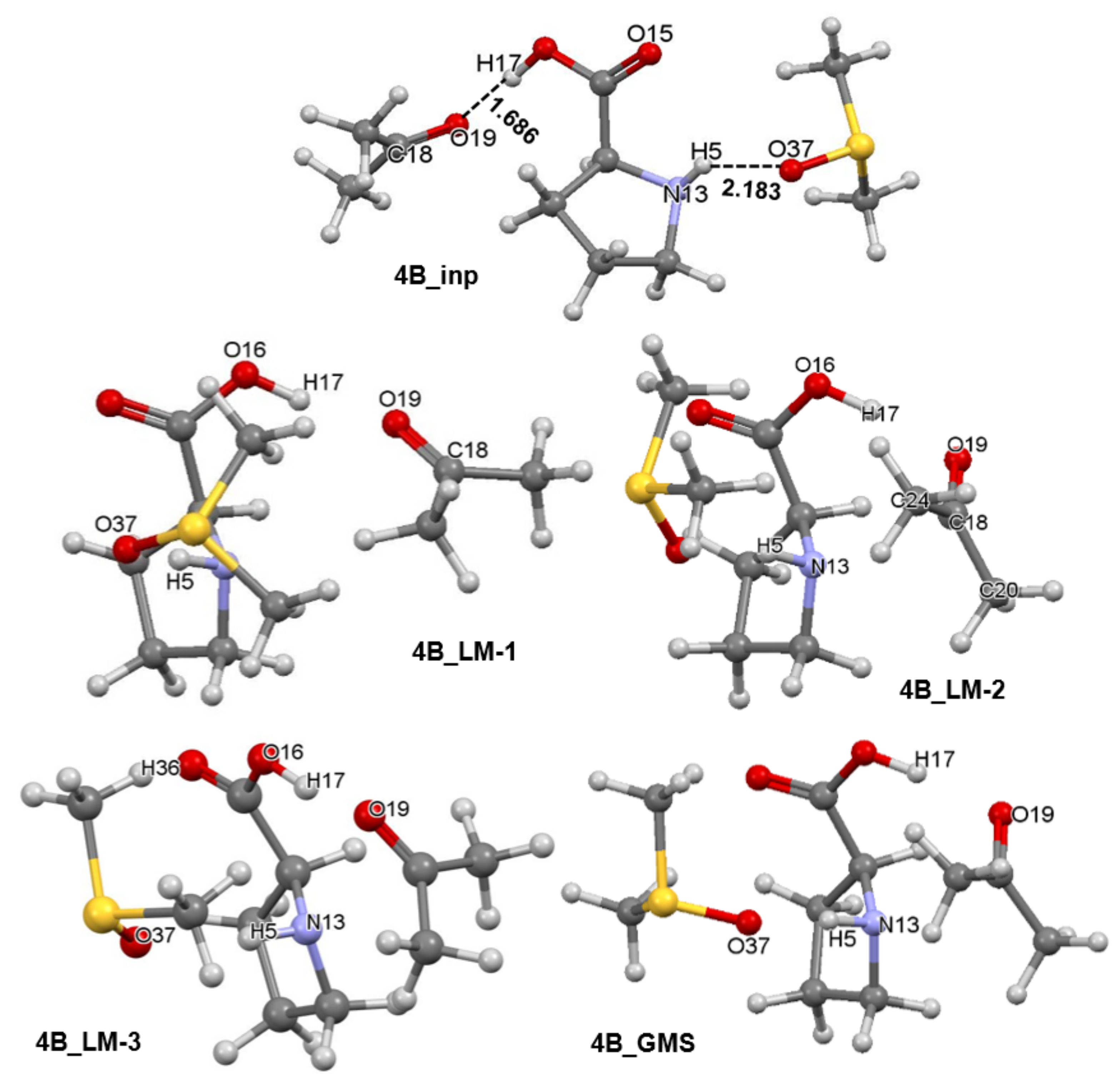
- Strengthening of interactions is a leading force in the 4_inp → 4_GMS structural re-arrangement.
- To gain a deeper understanding of a chemical change, one must explore different modes of interactions.
- Much stronger interactions found for 4B_GMS already point to the 1b-containing complex for which a smaller energy barrier at TS should be expected, and this agrees with trends seen in Figure 5 very well.
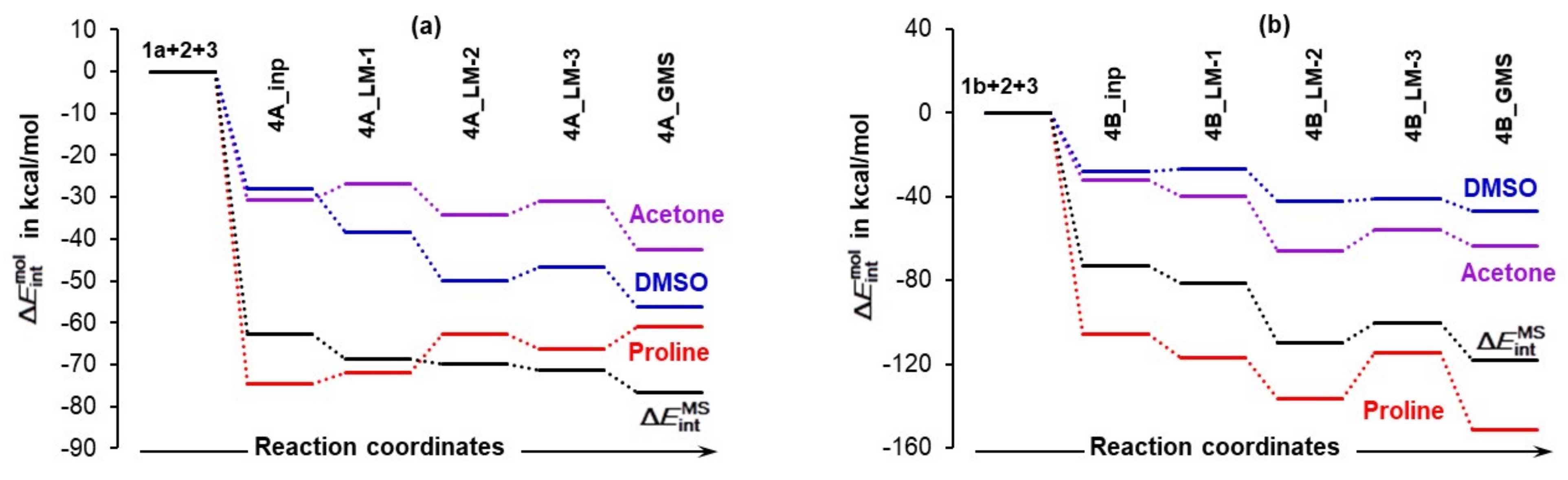
4.3.2. Total Intramolecular and Intermolecular Interaction Energies Computed for Individual Molecules
- Quite surprisingly, 1a is the only molecule for which interactions weakened as the term changed from –74.5 (in 4A_inp) to –61.1 kcal/mol (in 4A_GMS).
- Molecular interactions computed for 1b are not only much stronger when compared with 1a, but they strengthened immensely, from of –105.9 (in 4B_inp) to –151.6 (in 4B_GMS) kcal/mol. This means that molecular interactions computed for 4B_GMS are more than twice as strong as those obtained for 4A_GMS.
- Only trends computed for individual molecules of 1b-containing complexes follow the trend for throughout.
- Looking at data obtained for a DMSO molecule in 1a-containing complexes, its interactions strengthened more than that found for (i) a molecule of acetone and (ii) a DMSO molecule in 1b-containing complexes.
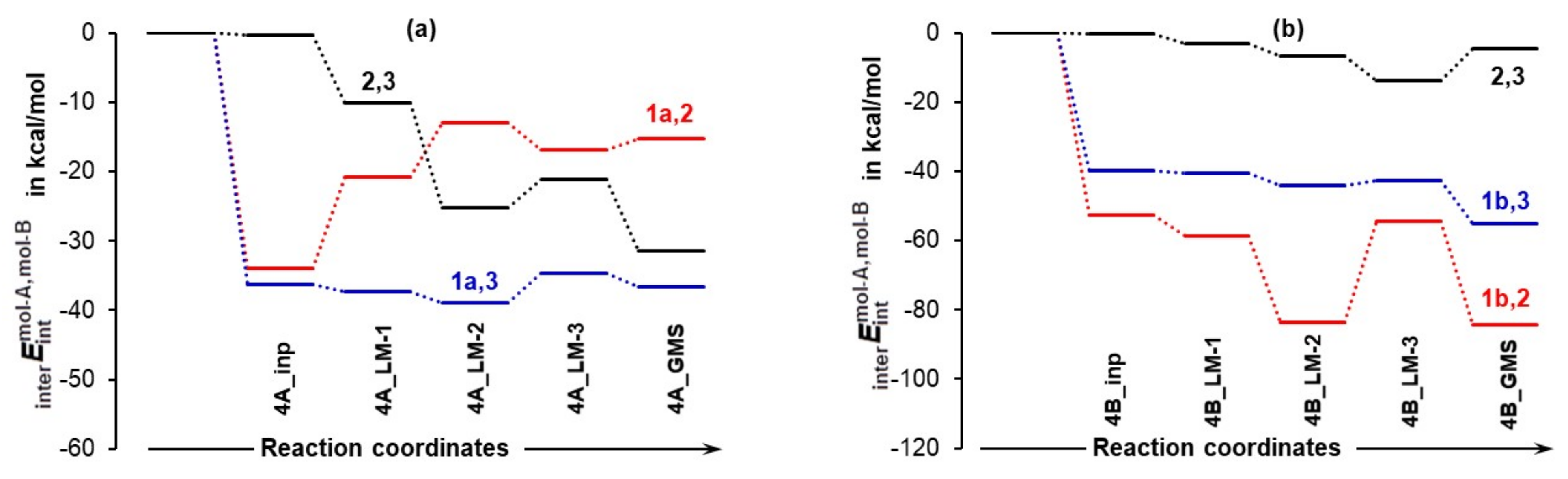
4.3.3. Intermolecular Interaction Energies between Individual Molecules
- The strongest intermolecular interactions are not between 1a and acetone 2, but between 1a and a DMSO solvent molecule 3; this is not what one would like to see at all.
- Significant weakening of interactions between 1a and 2 (from –34.0 to –15.3 kcal/mol) took place on the transition from 4A_inp → 4A_GMS—see red trace. This shows that ability to drive a catalytic process weakens when 1a spontaneously reaches its global minimum structure.
- Relative to the input structure 4A_inp, the = + term increased (interactions weakened) by about 19 kcal/mol in 4a_GMS, whereas the term became more negative, i.e., interactions strengthened by about –47 kcal/mol when in 4B_GMS.
- The combined intermolecular interactions between 1b and {2+3} of –139.6 kcal/mol in 4B_GMS are 2.7 times stronger than the interactions between 1a and {2+3} in 4A_GMS.
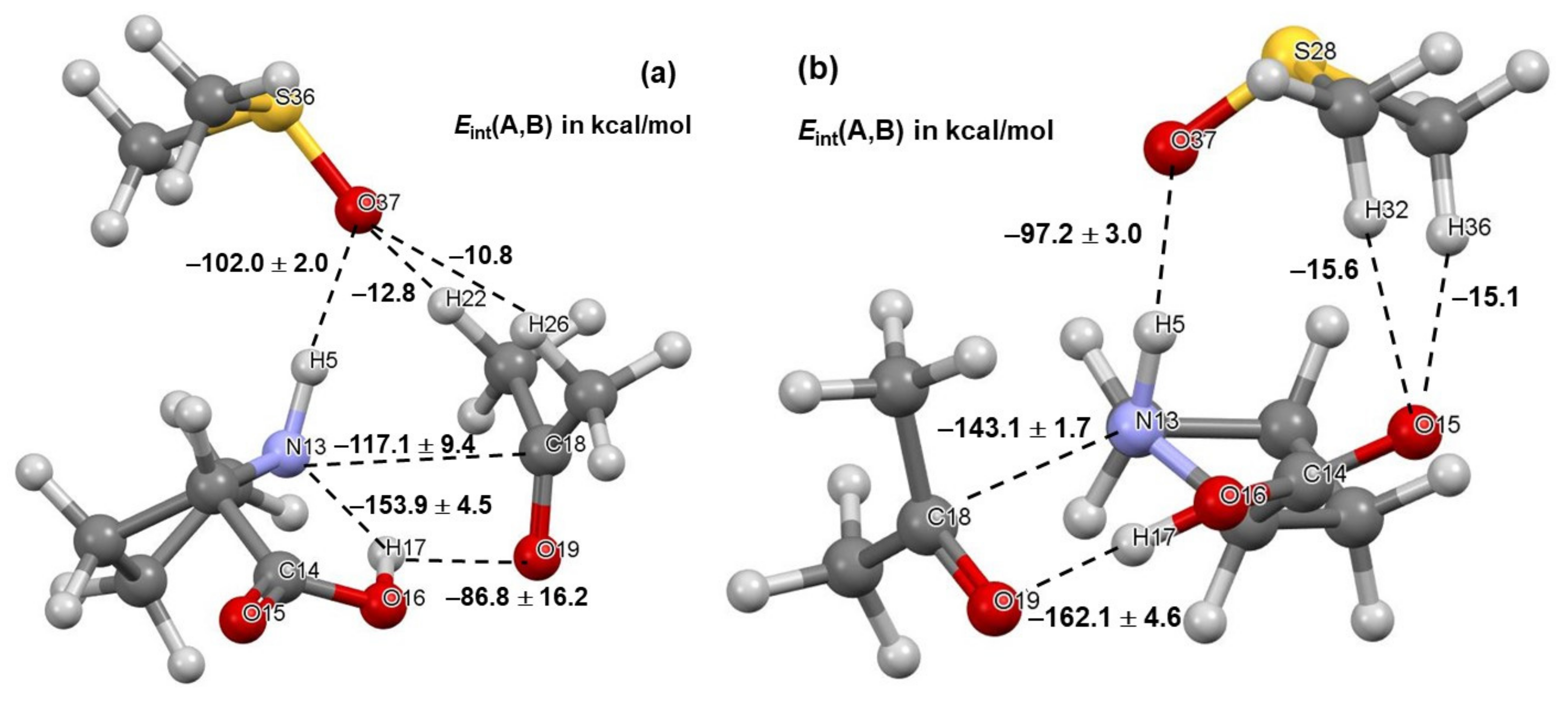
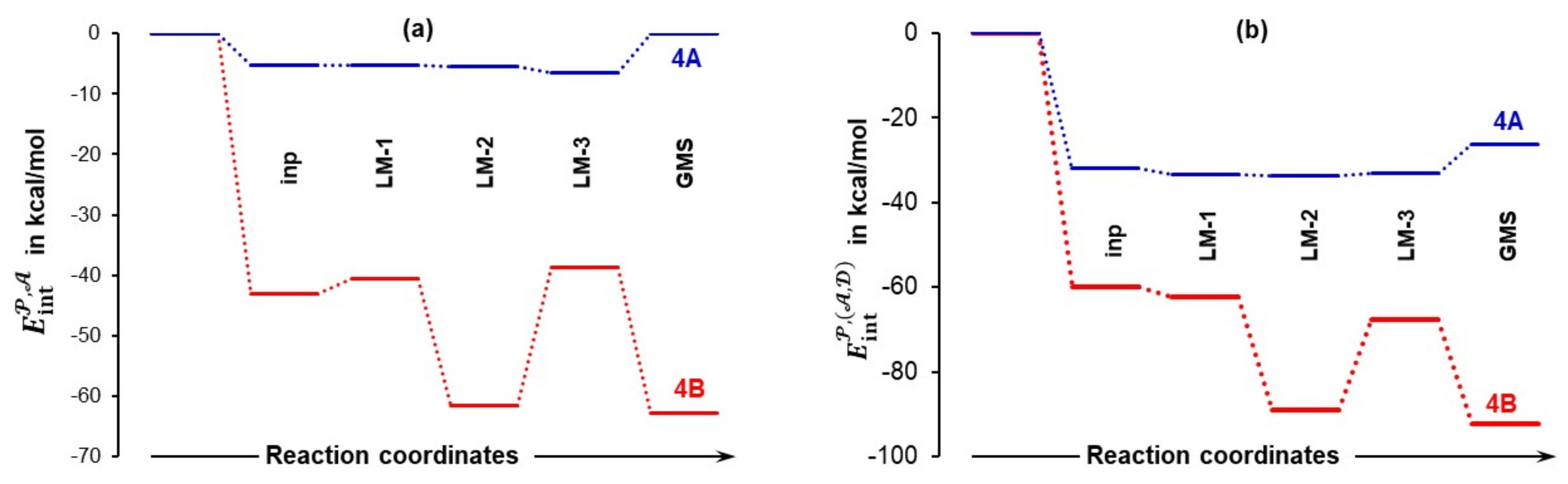
4.4. Molecular Fragments Driving a Chemical Change
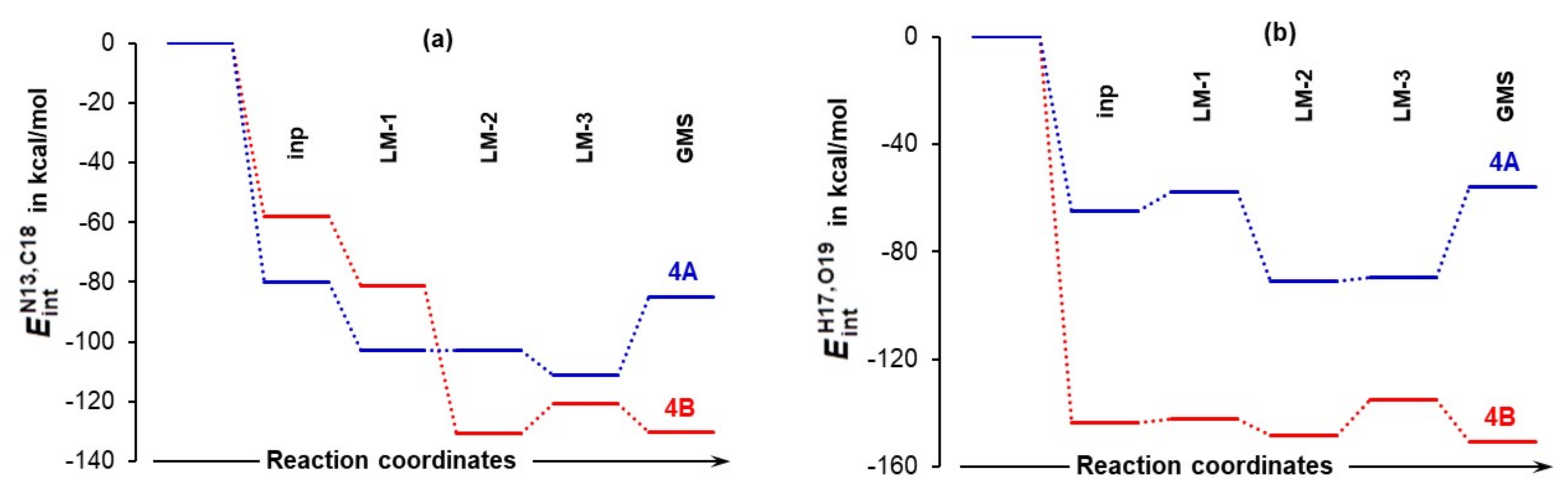
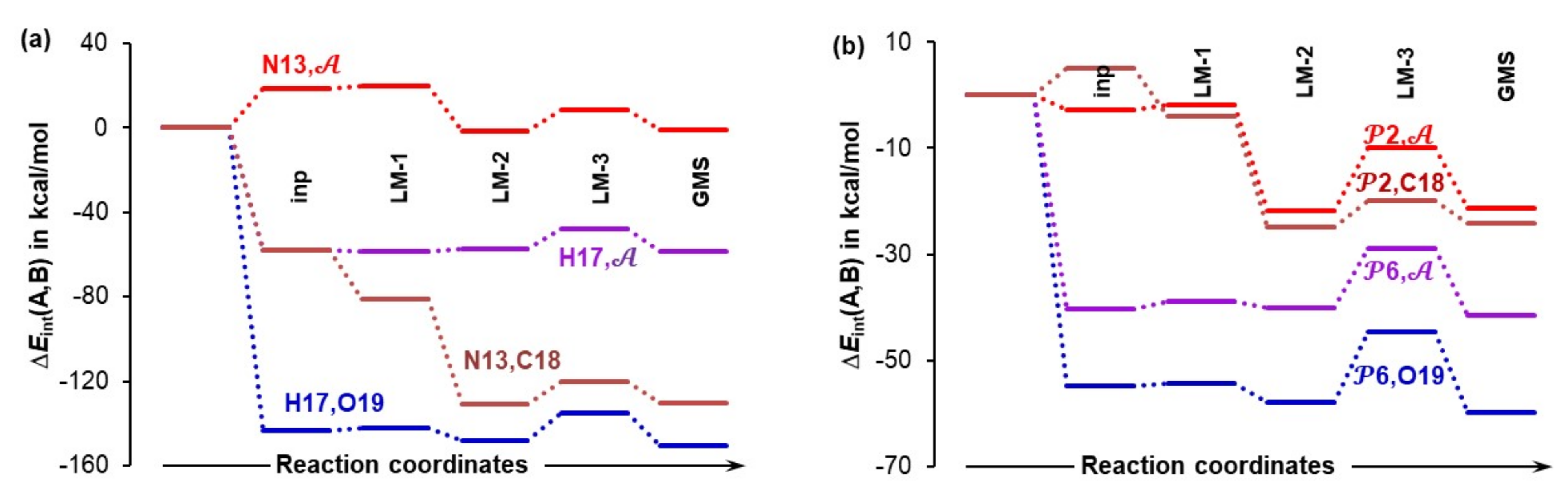
4.5. Importance of N13,C18 and H17,O19 Atom-Pairs
- The attractive interaction between N13 and C18 is far from being the strongest between atoms of proline 1 and acetone 2; so, why is only this interaction considered?
- There are also very strong repulsive interactions between atoms of proline 1 and acetone 2; why are they not considered at all?
4.6. Special Role Played by H17 in 1b-Containing Complexes
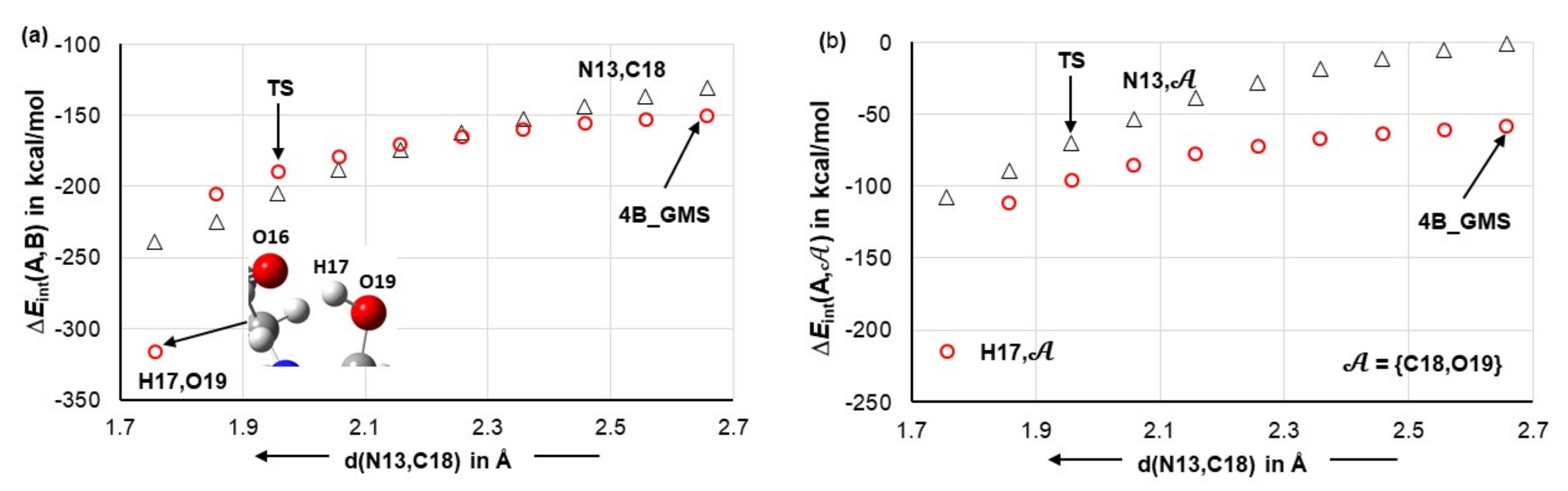
4.7. Forces in the Driving Seat of a Simulated CN-Bond Formation
5. Conclusions
- clearly both the pyrrolidine ring and the carboxylate are essential for efficient catalysis to occur and, based on experimental data, noted that
- after screening several solvents, we found anhydrous DMSO at room temperature to be the most suitable condition regarding reaction times and enantioselectivity.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A
| Terms Applicable to Entire Molecular System (MS) | |
| Accounts for all possible intra- and intermolecular interactions in a MS = + | |
| Sum of all intramolecular interaction energies in a MS = + | |
| Sum of interaction energies computed for all covalently bonded atom-pairs A,B in individual molecules of a MS | |
| Sum of long-distance (L−D) intramolecular interaction energies computed for individual molecules in a MS | |
| Sum of all intermolecular interaction energies in a MS. Here MS = 1 (proline) + 2 (acetone) + 3 (DMSO); hence, = + + | |
| Sum of diatomic interactions energies computed between all atoms of molecule A and all atoms of molecule B, e.g., | |
| Terms applicable to a molecule (mol) | |
| The total interaction energy computed for a molecule in an n-component MS. It is a sum of intra- and intermolecular contributions: = + | |
| Accounts for all intramolecular interactions: = + | |
| Sum of interaction energies computed for all covalently bonded atom-pairs A,B in an individual molecule, e.g., describes combined strength of covalent bonds in a molecule numbered 1, i.e., proline in this work. | |
| Sum of interaction energies computed for long-distance, L−D, covalently non-bonded atom-pairs A,B in individual molecule. | |
| The total intermolecular interaction energy between atoms of the specified molecule and atoms of remaining molecules in a MS. Here MS = 1 + 2 + 3; hence, for acetone 2, = + | |
| Sum of intermolecular interaction energies between atoms of a molecule (here LEC of proline 1a) and atoms of other two molecules (here acetone 2 and DMSO 3) = + | |
References
- List, B. Emil Knoevenagel and the roots of aminocatalysis. Angew. Chem. Int. Ed. 2010, 49, 1730–1734. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Hajos, Z.G.; Parrish, D.R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Eder, U.; Sauer, G.; Wiechert, R. New type of asymmetric cyclization to optically active steroid CD partial structures. Angew. Chem. Int. Ed. 1971, 10, 496–497. [Google Scholar] [CrossRef]
- Guillena, G.; Nájera, C.; Ramón, D.J. Enantioselective direct aldol reaction: The blossoming of modern organocatalysis. Tetrahedron Asymmetry 2007, 18, 2249–2293. [Google Scholar] [CrossRef]
- Pellissier, H. Asymmetric organocatalysis. Tetrahedron 2007, 63, 9267–9331. [Google Scholar] [CrossRef]
- Guillena, G.; Ramón, D.J. Enantioselective α-heterofunctionalisation of carbonyl compounds: Organocatalysis is the simplest approach. Tetrahedron Asymmetry 2006, 17, 1465–1492. [Google Scholar] [CrossRef]
- Sunoj, R.B. Proline-derived organocatalysis and synergism between theory and experiments. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 920–931. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Noto, R. Supported proline and proline-derivatives as recyclable organocatalysts. Chem. Soc. Rev. 2008, 37, 1666–1688. [Google Scholar] [CrossRef]
- Cobb, A.J.; Shaw, D.M.; Longbottom, D.A.; Gold, J.B.; Ley, S.V. Organocatalysis with proline derivatives: Improved catalysts for the asymmetric Mannich, nitro-Michael and aldol reactions. Org. Biom. Chem. 2005, 3, 84–96. [Google Scholar] [CrossRef]
- Notz, W.; Tanaka, F.; Barbas, C.F. Enamine-based organocatalysis with proline and diamines: The development of direct catalytic asymmetric aldol, Mannich, Michael, and Diels−Alder reactions. Acc. Chem. Res. 2004, 37, 580–591. [Google Scholar] [CrossRef] [PubMed]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- List, B. Proline-catalyzed asymmetric reactions. Tetrahedron 2002, 28, 5573–5590. [Google Scholar] [CrossRef]
- Córdova, A.; Notz, W.; Barbas III, C.F. Direct organocatalytic aldol reactions in buffered aqueous media. Chem. Commun. 2002, 24, 3024–3025. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C.F. Amino acid catalyzed direct asymmetric aldol reactions: A bioorganic approach to catalytic asymmetric carbon-carbon bond-forming reactions. J. Am. Chem. Soc. 2001, 123, 5260–5267. [Google Scholar] [CrossRef]
- Rankin, K.N.; Gauld, J.W.; Boyd, R.J. Density functional study of the proline-catalyzed direct aldol reaction. J. Phys. Chem. A 2002, 106, 5155–5159. [Google Scholar] [CrossRef]
- Clemente, F.R.; Houk, K. Computational evidence for the enamine mechanism of intramolecular aldol reactions catalyzed by proline. Angew. Chem. 2004, 116, 5890–5892. [Google Scholar] [CrossRef]
- Allemann, C.; Gordillo, R.; Clemente, F.R.; Cheong, P.H.-Y.; Houk, K.N. Theory of asymmetric organocatalysis of aldol and related reactions: Rationalizations and predictions. Acc. Chem. Res. 2004, 37, 558–569. [Google Scholar] [CrossRef]
- Sharma, A.K.; Sunoj, R.B. Enamine versus Oxazolidinone: What Controls Stereoselectivity in Proline-Catalyzed Asymmetric Aldol Reactions? Angew. Chem. Int. Ed. 2010, 49, 6373–6377. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, L. Mechanisms and reactivity differences of proline-mediated catalysis in water and organic solvents. Catal. Sci. Technol. 2016, 6, 3378–3385. [Google Scholar] [CrossRef]
- Ajitha, M.J.; Suresh, C.H. A higher energy conformer of (S)-proline is the active catalyst in intermolecular aldol reaction: Evidence from DFT calculations. J. Mol. Catal. A Chem. 2011, 345, 37–43. [Google Scholar] [CrossRef]
- Czinki, E.; Császár, A.G. Conformers of gaseous proline. Chem. Eur. J. 2003, 9, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Cukrowski, I.; Dhimba, G.; Riley, D.L. A reaction energy profile and fragment attributed molecular system energy change (FAMSEC)-based protocol designed to uncover reaction mechanisms: A case study of the proline-catalysed aldol reaction. Phys. Chem. Chem. Phys. 2019, 21, 16694–16705. [Google Scholar] [CrossRef] [PubMed]
- Mdhluli, B.K.; Nxumalo, W.; Cukrowski, I. A REP-FAMSEC Method as a Tool in Explaining Reaction Mechanisms: A Nucleophilic Substitution of 2-Phenylquinoxaline as a DFT Case Study. Molecules 2021, 26, 1570. [Google Scholar] [CrossRef] [PubMed]
- Arnó, M.; Domingo, L.R. Density functional theory study of the mechanism of the proline-catalyzed intermolecular aldol reaction. Theor. Chem. Acc. 2002, 108, 232–239. [Google Scholar] [CrossRef]
- Arnó, M.; Zaragozá, R.J.; Domingo, L.R. Density functional theory study of the 5-pyrrolidin-2-yltetrazole-catalyzed aldol reaction. Tetrahedron Asymmetry 2005, 16, 2764–2770. [Google Scholar] [CrossRef]
- Yang, G.; Yang, Z.; Zhou, L.; Zhu, R.; Liu, C. A revisit to proline-catalyzed aldol reaction: Interactions with acetone and catalytic mechanisms. J. Mol. Catal. A Chem. 2010, 316, 112–117. [Google Scholar] [CrossRef]
- List, B.; Hoang, L.; Martin, H.J. New mechanistic studies on the proline-catalyzed aldol reaction. Proc. Nat. Acad. Sci. USA 2004, 101, 5839–5842. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Li, S.J.; Fang, D.C. A Theoretical Study of Ene Reactions in Solution: A Solution-Phase Translational Entropy Model. ChemPhysChem 2015, 16, 3711–3718. [Google Scholar] [CrossRef]
- Han, L.-L.; Li, S.-J.; Fang, D.-C. Theoretical estimation of kinetic parameters for nucleophilic substitution reactions in solution: An application of a solution translational entropy model. Phys. Chem. Chem. Phys. 2016, 18, 6182–6190. [Google Scholar] [CrossRef]
- Varghese, J.J.; Mushrif, S.H. Origins of complex solvent effects on chemical reactivity and computational tools to investigate them: A review. React. Chem. Eng. 2019, 4, 165–206. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Han, K.-L.; Peng, X.-J. A DFT study of Diels−Alder reactions of o-quinone methides and various substituted ethenes: Selectivity and reaction mechanism. J. Org. Chem. 2005, 70, 4910–4917. [Google Scholar] [CrossRef] [PubMed]
- Zeifman, A.A.; Novikov, F.N.; Stroylov, V.S.; Stroganov, O.V.; Svitanko, I.V.; Chilov, G.G. An explicit account of solvation is essential for modeling Suzuki–Miyaura coupling in protic solvents. Dalton Trans. 2015, 44, 17795–17799. [Google Scholar] [CrossRef] [PubMed]
- Cukrowski, I. A unified molecular-wide and electron density based concept of chemical bonding. WIREs Comput. Mol. Sci. 2021, e1579. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Thapa, B.; Schlegel, H.B. Density functional theory calculation of pKa’s of thiols in aqueous solution using explicit water molecules and the polarizable continuum model. J. Phys. Chem. A 2016, 120, 5726–5735. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. AIMAll, version 19.10.13; TK Gristmill Software: Overland Park, KS, USA, 2019. Available online: Aim.tkgristmill.com (accessed on 2 February 2021).
- Cukrowski, I. Reliability of HF/IQA, B3LYP/IQA, and MP2/IQA data in interpreting the nature and strength of interactions. Phys. Chem. Chem. Phys. 2019, 21, 10244–10260. [Google Scholar] [CrossRef]
- Spartan’10, version 1.1.0; Wavefunction, Inc.: Irvine, CA, USA, 2010.
- Blanco, M.; Pendás, A.M.; Francisco, E. Interacting quantum atoms: A correlated energy decomposition scheme based on the quantum theory of atoms in molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef]
- Francisco, E.; Pendás, A.M.; Blanco, M. A molecular energy decomposition scheme for atoms in molecules. J. Chem. Theory Comput. 2006, 2, 90–102. [Google Scholar] [CrossRef]
- Bachrach, S.M. Challenges in computational organic chemistry. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 482–487. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Part (a) | |||||
| Atoms in | Di-Atomic Interaction Energies in kcal/mol | ||||
| 1b | 2 | 3-MC | 4-MC | 5-MC | Average |
| C14 | O19 | −188.5 | −178.1 | −190.1 | −185.6 ± 7 |
| H17 | O19 | −164.8 | −156.8 | −164.6 | −162.1 ± 5 |
| N13 | C18 | −145.1 | −142.1 | −147.4 | −144.9 ± 3 |
| O16 | C18 | −135.0 | −131.7 | −132.0 | −132.9 ± 2 |
| O15 | C18 | −88.8 | −85.0 | −87.1 | −87.0 ± 2 |
| H5 | O19 | −54.5 | −58.4 | −57.9 | −57.0 ± 2 |
| Part (b) | |||||
| Atoms in | Di-Atomic Interaction Energies in kcal/mol | ||||
| 1b | 2 | 3-MC | 4-MC | 5-MC | Average |
| H5 | C18 | 46.7 | 47.6 | 48.7 | 47.7 ± 1 |
| H17 | C18 | 100.2 | 98.7 | 97.6 | 98.8 ± 1 |
| O15 | O19 | 115.4 | 110.2 | 116.1 | 113.9 ± 3 |
| C14 | C18 | 142.5 | 135.6 | 140.1 | 139.4 ± 4 |
| N13 | O19 | 145.7 | 149.5 | 147.7 | 147.6 ± 2 |
| O16 | O19 | 174.4 | 171.9 | 176.0 | 174.1 ± 2 |
| Part (a) | ||||||||
| E | ∆E | EZPVE | ΔEZPVE | H | ∆H | G | ∆G | |
| 3-MCs | ||||||||
| input | −1147.6522 | 0.0 | −1147.3418 | 0.0 | −1147.3193 | 0.0 | −1147.3962 | 0.0 |
| TS | −1147.6413 | 6.8 | −1147.3295 | 7.8 | −1147.3093 | 6.3 | −1147.3786 | 11.1 |
| EQ | −1147.6595 | −4.6 | −1147.3430 | −0.7 | −1147.3232 | −2.5 | −1147.3902 | 3.8 |
| 4-MCs | ||||||||
| input | −1700.8866 | 0.0 | −1700.4953 | 0.0 | −1700.4654 | 0.0 | −1700.5614 | 0.0 |
| TS | −1700.8767 | 6.2 | −1700.4841 | 7.0 | −1700.4565 | 5.6 | −1700.5451 | 10.3 |
| EQ | −1700.8940 | −4.6 | −1700.4968 | −0.9 | −1700.4695 | −2.6 | −1700.5557 | 3.6 |
| 5-MCs | ||||||||
| input | −2254.1254 | 0.0 | −2253.6530 | 0.0 | −2253.6158 | 0.0 | −2253.7275 | 0.0 |
| TS | −2254.1111 | 9.0 | −2253.6371 | 10.0 | −2253.6026 | 8.3 | −2253.7051 | 14.0 |
| EQ | −2254.1271 | −1.1 | −2253.6491 | 2.5 | −2253.6143 | 0.9 | −2253.7190 | 5.3 |
| Part (b) | ||||||||
| E | ∆ | EZPVE | Δ | H | ∆ | G | ∆ | |
| 3-MCs | ||||||||
| input | −1147.6474 | 0.0 | −1147.3360 | 0.0 | −1147.3140 | 0.0 | −1147.3880 | 0.0 |
| TS | −1147.6450 | 1.5 | −1147.3325 | 2.2 | −1147.3130 | 0.9 | −1147.3800 | 5.0 |
| EQ | −1147.6613 | −8.7 | −1147.3454 | −5.9 | −1147.3260 | −7.3 | −1147.3930 | −3.0 |
| 4-MCs | ||||||||
| input | −1700.8798 | 0.0 | −1700.4884 | 0.0 | −1700.4580 | 0.0 | −1700.5530 | 0.0 |
| TS | −1700.8798 | 0.0 | −1700.4868 | 1.0 | −1700.4590 | −0.6 | −1700.5460 | 4.6 |
| EQ | −1700.8969 | −10.7 | −1700.4999 | −7.3 | −1700.4730 | −8.9 | −1700.5590 | −3.4 |
| 5-MCs | ||||||||
| input | −2254.1236 | 0.0 | −2253.6494 | 0.00 | −2253.6130 | 0.0 | −2253.7210 | 0.0 |
| TS | −2254.1223 | 0.8 | −2253.6471 | 1.5 | −2253.6130 | 0.3 | −2253.7140 | 4.0 |
| EQ | −2254.1362 | −7.9 | −2253.6577 | −5.2 | −2253.6240 | −6.5 | −2253.7240 | −2.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cukrowski, I.; Dhimba, G.; Riley, D.L. A Molecular-Wide and Electron Density-Based Approach in Exploring Chemical Reactivity and Explicit Dimethyl Sulfoxide (DMSO) Solvent Molecule Effects in the Proline Catalyzed Aldol Reaction. Molecules 2022, 27, 962. https://doi.org/10.3390/molecules27030962
Cukrowski I, Dhimba G, Riley DL. A Molecular-Wide and Electron Density-Based Approach in Exploring Chemical Reactivity and Explicit Dimethyl Sulfoxide (DMSO) Solvent Molecule Effects in the Proline Catalyzed Aldol Reaction. Molecules. 2022; 27(3):962. https://doi.org/10.3390/molecules27030962
Chicago/Turabian StyleCukrowski, Ignacy, George Dhimba, and Darren L. Riley. 2022. "A Molecular-Wide and Electron Density-Based Approach in Exploring Chemical Reactivity and Explicit Dimethyl Sulfoxide (DMSO) Solvent Molecule Effects in the Proline Catalyzed Aldol Reaction" Molecules 27, no. 3: 962. https://doi.org/10.3390/molecules27030962
APA StyleCukrowski, I., Dhimba, G., & Riley, D. L. (2022). A Molecular-Wide and Electron Density-Based Approach in Exploring Chemical Reactivity and Explicit Dimethyl Sulfoxide (DMSO) Solvent Molecule Effects in the Proline Catalyzed Aldol Reaction. Molecules, 27(3), 962. https://doi.org/10.3390/molecules27030962