
On the Photostability of Cyanuric Acid and Its Candidature as a Prebiotic Nucleobase


Abstract
:1. Introduction
2. Results and Discussion
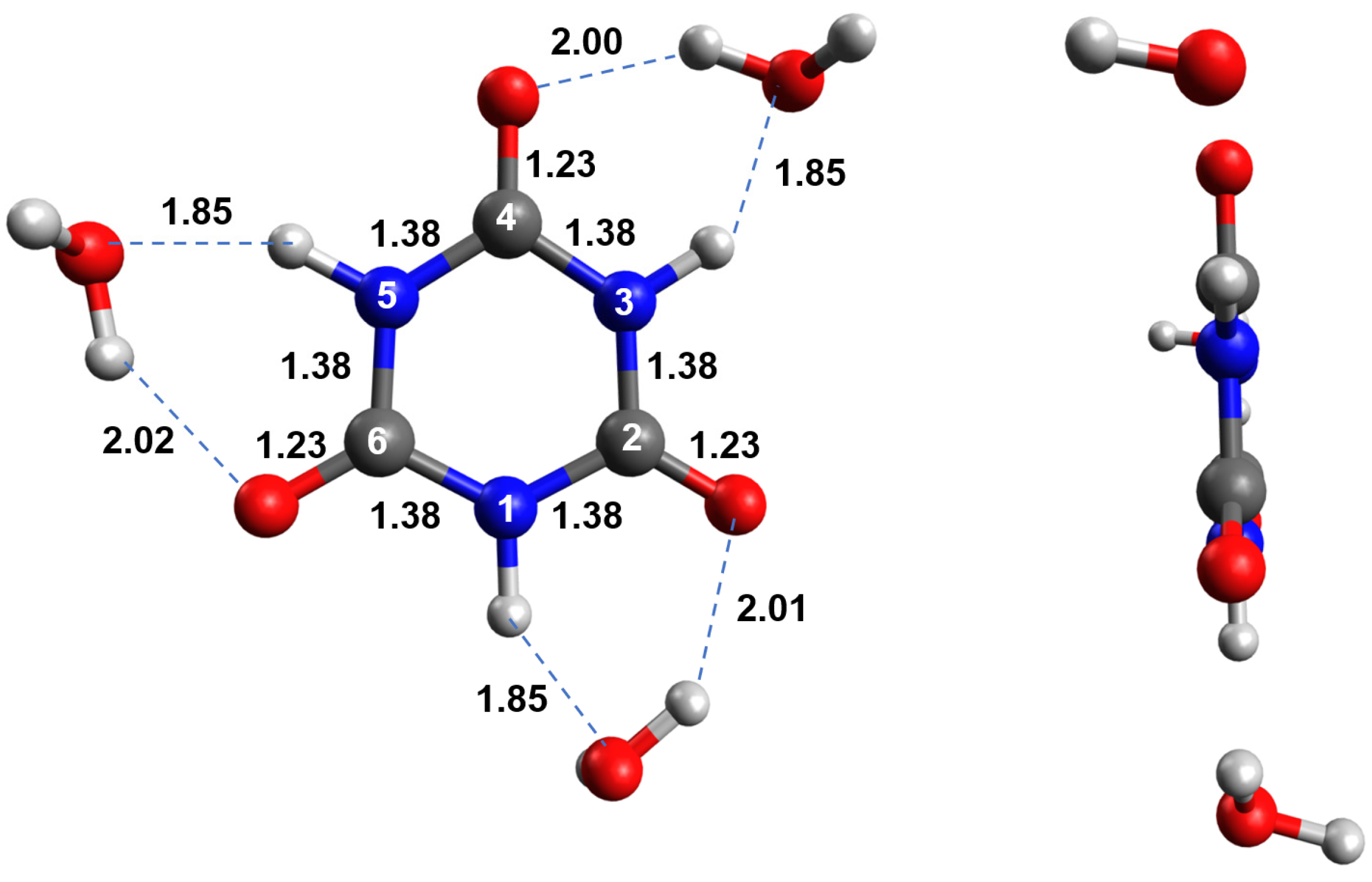
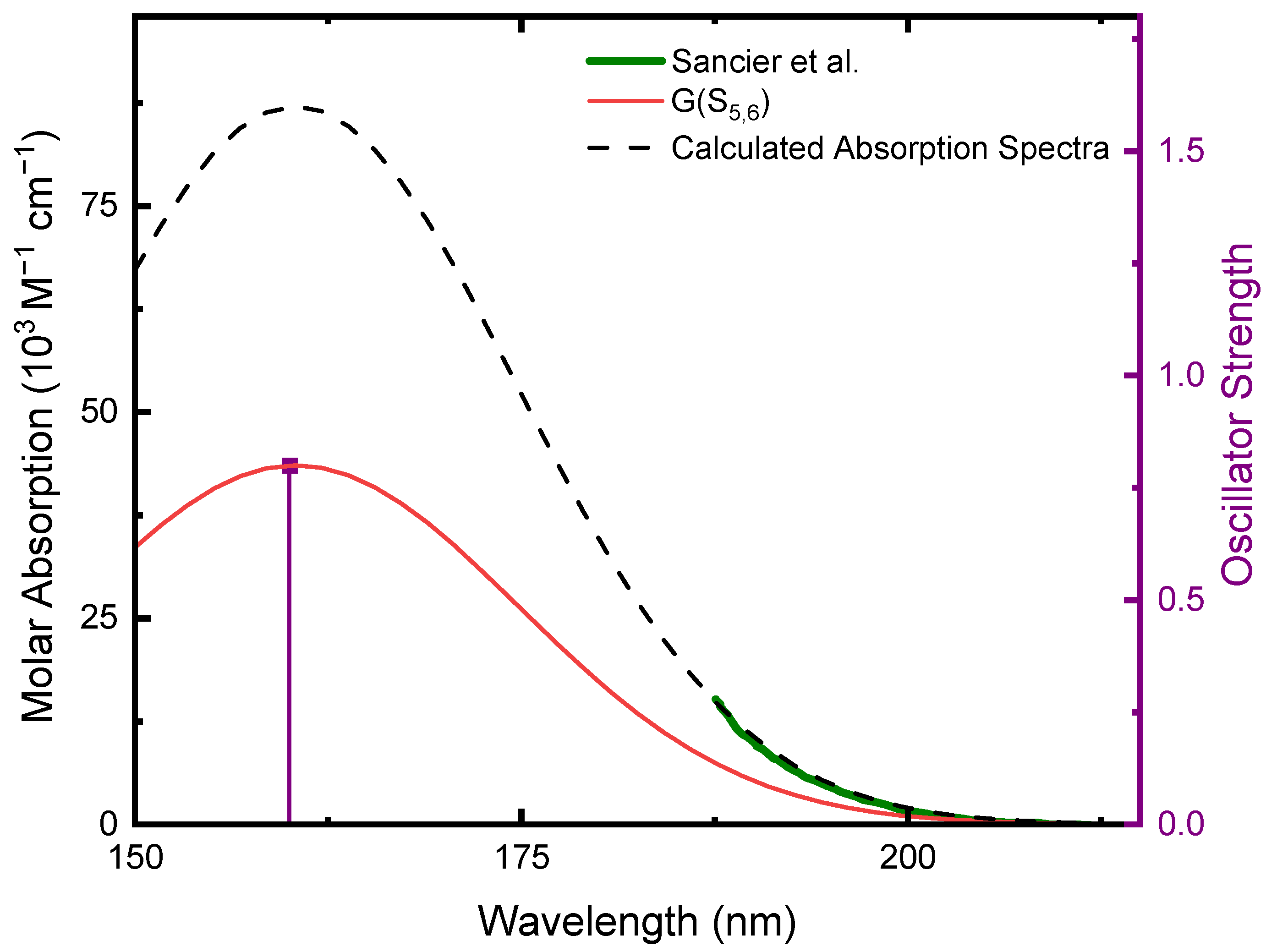
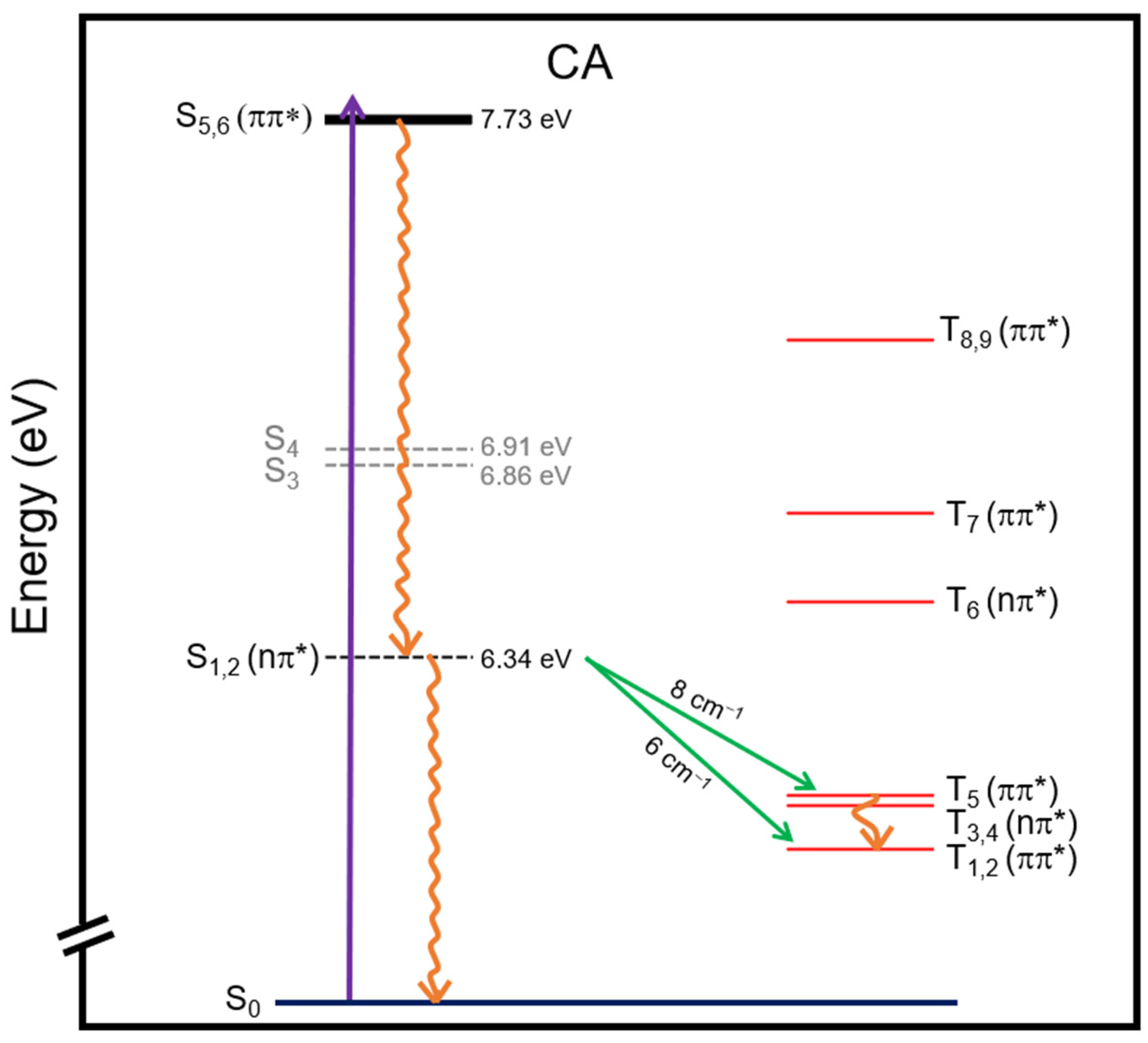
2.1. Ground-State Structure and Absorption Spectrum of CA
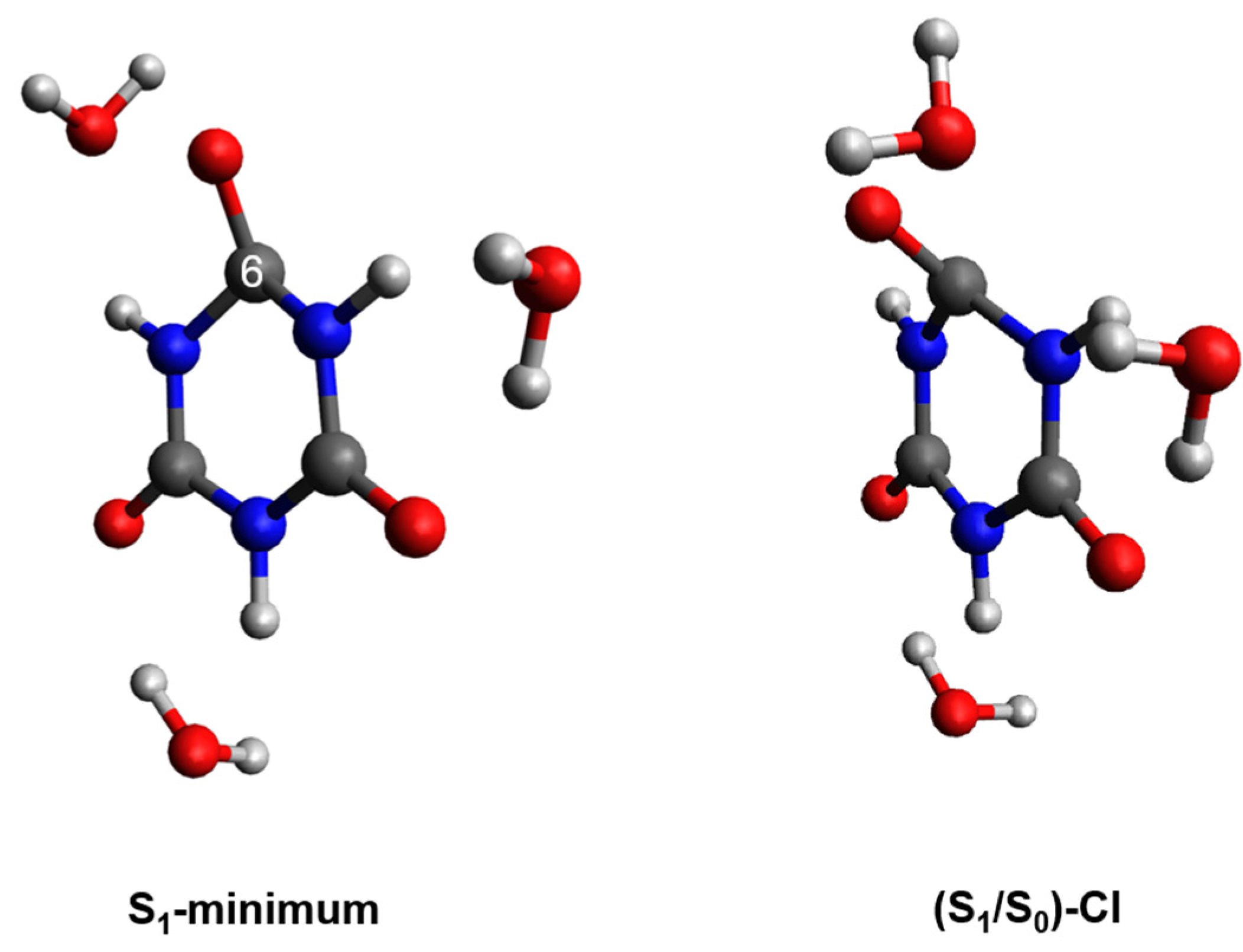
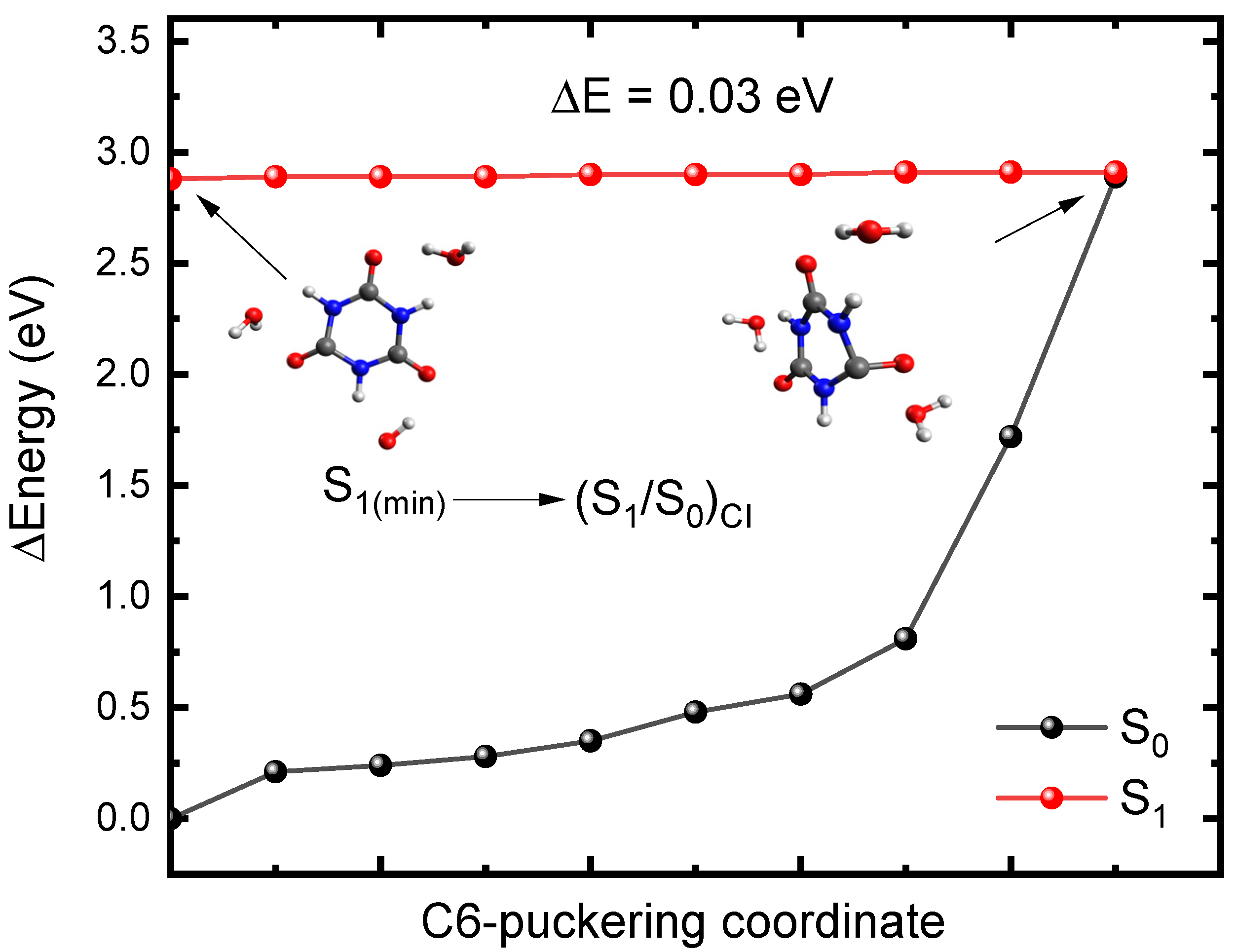
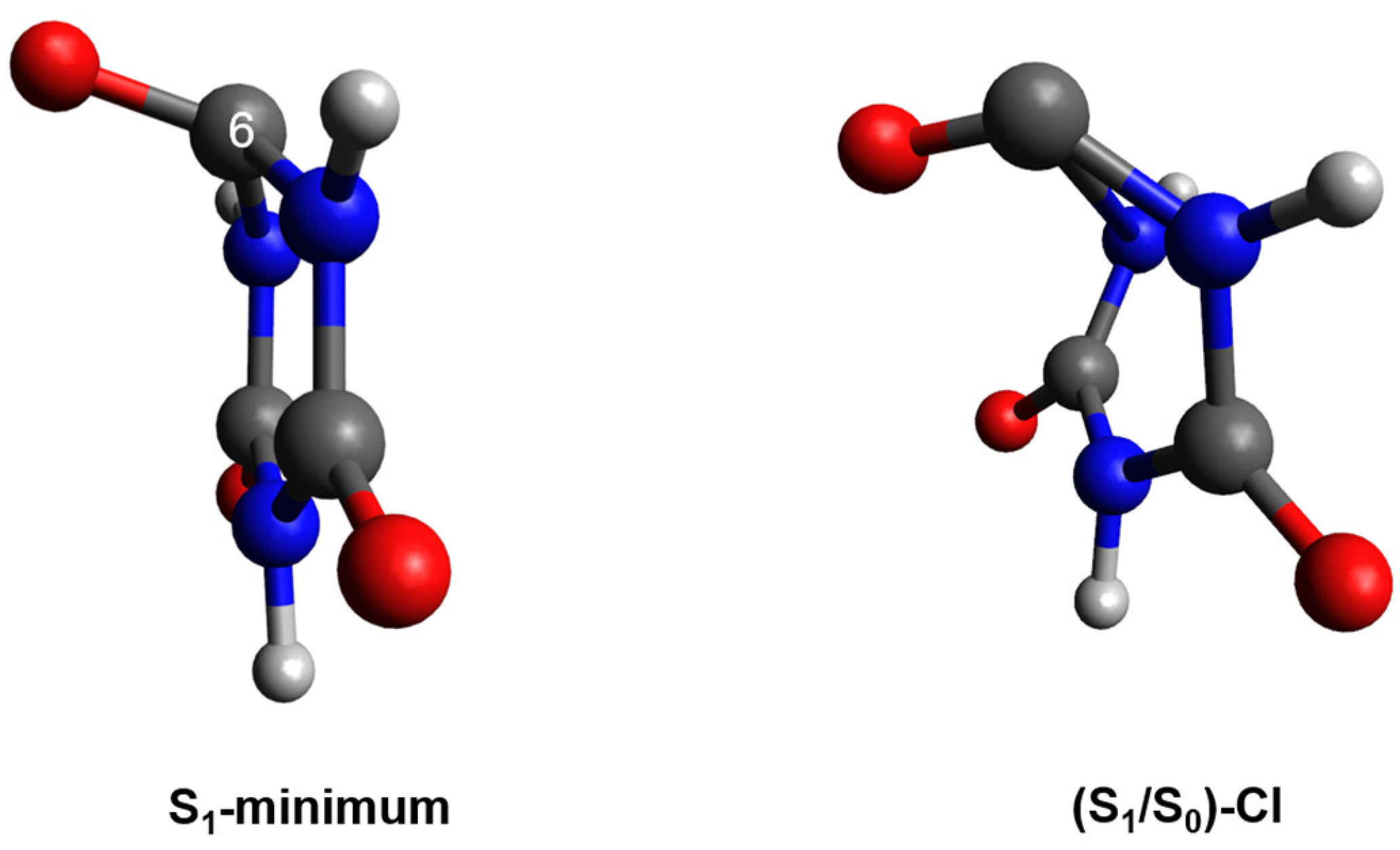
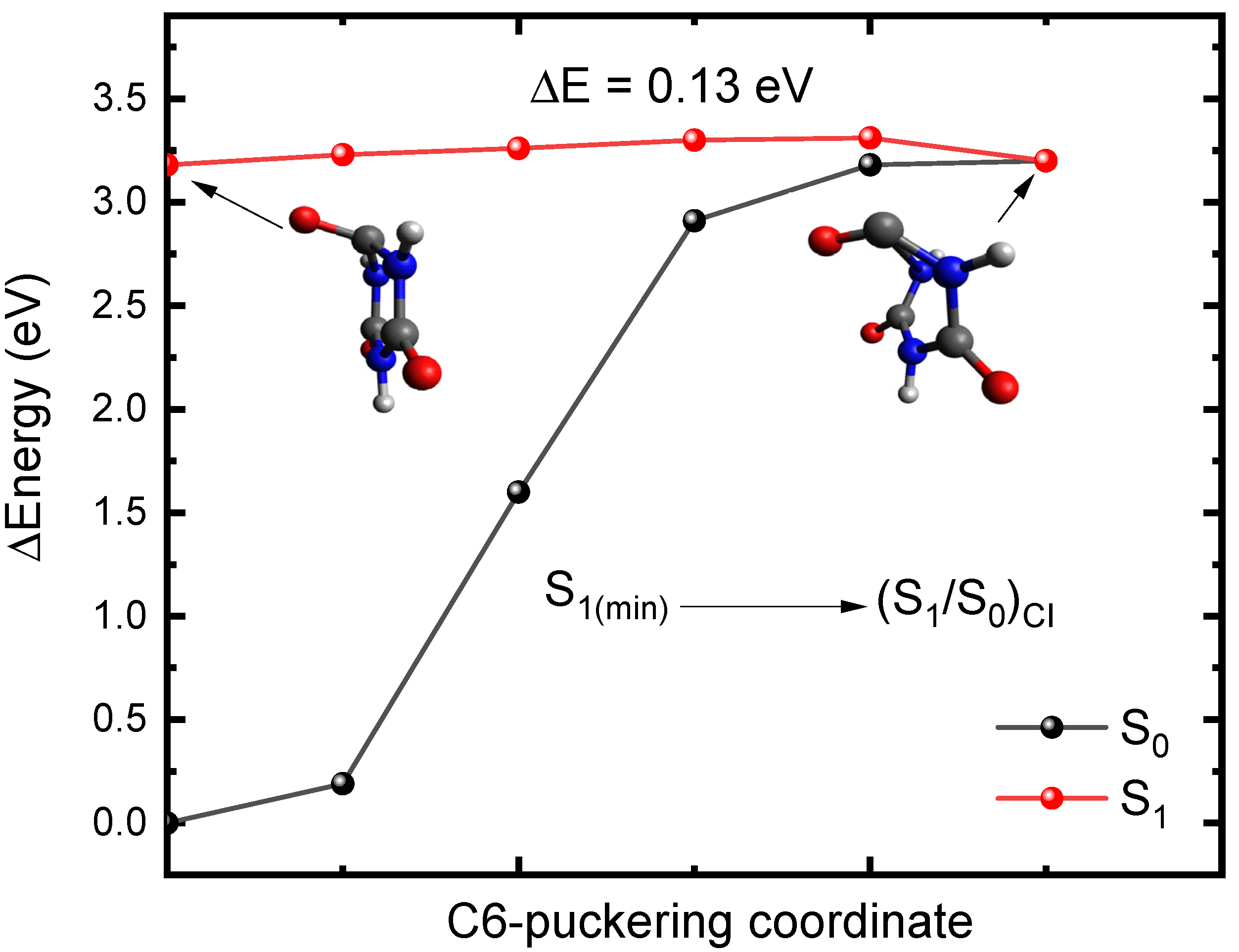
2.2. Plausible Photochemical Deactivation Pathways of CA in Water
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hayatsu, R.; Studier, M.H.; Matsuoka, S.; Anders, E. Origin of organic matter in early solar system—VI. Catalytic synthesis of nitriles, nitrogen bases and porphyrin-like pigments. Geochim. Cosmochim. Acta 1972, 36, 555–571. [Google Scholar] [CrossRef]
- Hayatsu, R.; Studier, M.H.; Moore, L.P.; Anders, E. Purines and triazines in the Murchison meteorite. Geochim. Cosmochim. Acta 1975, 39, 471–488. [Google Scholar] [CrossRef]
- Schaber, P.M.; Colson, J.; Higgins, S.; Thielen, D.; Anspach, B.; Brauer, J. Thermal decomposition (pyrolysis) of urea in an open reaction vessel. Thermochim. Acta 2004, 424, 131–142. [Google Scholar] [CrossRef]
- Hayatsu, R.; Studier, M.H.; Oda, A.; Fuse, K.; Anders, E. Origin of organic matter in early solar system—II. Nitrogen compounds. Geochim. Cosmochim. Acta 1968, 32, 175–190. [Google Scholar] [CrossRef]
- Jeilani, Y.A.; Orlando, T.M.; Pope, A.; Pirim, C.; Nguyen, M.T. Prebiotic synthesis of triazines from urea: A theoretical study of free radical routes to melamine, ammeline, ammelide and cyanuric acid. RSC Adv. 2014, 4, 32375–32382. [Google Scholar] [CrossRef]
- Brister, M.M.; Pollum, M.; Crespo-Hernández, C.E. Photochemical etiology of promising ancestors of the RNA nucleobases. Phys. Chem. Chem. Phys. 2016, 18, 20097–20103. [Google Scholar] [CrossRef]
- Cafferty, B.J.; Hud, N.V. Was a Pyrimidine-Pyrimidine Base Pair the Ancestor of Watson-Crick Base Pairs? Insights from a Systematic Approach to the Origin of RNA. Isr. J. Chem. 2015, 55, 891–905. [Google Scholar] [CrossRef]
- Seto, C.; Whitesides, G. Self-Assembly Based on the Cyanuric Acid-Melamine Lattice1. J. Am. Chem. Soc. 1990, 112, 6409–6411. [Google Scholar] [CrossRef]
- Cafferty, B.J.; Gállego, I.; Chen, M.C.; Farley, K.I.; Eritja, R.; Hud, N.V. Efficient self-assembly in water of long noncovalent polymers by nucleobase analogues. J. Am. Chem. Soc. 2013, 135, 2447–2450. [Google Scholar] [CrossRef]
- Chen, M.C.; Cafferty, B.J.; Mamajanov, I.; Gállego, I.; Khanam, J.; Krishnamurthy, R.; Hud, N.V. Spontaneous prebiotic formation of a β-ribofuranoside that self-assembles with a complementary heterocycle. J. Am. Chem. Soc. 2014, 136, 5640–5646. [Google Scholar] [CrossRef]
- Karunakaran, S.C.; Cafferty, B.J.; Weigert-Muñoz, A.; Schuster, G.B.; Hud, N.V. Spontaneous Symmetry Breaking in the Formation of Supramolecular Polymers: Implications for the Origin of Biological Homochirality. Angew. Chem. Int. Ed. 2019, 58, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Sagan, C. Ultraviolet selection pressure on the earliest organisms. J. Theor. Biol. 1973, 39, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, S.; Sasselov, D.D. Influence of the UV Environment on the Synthesis of Prebiotic Molecules. Astrobiology 2016, 16, 68–88. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, S.; Sasselov, D.D. Constraints on the Early Terrestrial Surface UV Environment Relevant to Prebiotic Chemistry. Astrobiology 2017, 17, 169–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, M.S.; Papineau, D.; Grenne, T.; Slack, J.F.; Rittner, M.; Pirajno, F.; O’Neil, J.; Little, C.T.S. Evidence for early life in Earth’s oldest hydrothermal vent precipitates. Nature 2017, 543, 60–64. [Google Scholar] [CrossRef]
- Cavalazzi, B.; Lemelle, L.; Simionovici, A.; Cady, S.L.; Russell, M.J.; Bailo, E.; Canteri, R.; Enrico, E.; Manceau, A.; Maris, A.; et al. Cellular remains in a ~3.42-billion-year-old subseafloor hydrothermal environment. Sci. Adv. 2021, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Betts, H.C.; Puttick, M.N.; Clark, J.W.; Williams, T.A.; Donoghue, P.C.J.; Pisani, D. Integrated genomic and fossil evidence illuminates life’s early evolution and eukaryote origin. Nat. Ecol. Evol. 2018, 2, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Krissansen-Totton, J.; Arney, G.N.; Catling, D.C. Constraining the climate and ocean pH of the early Earth with a geological carbon cycle model. Proc. Natl. Acad. Sci. USA 2018, 115, 4105–4110. [Google Scholar] [CrossRef] [Green Version]
- Sancier, K.M.; Brady, A.P.; Lee, W.W. Absorption spectra of solutions of cyanuric acid and its chlorinated derivatives. Spectrochim. Acta 1964, 20, 397–403. [Google Scholar] [CrossRef]
- Xu, X.; Goddard, W.A. The X3LYP extended density functional for accurate descriptions of nonbond interactions, spin states, and thermochemical properties. Proc. Natl. Acad. Sci. USA 2004, 101, 2673–2677. [Google Scholar] [CrossRef] [Green Version]
- Rao, L.; Ke, H.; Fu, G.; Xu, X.; Yan, Y. Performance of several density functional theory methods on describing hydrogen-bond interactions. J. Chem. Theory Comput. 2009, 5, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange--correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 84106. [Google Scholar] [CrossRef] [PubMed]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Silva-Junior, M.R.; Schreiber, M.; Sauer, S.P.A.; Thiel, W. Benchmarks for electronically excited states: Time-dependent density functional theory and density functional theory based multireference configuration interaction. J. Chem. Phys. 2008, 129, 104103. [Google Scholar] [CrossRef] [Green Version]
- Winter, N.O.C.; Graf, N.K.; Leutwyler, S.; Hättig, C. Benchmarks for 0--0 transitions of aromatic organic molecules: DFT/B3LYP, ADC (2), CC2, SOS-CC2 and SCS-CC2 compared to high-resolution gas-phase data. Phys. Chem. Chem. Phys. 2013, 15, 6623–6630. [Google Scholar] [CrossRef]
- El-Sayed, M.A. The triplet state: Its radiative and nonradiative properties. Acc. Chem. Res. 1968, 1, 8–16. [Google Scholar] [CrossRef]
- El-Sayed, M.A. Spin-orbit coupling and the radiationless processes in nitrogen heterocyclics. J. Chem. Phys. 1963, 38, 2834–2838. [Google Scholar] [CrossRef]
- Kasha, M. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 1950, 9, 14–19. [Google Scholar] [CrossRef]
- Rankine, C.D. Ultrafast excited-state dynamics of promising nucleobase ancestor 2, 4, 6-triaminopyrimidine. Phys. Chem. Chem. Phys. 2021, 23, 4007–4017. [Google Scholar] [CrossRef]
- Serrano-Andres, L.; Merchán, M. Are the five natural DNA/RNA base monomers a good choice from natural selection? A photochemical perspective. J. Photochem. Photobiol. C Photochem. Rev. 2009, 10, 21–32. [Google Scholar] [CrossRef]
- Giussani, A.; Serra-Martí, J.; Roca-Sanjuán, D.; Merchán, M. Excitation of Nucleobases from a Computational Perspective I: Reaction Paths. Top. Curr. Chem. 2015, 355, 57–98. [Google Scholar]
- Pepino, A.J.; Segarra-Martí, J.; Nenov, A.; Improta, R.; Garavelli, M. Resolving ultrafast photoinduced deactivation in water-solvated pyrimidines nucleosides. J. Phys. Chem. Lett. 2017, 8, 1777–1783. [Google Scholar] [CrossRef]
- Brister, M.M.; Crespo-Hernández, C.E. Excited-state dynamics in the RNA nucleotide uridine 5′-monophosphate investigated using femtosecond broadband transient absorption spectroscopy. J. Phys. Chem. Lett. 2019, 10, 2156–2161. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, S.J.; Krul, S. Crespo-Hernández, Increased Photostability of the Integral mRNA Vaccine Component N1-Methylpseudouridine Compared to Uridine. Chem. A Eur. J. 2021, 28, e202103667. [Google Scholar] [CrossRef]
- Hoehn, S.J.; Caldero-Rodríguez, N.E.; Crespo-Hernández, C.E. Photochemistry of RNA, RNA Monomers and their Plausible Prebiotic Precursors. Photochem. Photobiol. Sci. 2021, 9, 197–226. [Google Scholar]
- Pollum, M.; Martinez-Fernandez, L.; Crespo-Hernandez, C.E. Photochemistry of nucleic acid bases and their thio- and aza-analogues in solution. Top. Curr. Chem. 2015, 47, 245–327. [Google Scholar]
- Matsika, S. Electronic Structure Methods for the Description of Nonadiabatic Effects and Conical Intersections. Chem. Rev. 2021, 121, 9407–9449. [Google Scholar] [CrossRef]
- Krylov, A.I. Size-consistent wave functions for bond-breaking: The equation-of-motion spin-flip model. Chem. Phys. Lett. 2001, 338, 375–384. [Google Scholar] [CrossRef]
- Krylov, A.I. Spin-flip equation-of-motion coupled-cluster electronic structure method for a description of excited states, bond breaking, diradicals, and triradicals. Acc. Chem. Res. 2006, 39, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Slipchenko, L.V.; Krylov, A.I. Singlet-triplet gaps in diradicals by the spin-flip approach: A benchmark study. J. Chem. Phys. 2002, 117, 4694–4708. [Google Scholar] [CrossRef] [Green Version]
- Huix-Rotllant, M.; Natarajan, B.; Ipatov, A.; Wawire, C.M.; Deutsch, T.; Casida, M.E. Assessment of noncollinear spin-flip Tamm-Dancoff approximation time-dependent density-functional theory for the photochemical ring-opening of oxirane. Phys. Chem. Chem. Phys. 2010, 12, 12811–12825. [Google Scholar] [CrossRef] [PubMed]
- Minezawa, N.; Gordon, M.S. Optimizing conical intersections by spin- flip density functional theory: Application to ethylene. J. Phys. Chem. A 2009, 113, 12749–12753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree--Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence ad quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Ásgeirsson, V.; Birgisson, B.O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jónsson, H. Nudged elastic band method for molecular reactions using energy-weighted springs combined with eigenvector following. J. Chem. Theory Comput. 2021, 17, 4929–4945. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Character | Energy (eV) |
|---|---|---|
| S1 | nπ* | 6.34 (0.000) |
| S2 | nπ* | 6.34 (0.000) |
| S3 | ππ* | 6.86 (0.000) |
| S4 | ππ* | 6.91 (0.000) |
| S5 | ππ* | 7.73 (0.80) |
| S6 | ππ* | 7.73 (0.80) |
| T1 | ππ* | 5.74 |
| T2 | ππ* | 5.74 |
| T3 | nπ* | 5.76 |
| T4 | nπ* | 5.76 |
| T5 | ππ* | 5.81 |
| T6 | nπ* | 6.32 |
| T7 | ππ* | 6.49 |
| T8 | ππ* | 6.69 |
| T9 | ππ* | 6.70 |
| Transition | ΔE (eV) | SOCs (cm−1) |
|---|---|---|
| S1,2 → T1,2 | 0.48 | 6.3 |
| S1,2 → T5 | 0.36 | 8.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Rodríguez, L.A.; Hoehn, S.J.; Crespo-Hernández, C.E. On the Photostability of Cyanuric Acid and Its Candidature as a Prebiotic Nucleobase. Molecules 2022, 27, 1184. https://doi.org/10.3390/molecules27041184
Ortiz-Rodríguez LA, Hoehn SJ, Crespo-Hernández CE. On the Photostability of Cyanuric Acid and Its Candidature as a Prebiotic Nucleobase. Molecules. 2022; 27(4):1184. https://doi.org/10.3390/molecules27041184
Chicago/Turabian StyleOrtiz-Rodríguez, Luis A., Sean J. Hoehn, and Carlos E. Crespo-Hernández. 2022. "On the Photostability of Cyanuric Acid and Its Candidature as a Prebiotic Nucleobase" Molecules 27, no. 4: 1184. https://doi.org/10.3390/molecules27041184
APA StyleOrtiz-Rodríguez, L. A., Hoehn, S. J., & Crespo-Hernández, C. E. (2022). On the Photostability of Cyanuric Acid and Its Candidature as a Prebiotic Nucleobase. Molecules, 27(4), 1184. https://doi.org/10.3390/molecules27041184