
Computational Design of Single-Peptide Nanocages with Nanoparticle Templating

 , ,
, ,
Abstract
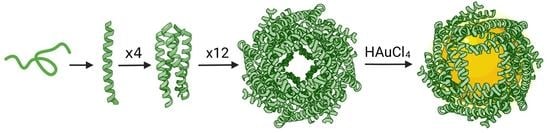
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
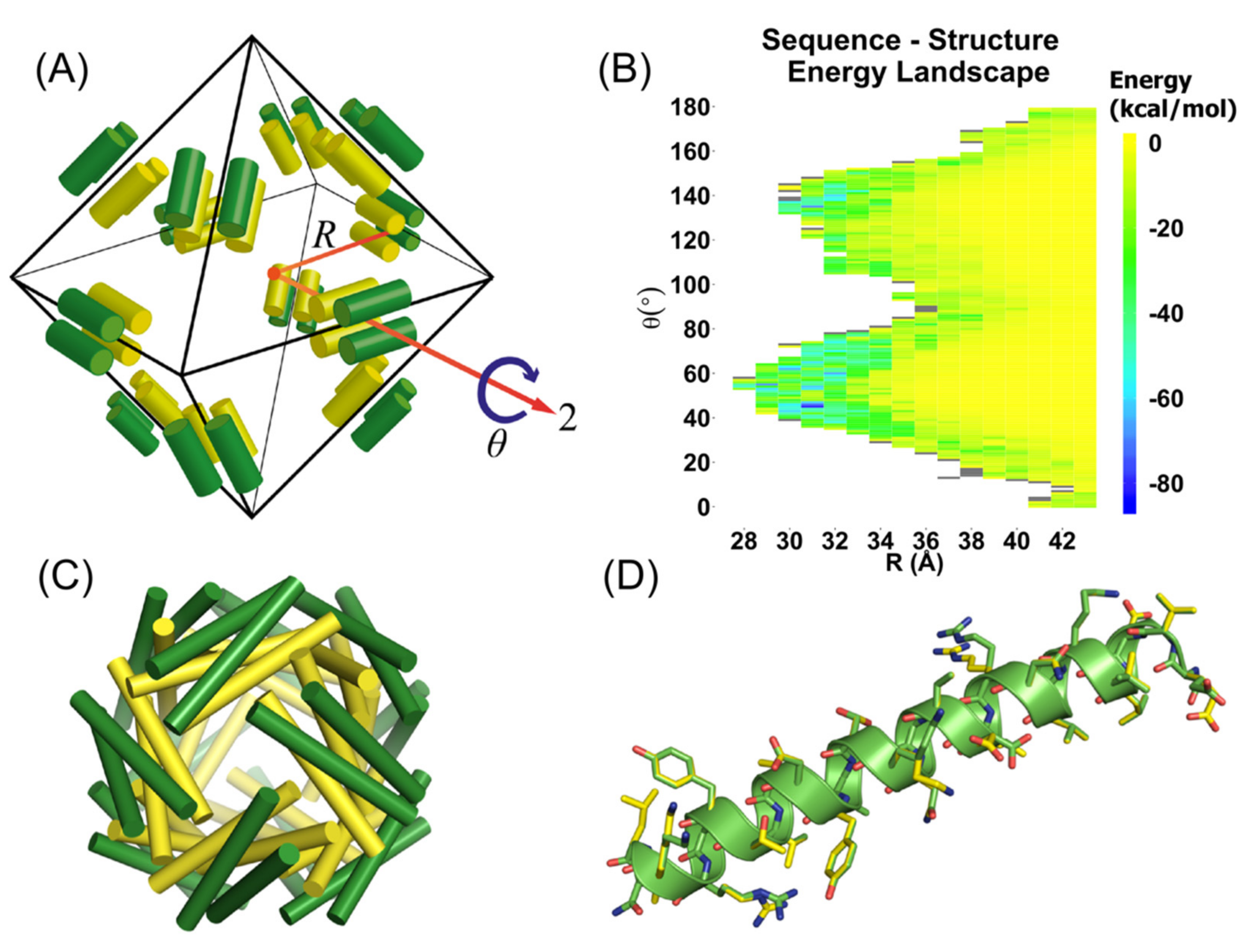
2.1. Computational Design of Self-Assembling Peptides
2.2. Experimental Methods
Peptide Synthesis and Purification
2.3. Nanocage Self-Assembly
2.4. Transmission Electron Microscopy (TEM)
2.5. Dynamic Light Scattering
2.6. Incubation of Gold Inside Peptide Nanocages
3. Results
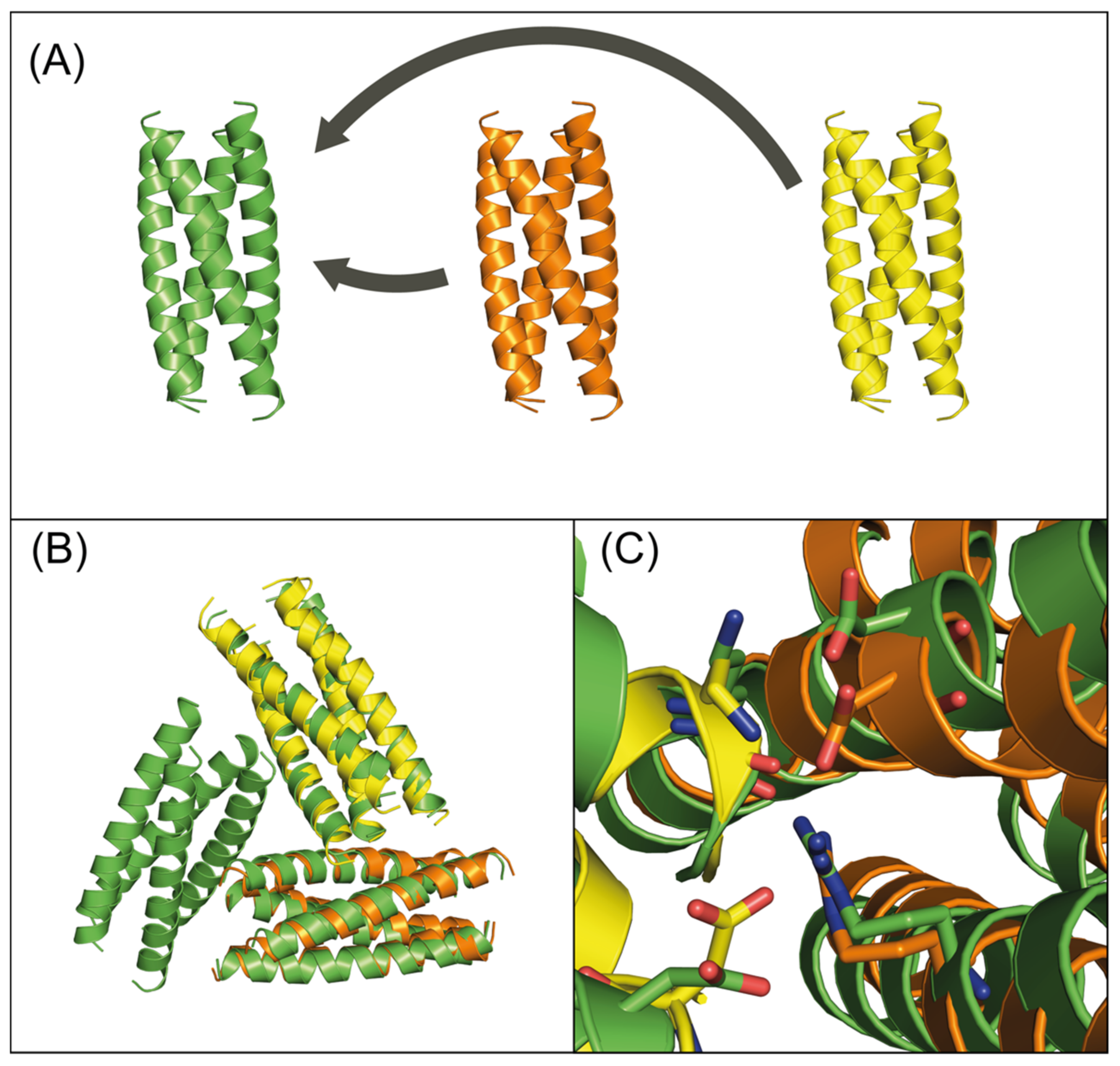
3.1. Computational Design of Peptide Cages
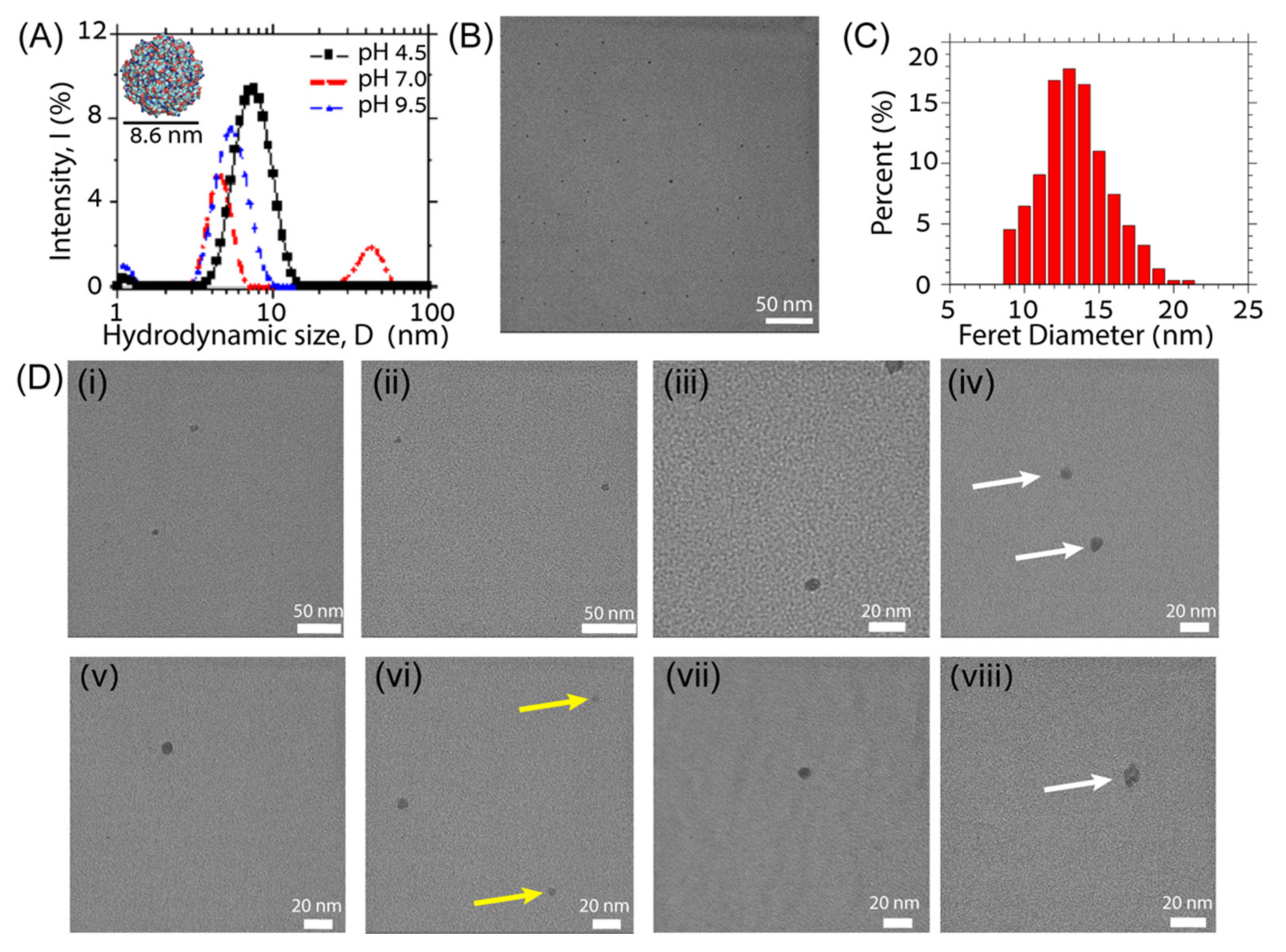
3.2. Experimental Verification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edwardson, T.G.W.; Hilvert, D. Virus-Inspired Function in Engineered Protein Cages. J. Am. Chem. Soc. 2019, 141, 9432–9443. [Google Scholar] [CrossRef] [PubMed]
- Stupka, I.; Heddle, J.G. Artificial Protein Cages—Inspiration, Construction, and Observation. Curr. Opin. Struct. Biol 2020, 64, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Khmelinskaia, A.; Wargacki, A.; King, N.P. Structure-Based Design of Novel Polyhedral Protein Nanomaterials. Curr. Opin. Microbiol. 2021, 61, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Cannon, K.A.; Nguyen, V.N.; Morgan, C.; Yeates, T.O. Design and Characterization of an Icosahedral Protein Cage Formed by a Double-Fusion Protein Containing Three Distinct Symmetry Elements. ACS Synth. Biol. 2020, 9, 517–524. [Google Scholar] [CrossRef]
- Golub, E.; Subramanian, R.H.; Esselborn, J.; Alberstein, R.G.; Bailey, J.B.; Chiong, J.A.; Yan, X.; Booth, T.; Baker, T.S.; Tezcan, F.A. Constructing Protein Polyhedra via Orthogonal Chemical Interactions. Nature 2020, 578, 172–176. [Google Scholar] [CrossRef]
- Laniado, J.; Cannon, K.A.; Miller, J.E.; Sawaya, M.R.; McNamara, D.E.; Yeates, T.O. Geometric Lessons and Design Strategies for Nanoscale Protein Cages. ACS Nano 2021, 15, 4277–4286. [Google Scholar] [CrossRef]
- Douglas, T.; Young, M. Host–guest Encapsulation of Materials by Assembled Virus Protein Cages. Nature 1998, 393, 152. [Google Scholar] [CrossRef]
- Edwardson, T.G.W.; Tetter, S.; Hilvert, D. Two-Tier Supramolecular Encapsulation of Small Molecules in a Protein Cage. Nat. Commun. 2020, 11, 5410. [Google Scholar] [CrossRef]
- Chakraborti, S.; Lin, T.-Y.; Glatt, S.; Heddle, J.G. Enzyme Encapsulation by Protein Cages. RSC Adv. 2020, 10, 13293–13301. [Google Scholar] [CrossRef] [Green Version]
- Douglas, T.; Strable, E.; Willits, D.; Aitouchen, A.; Libera, M.; Young, M. Protein Engineering of a Viral Cage for Constrained Nanomaterials Synthesis. Adv. Mater. 2002, 14, 415–418. [Google Scholar] [CrossRef]
- Yamashita, I.; Hayashi, J.; Hara, M. Bio-Template Synthesis of Uniform CdSe Nanoparticles Using Cage-Shaped Protein, Apoferritin. Chem. Lett. 2004, 33, 1158–1159. [Google Scholar] [CrossRef]
- McConnell, S.A.; Cannon, K.A.; Morgan, C.; McAllister, R.; Amer, B.R.; Clubb, R.T.; Yeates, T.O. Designed Protein Cages as Scaffolds for Building Multienzyme Materials. ACS Synth. Biol. 2020, 9, 381–391. [Google Scholar] [CrossRef]
- Bellomo, E.G.; Wyrsta, M.D.; Pakstis, L.; Pochan, D.J.; Deming, T.J. Stimuli-Responsive Polypeptide Vesicles by Conformation-Specific Assembly. Nat. Mater. 2004, 3, 244–248. [Google Scholar] [CrossRef]
- Choi, S.-H.; Choi, K.; Chan Kwon, I.; Ahn, H.J. The Incorporation of GALA Peptide into a Protein Cage for an Acid-Inducible Molecular Switch. Biomaterials 2010, 31, 5191–5198. [Google Scholar] [CrossRef]
- Szyszka, T.N.; Jenner, E.N.; Tasneem, N.; Lau, Y.H. Molecular Display on Protein Nanocompartments: Design Strategies and Systems Applications. ChemSystemsChem 2021, 4, 202100025. [Google Scholar] [CrossRef]
- King, N.P.; Bale, J.B.; Sheffler, W.; McNamara, D.E.; Gonen, S.; Gonen, T.; Yeates, T.O.; Baker, D. Accurate Design of Co-Assembling Multi-Component Protein Nanomaterials. Nature 2014, 510, 103–108. [Google Scholar] [CrossRef]
- Hsia, Y.; Bale, J.B.; Gonen, S.; Shi, D.; Sheffler, W.; Fong, K.K.; Nattermann, U.; Xu, C.; Huang, P.-S.; Ravichandran, R.; et al. Design of a Hyperstable 60-Subunit Protein Dodecahedron. [corrected]. Nature 2016, 535, 136–139. [Google Scholar] [CrossRef]
- Lanci, C.J.; MacDermaid, C.M.; Kang, S.-G.; Acharya, R.; North, B.; Yang, X.; Qiu, X.J.; DeGrado, W.F.; Saven, J.G. Computational Design of a Protein Crystal. Proc. Natl. Acad. Sci. USA 2012, 109, 7304–7309. [Google Scholar] [CrossRef] [Green Version]
- King, N.P.; Sheffler, W.; Sawaya, M.R.; Vollmar, B.S.; Sumida, J.P.; André, I.; Gonen, T.; Yeates, T.O.; Baker, D. Computational Design of Self-Assembling Protein Nanomaterials with Atomic Level Accuracy. Science 2012, 336, 1171–1174. [Google Scholar] [CrossRef] [Green Version]
- Yeates, T.O. Geometric Principles for Designing Highly Symmetric Self-Assembling Protein Nanomaterials. Annu. Rev. Biophys. 2017, 46, 23–42. [Google Scholar] [CrossRef]
- Mosayebi, M.; Shoemark, D.K.; Fletcher, J.M.; Sessions, R.B.; Linden, N.; Woolfson, D.N.; Liverpool, T.B. Beyond Icosahedral Symmetry in Packings of Proteins in Spherical Shells. Proc. Natl. Acad. Sci. USA 2017, 114, 9014–9019. [Google Scholar] [CrossRef] [Green Version]
- Bale, J.B.; Gonen, S.; Liu, Y.; Sheffler, W.; Ellis, D.; Thomas, C.; Cascio, D.; Yeates, T.O.; Gonen, T.; King, N.P.; et al. Accurate Design of Megadalton-Scale Two-Component Icosahedral Protein Complexes. Science 2016, 353, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.V.; Polzer, F.; Haider, M.J.; Tian, Y.; Villegas, J.A.; Kiick, K.L.; Pochan, D.J.; Saven, J.G. Computationally Designed Peptides for Self-Assembly of Nanostructured Lattices. Sci. Adv. 2016, 2, e1600307. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.-T.; Lai, Y.; Cascio, D.; Yeates, T.O. Structure of a 16-Nm Cage Designed by Using Protein Oligomers. Science 2012, 336, 1129. [Google Scholar] [CrossRef]
- Sinha, N.J.; Langenstein, M.G.; Pochan, D.J.; Kloxin, C.J.; Saven, J.G. Peptide Design and Self-Assembly into Targeted Nanostructure and Functional Materials. Chem. Rev. 2021, 121, 13915–13935. [Google Scholar] [CrossRef]
- Der, B.S.; Kuhlman, B. Cages from Coils. Nat. Biotechnol. 2013, 31, 809–810. [Google Scholar] [CrossRef]
- Beesley, J.L.; Woolfson, D.N. The de Novo Design of α-Helical Peptides for Supramolecular Self-Assembly. Curr. Opin. Biotechnol. 2019, 58, 175–182. [Google Scholar] [CrossRef]
- Gradišar, H.; Božič, S.; Doles, T.; Vengust, D.; Hafner-Bratkovič, I.; Mertelj, A.; Webb, B.; Šali, A.; Klavžar, S.; Jerala, R. Design of a Single-Chain Polypeptide Tetrahedron Assembled from Coiled-Coil Segments. Nat. Chem. Biol. 2013, 9, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Lapenta, F.; Aupič, J.; Strmšek, Ž.; Jerala, R. Coiled Coil Protein Origami: From Modular Design Principles towards Biotechnological Applications. Chem. Soc. Rev. 2018, 47, 3530–3542. [Google Scholar] [CrossRef] [Green Version]
- Lapenta, F.; Aupič, J.; Vezzoli, M.; Strmšek, Ž.; Da Vela, S.; Svergun, D.I.; Carazo, J.M.; Melero, R.; Jerala, R. Self-Assembly and Regulation of Protein Cages from Pre-Organised Coiled-Coil Modules. Nat. Commun. 2021, 12, 939. [Google Scholar] [CrossRef]
- Doll, T.A.P.F.; Tais, A.P.; Dey, R.; Burkhard, P. Design and Optimization of Peptide Nanoparticles. J. Nanobiotechnol. 2015, 13, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indelicato, G.; Wahome, N.; Ringler, P.; Müller, S.A.; Nieh, M.-P.; Burkhard, P.; Twarock, R. Principles Governing the Self-Assembly of Coiled-Coil Protein Nanoparticles. Biophys. J. 2016, 110, 646–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, J.M.; Harniman, R.L.; Barnes, F.R.H.; Boyle, A.L.; Collins, A.; Mantell, J.; Sharp, T.H.; Antognozzi, M.; Booth, P.J.; Linden, N.; et al. Self-Assembling Cages from Coiled-Coil Peptide Modules. Science 2013, 340, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Laniado, J.; Yeates, T.O. A Complete Rule Set for Designing Symmetry Combination Materials from Protein Molecules. Proc. Natl. Acad. Sci. USA 2020, 117, 31817–31823. [Google Scholar] [CrossRef]
- Levy, E.D.; Boeri Erba, E.; Robinson, C.V.; Teichmann, S.A. Assembly Reflects Evolution of Protein Complexes. Nature 2008, 453, 1262–1265. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, J.M.; Nguyen, V.N.; Nie, M.; Sawaya, M.R.; Bobik, T.A.; Yeates, T.O. Symmetry Breaking and Structural Polymorphism in a Bacterial Microcompartment Shell Protein for Choline Utilization. Protein Sci. 2020, 29, 2201–2212. [Google Scholar] [CrossRef]
- Rhys, G.G.; Wood, C.W.; Lang, E.J.M.; Mulholland, A.J.; Brady, R.L.; Thomson, A.R.; Woolfson, D.N. Maintaining and Breaking Symmetry in Homomeric Coiled-Coil Assemblies. Nat. Commun. 2018, 9, 4132. [Google Scholar] [CrossRef] [Green Version]
- Dawson, W.M.; Lang, E.J.M.; Rhys, G.G.; Shelley, K.L.; Williams, C.; Brady, R.L.; Crump, M.P.; Mulholland, A.J.; Woolfson, D.N. Structural Resolution of Switchable States of a de Novo Peptide Assembly. Nat. Commun. 2021, 12, 1530. [Google Scholar] [CrossRef]
- Sasaki, E.; Böhringer, D.; van de Waterbeemd, M.; Leibundgut, M.; Zschoche, R.; Heck, A.J.R.; Ban, N.; Hilvert, D. Structure and Assembly of Scalable Porous Protein Cages. Nat. Commun. 2017, 8, 14663. [Google Scholar] [CrossRef] [Green Version]
- Terasaka, N.; Azuma, Y.; Hilvert, D. Laboratory Evolution of Virus-like Nucleocapsids from Nonviral Protein Cages. Proc. Natl. Acad. Sci. USA 2018, 115, 5432–5437. [Google Scholar] [CrossRef] [Green Version]
- Pavone, V.; Zhang, S.-Q.; Merlino, A.; Lombardi, A.; Wu, Y.; DeGrado, W.F. Crystal Structure of an Amphiphilic Foldamer Reveals a 48-Mer Assembly Comprising a Hollow Truncated Octahedron. Nat. Commun. 2014, 5, 3581. [Google Scholar] [CrossRef] [Green Version]
- Pappas, C.G.; Mandal, P.K.; Liu, B.; Kauffmann, B.; Miao, X.; Komáromy, D.; Hoffmann, W.; Manz, C.; Chang, R.; Liu, K.; et al. Emergence of Low-Symmetry Foldamers from Single Monomers. Nat. Chem. 2020, 12, 1180–1186. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, L.-P.; Zheng, Y.; Wang, K.; Song, B.; Yan, X.; Wojtas, L.; Wang, X.-Q.; Jiang, X.; Wang, M.; et al. Double-layered Supramolecular Prisms Self-assembled by Geometrically Non-equivalent Tetratopic Subunits. Angew. Chem. Weinheim Bergstr. Ger. 2021, 133, 1318–1325. [Google Scholar] [CrossRef]
- Deng, Y.; Liu, J.; Zheng, Q.; Eliezer, D.; Kallenbach, N.R.; Lu, M. Antiparallel Four-Stranded Coiled Coil Specified by a 3-3-1 Hydrophobic Heptad Repeat. Structure 2006, 14, 247–255. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System. Available online: http://www.pymol.org (accessed on 9 February 2022).
- Hahn, T. (Ed.) International Tables for Crystallography. Volume A: Space-Group Symmetry; Springer: Berlin, Germany, 2005. [Google Scholar]
- Swift, J.; Wehbi, W.A.; Kelly, B.D.; Stowell, X.F.; Saven, J.G.; Dmochowski, I.J. Design of Functional Ferritin-like Proteins with Hydrophobic Cavities. J. Am. Chem. Soc. 2006, 128, 6611–6619. [Google Scholar] [CrossRef]
- Calhoun, J.R.; Kono, H.; Lahr, S.; Wang, W.; DeGrado, W.F.; Saven, J.G. Computational Design and Characterization of a Monomeric Helical Dinuclear Metalloprotein. J. Mol. Biol. 2003, 334, 1101–1115. [Google Scholar] [CrossRef]
- Pulsipher, K.W.; Villegas, J.A.; Roose, B.W.; Hicks, T.L.; Yoon, J.; Saven, J.G.; Dmochowski, I.J. Thermophilic Ferritin 24mer Assembly and Nanoparticle Encapsulation Modulated by Interdimer Electrostatic Repulsion. Biochemistry 2017, 56, 3596–3606. [Google Scholar] [CrossRef]
- Zou, J.; Saven, J.G. Statistical Theory of Combinatorial Libraries of Folding Proteins: Energetic Discrimination of a Target Structure. J. Mol. Biol. 2000, 296, 281–294. [Google Scholar] [CrossRef]
- Kono, H.; Saven, J.G. Statistical Theory for Protein Combinatorial Libraries. Packing Interactions, Backbone Flexibility, and the Sequence Variability of a Main-Chain Structure. J. Mol. Biol. 2001, 306, 607–628. [Google Scholar] [CrossRef]
- Fu, X.; Kono, H.; Saven, J.G. Probabilistic Approach to the Design of Symmetric Protein Quaternary Structures. Protein Eng. 2003, 16, 971–977. [Google Scholar] [CrossRef] [Green Version]
- Dunbrack, R.L., Jr. Rotamer Libraries in the 21st Century. Curr. Opin. Struct. Biol. 2002, 12, 431–440. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: A Fully Automated Algorithm for Protein-Protein Docking. Nucleic Acids Res. 2004, 32, W96–W99. [Google Scholar] [CrossRef] [Green Version]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: An Automated Docking and Discrimination Method for the Prediction of Protein Complexes. Bioinformatics 2004, 20, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-Based Protein Docking Program with Pairwise Potentials. Proteins Struct. Funct. Bioinform. 2006, 65, 392–406. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How Good Is Automated Protein Docking? Proteins Struct. Funct. Bioinform. 2013, 81, 2159–2166. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein-Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Sinha, N.J.; Guo, R.; Misra, R.; Fagan, J.; Faraone, A.; Kloxin, C.J.; Saven, J.G.; Jensen, G.V.; Pochan, D.J. Colloid-like Solution Behavior of Computationally Designed Coiled Coil Bundlemers. J. Colloid Interface Sci. 2022, 606, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Swift, J.; Butts, C.A.; Yerubandi, V.; Dmochowski, I.J. Structure and Activity of Apoferritin-Stabilized Gold Nanoparticles. J. Inorg. Biochem. 2007, 101, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.J.; Wu, D.; Kloxin, C.J.; Saven, J.G.; Jensen, G.V.; Pochan, D.J. Polyelectrolyte Character of Rigid Rod Peptide Bundlemer Chains Constructed via Hierarchical Self-Assembly. Soft Matter 2019, 15, 9858–9870. [Google Scholar] [CrossRef]
- Adolph, K.W.; Butler, P.J. Studies on the Assembly of a Spherical Plant Virus. I. States of Aggregation of the Isolated Protein. J. Mol. Biol. 1974, 88, 327–341. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Reddy, V.S.; Brooks, C.L., III. Deciphering the kinetic mechanism of spontaneous self-assembly of icosahedral capsids. Nano Lett. 2007, 7, 338. [Google Scholar] [CrossRef]
- Cannon, K.A.; Park, R.U.; Boyken, S.E.; Nattermann, U.; Yi, S.; Baker, D.; King, N.P.; Yeates, T.O. Design and Structure of Two New Protein Cages Illustrate Successes and Ongoing Challenges in Protein Engineering. Protein Sci. 2020, 29, 919–929. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villegas, J.A.; Sinha, N.J.; Teramoto, N.; Von Bargen, C.D.; Pochan, D.J.; Saven, J.G. Computational Design of Single-Peptide Nanocages with Nanoparticle Templating. Molecules 2022, 27, 1237. https://doi.org/10.3390/molecules27041237
Villegas JA, Sinha NJ, Teramoto N, Von Bargen CD, Pochan DJ, Saven JG. Computational Design of Single-Peptide Nanocages with Nanoparticle Templating. Molecules. 2022; 27(4):1237. https://doi.org/10.3390/molecules27041237
Chicago/Turabian StyleVillegas, José A., Nairiti J. Sinha, Naozumi Teramoto, Christopher D. Von Bargen, Darrin J. Pochan, and Jeffery G. Saven. 2022. "Computational Design of Single-Peptide Nanocages with Nanoparticle Templating" Molecules 27, no. 4: 1237. https://doi.org/10.3390/molecules27041237
APA StyleVillegas, J. A., Sinha, N. J., Teramoto, N., Von Bargen, C. D., Pochan, D. J., & Saven, J. G. (2022). Computational Design of Single-Peptide Nanocages with Nanoparticle Templating. Molecules, 27(4), 1237. https://doi.org/10.3390/molecules27041237