
Tuning Photophysical Properties by p-Functional Groups in Zn(II) and Cd(II) Complexes with Piperonylic Acid

 ,
,  , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
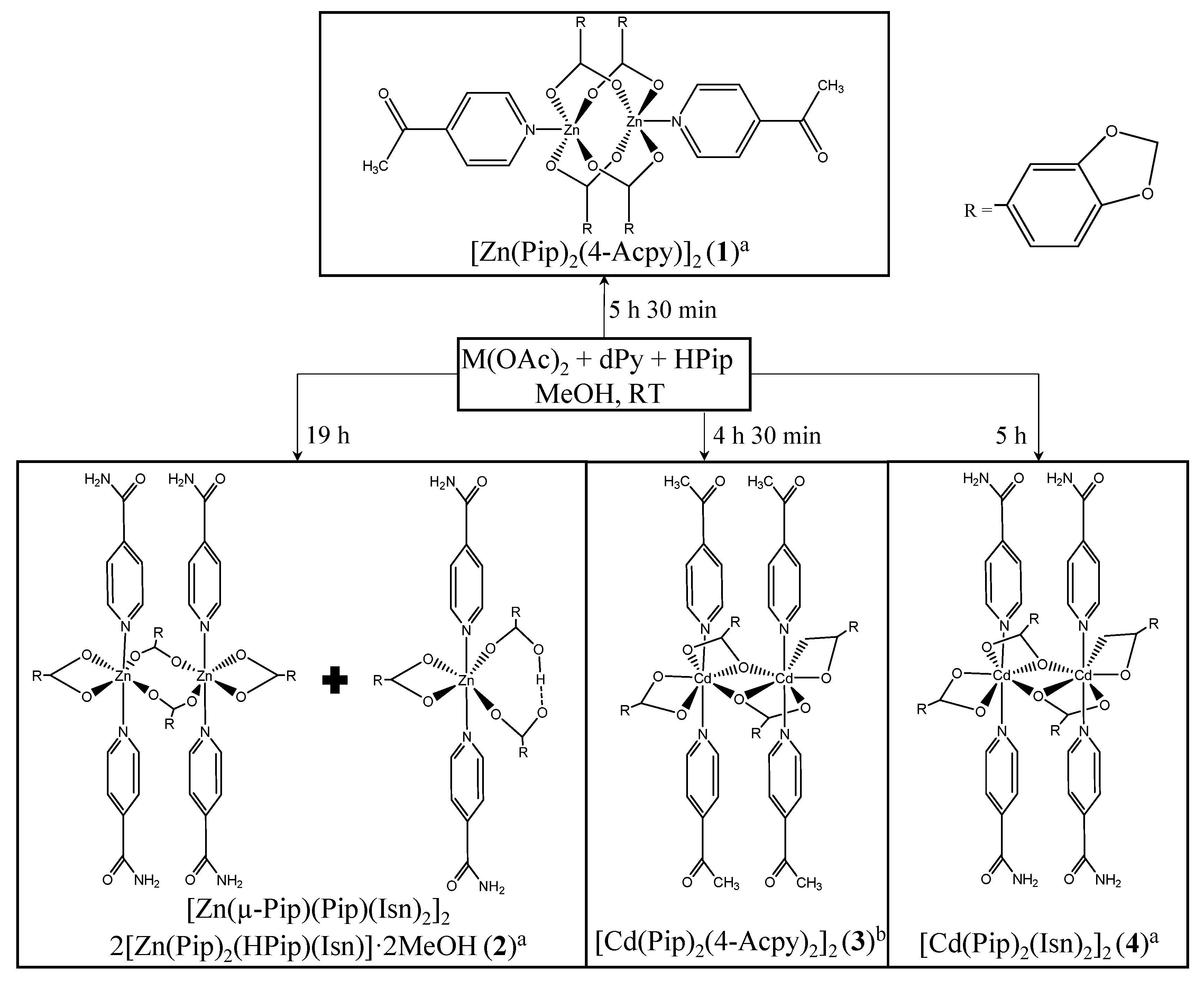
2.1. Synthesis and General Characterization
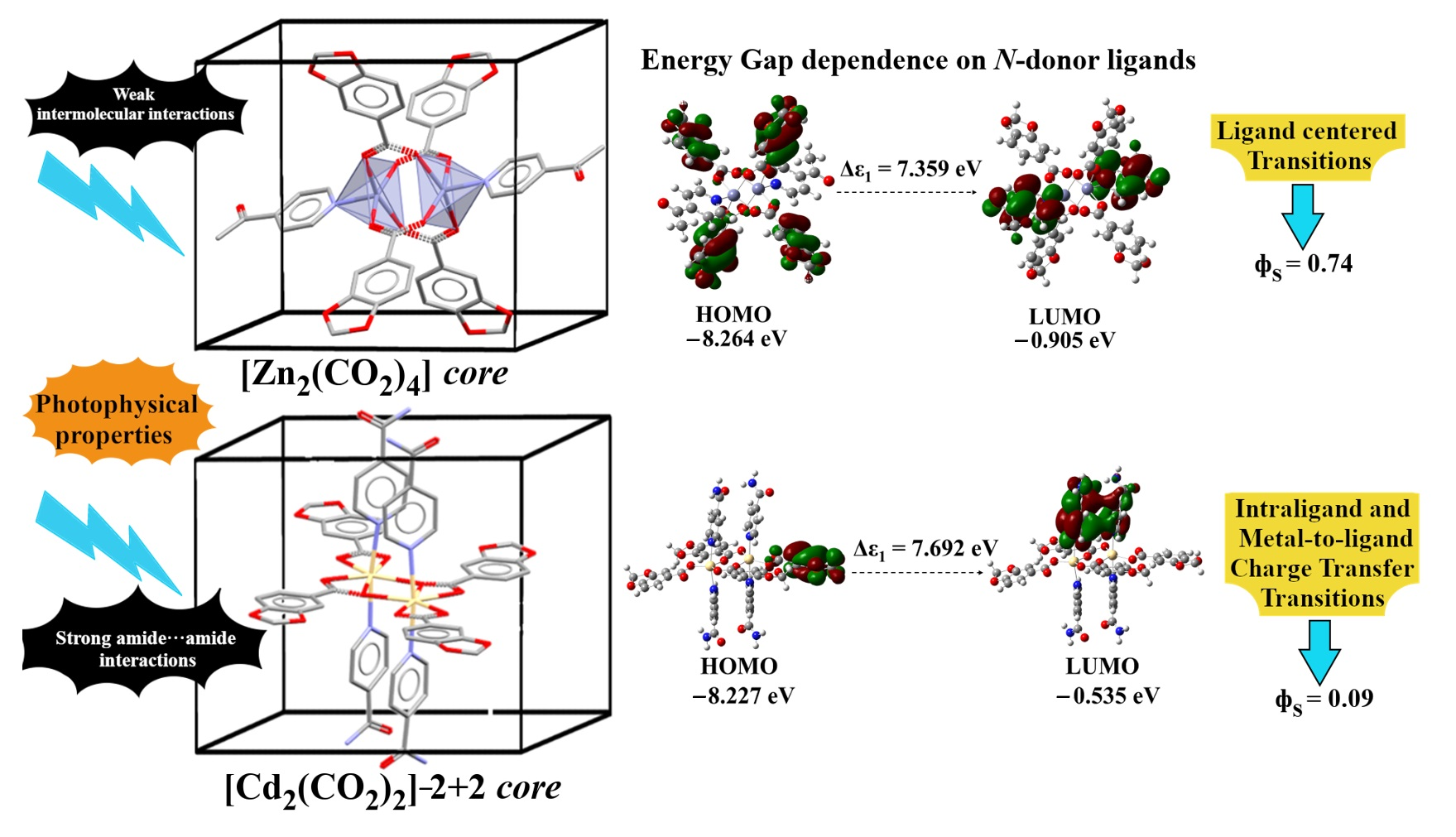
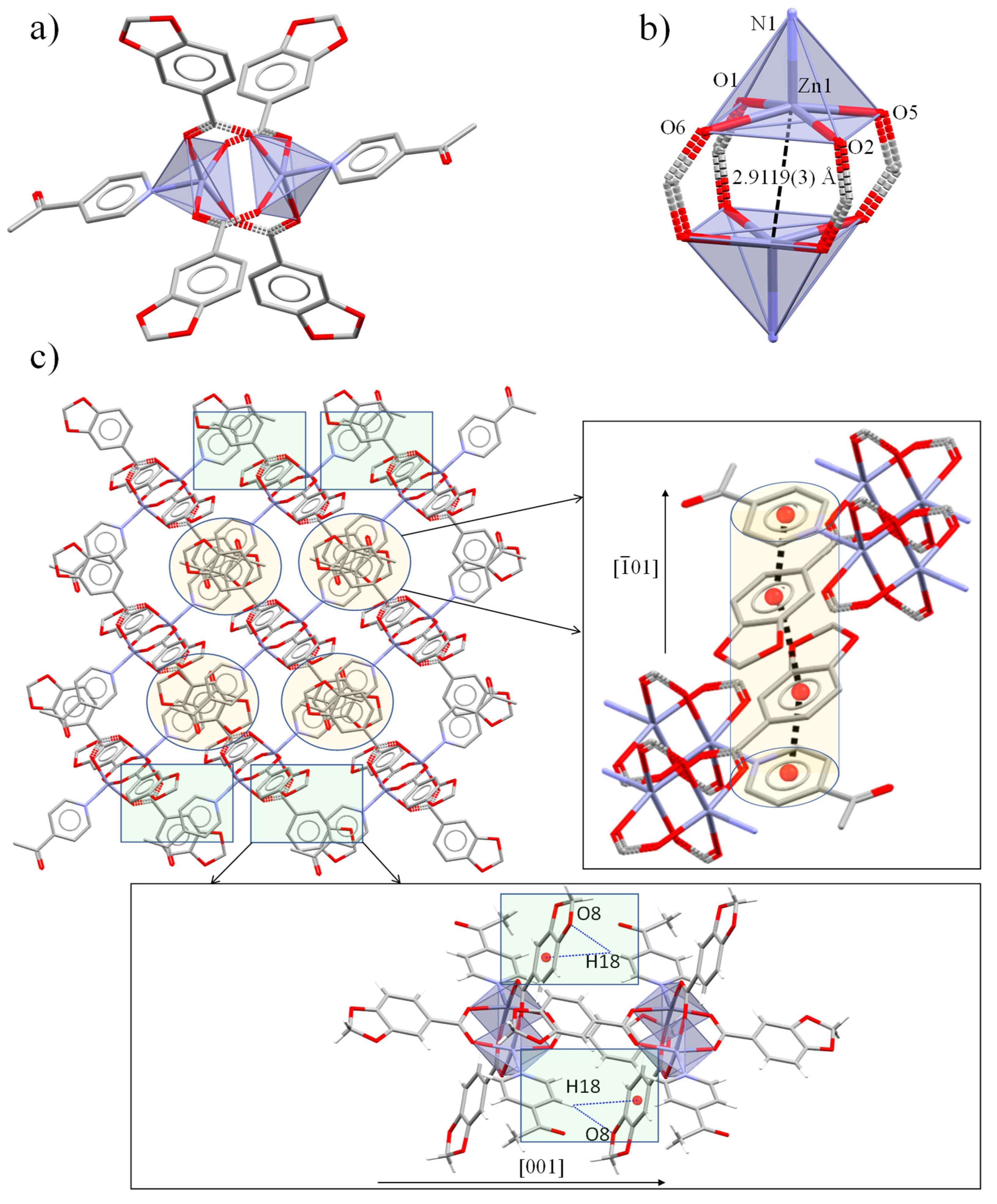
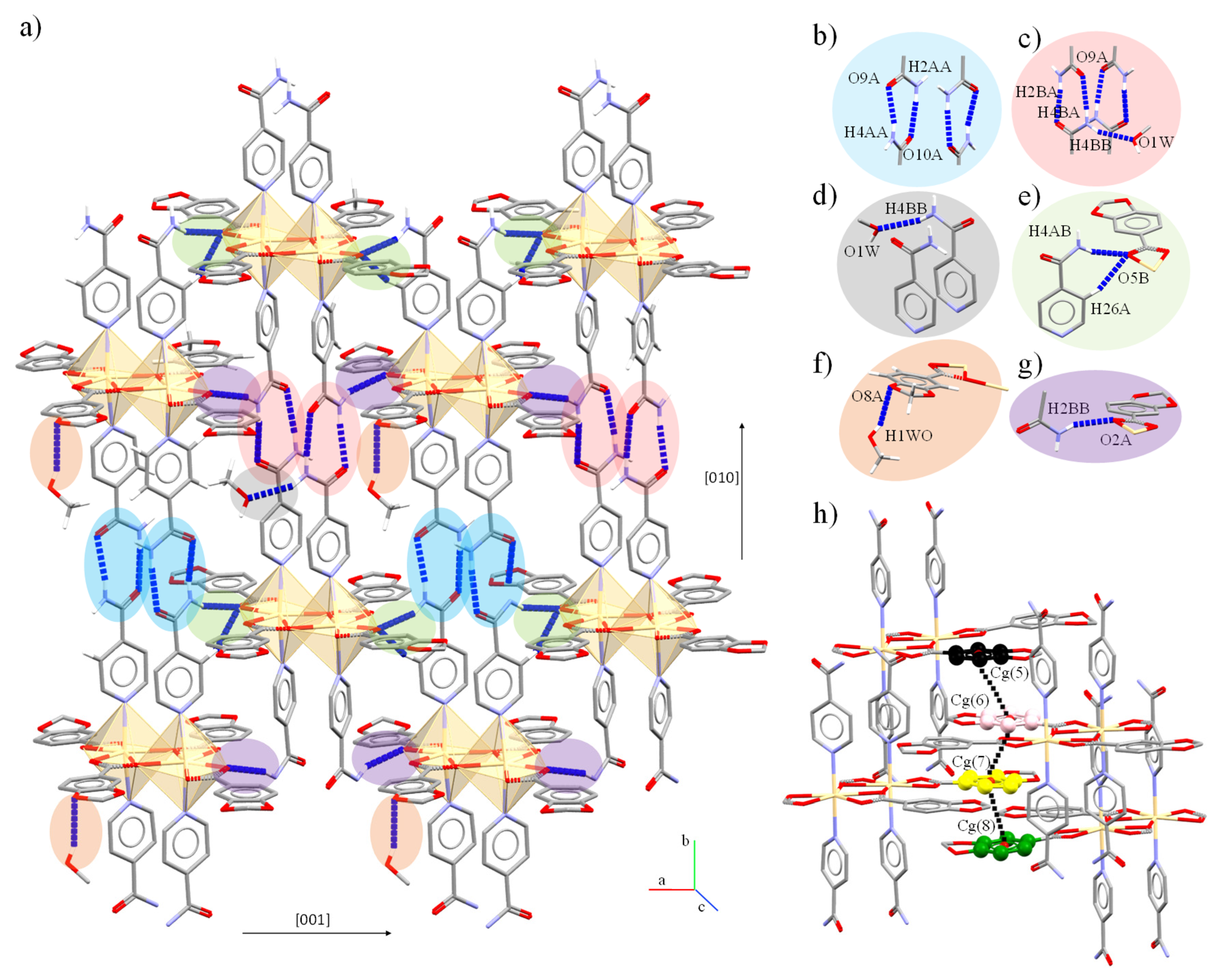
2.2. Crystal and Extended Structure of Zn(II) Complexes
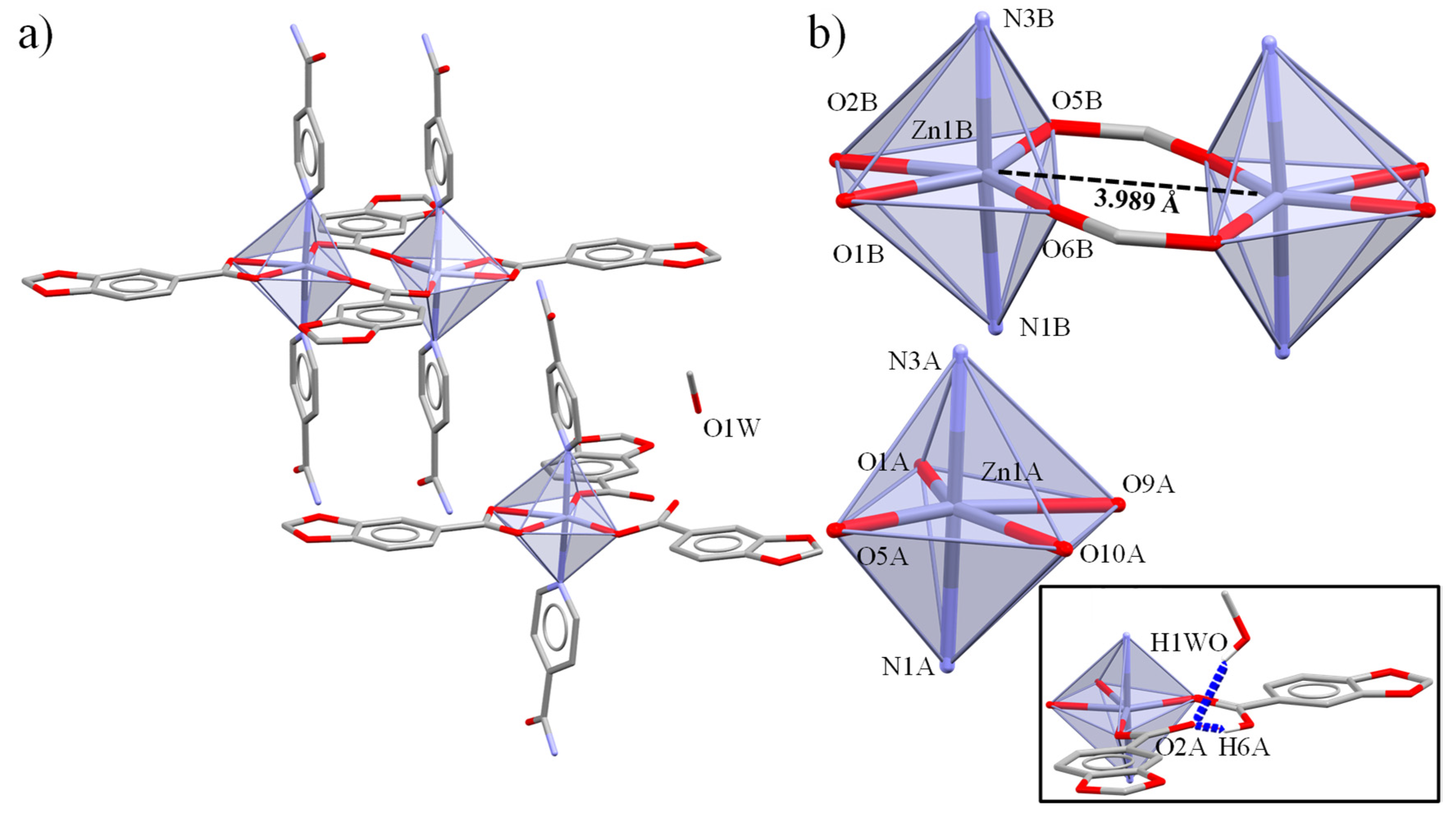
2.3. Crystal and Extended Structure of Cd(II) Compounds
2.4. Structure and Geometric Evaluation
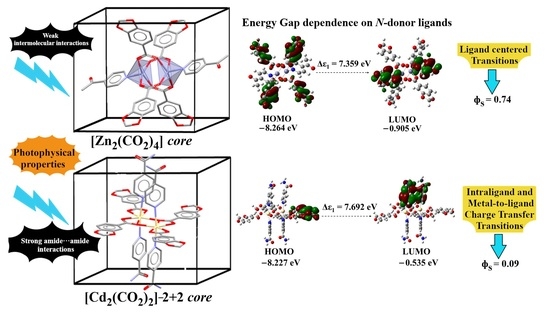
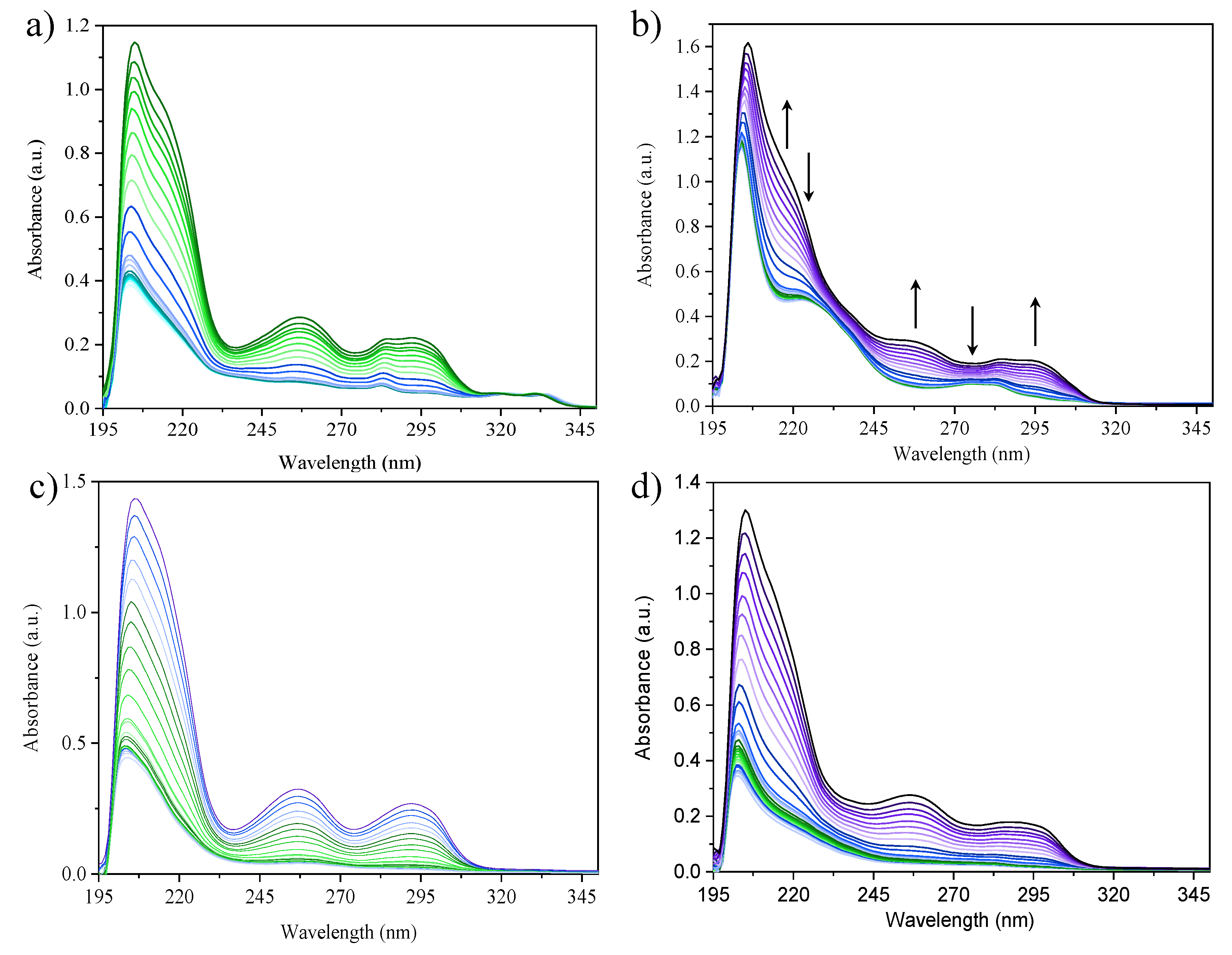
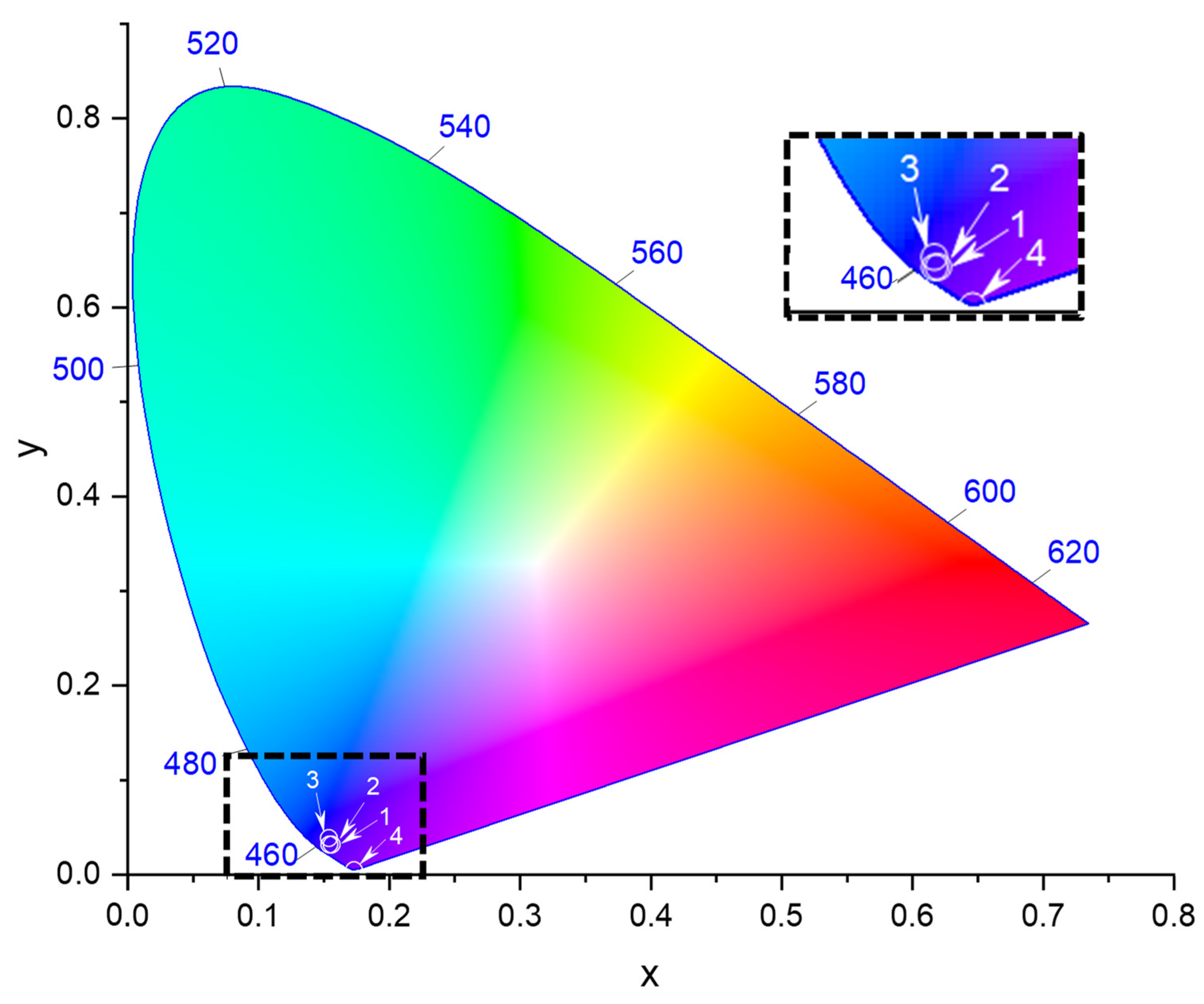
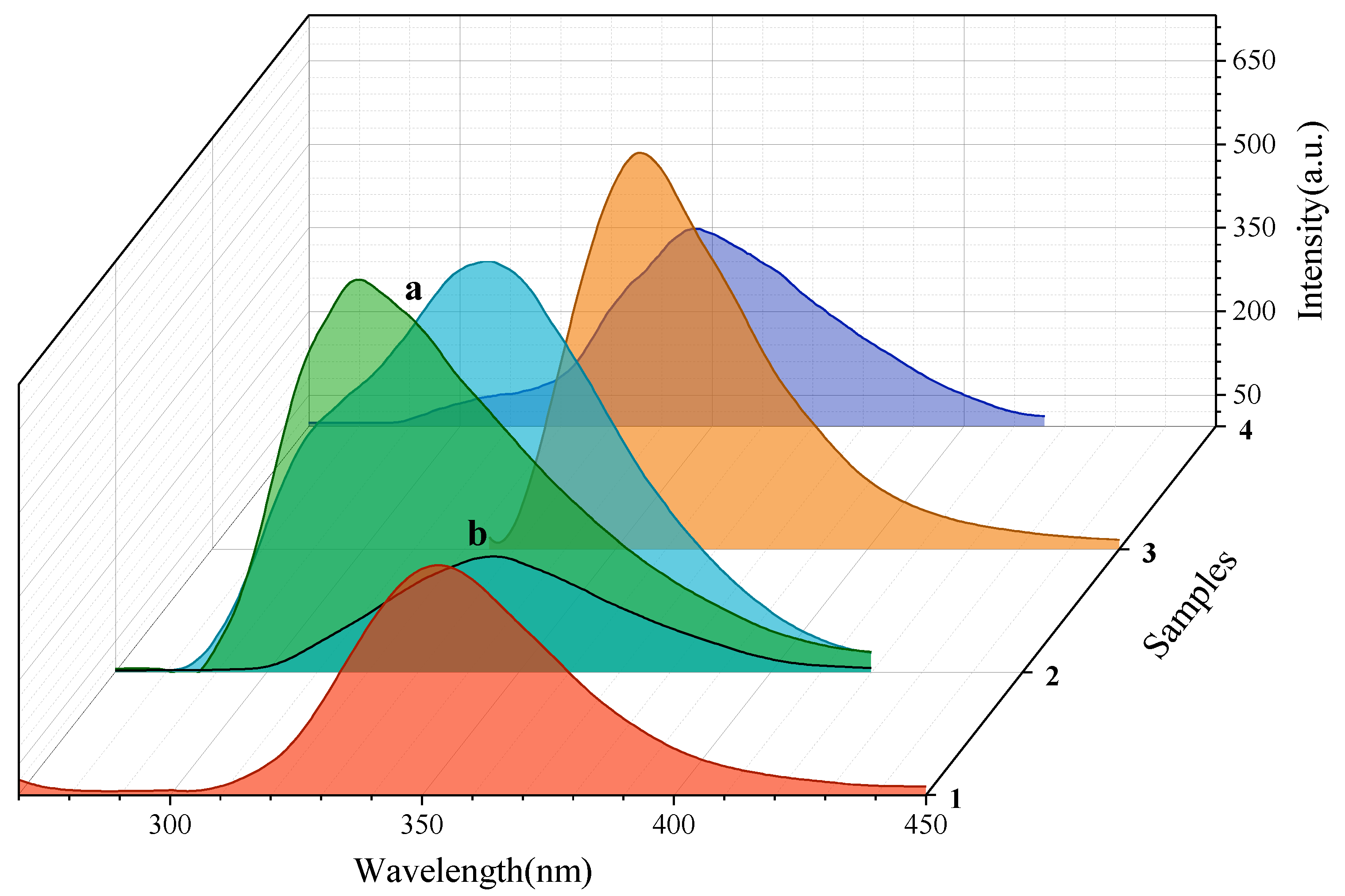
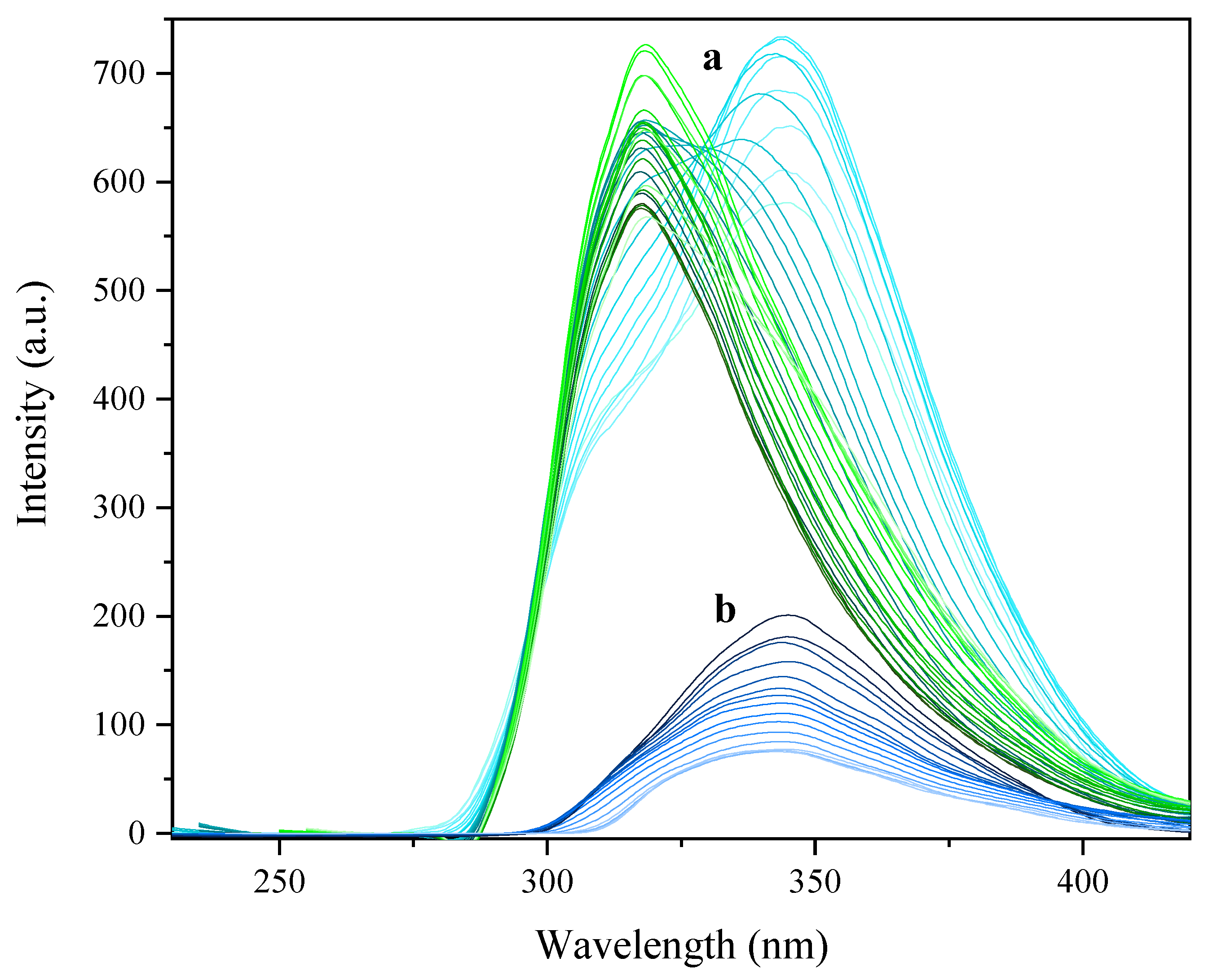
2.5. Photophysical Properties
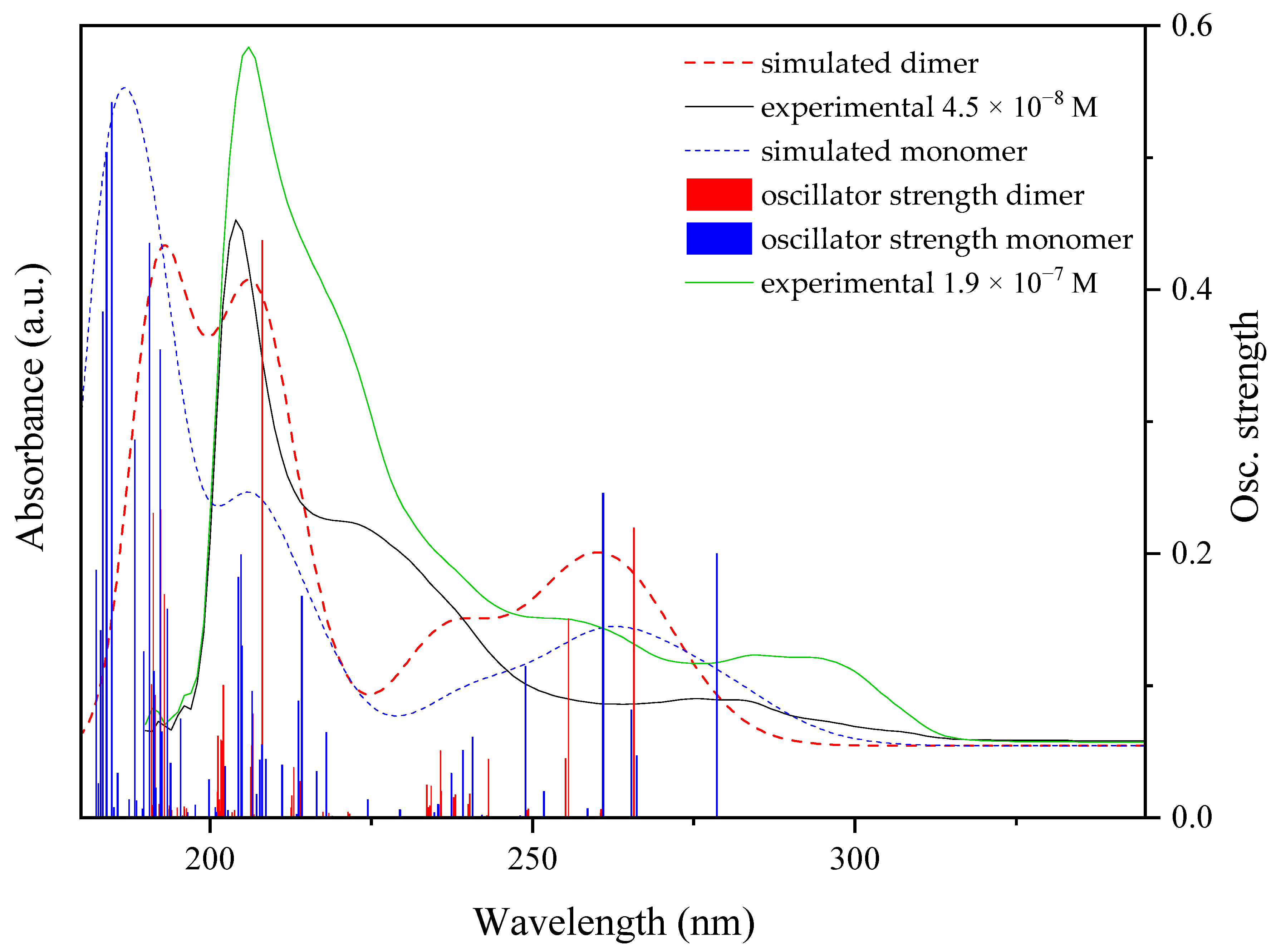
2.6. Electronic Calculations
3. Conclusions
4. Experimental
4.1. Materials and General Details
4.2. Synthesis of Complexes 1–4
4.3. X-ray Crystallography
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gusev, A.; Shul, V.; Braga, E.; Zamnius, E.; Kryukova, M.; Linert, W. Luminescent Properties of Zn Complexes Based on Tetradentate N2O2-Donor Pyrazolone Schiff Bases. Dyes Pigment. 2020, 183, 108626. [Google Scholar] [CrossRef]
- Lu, C.; Xu, Z.; Cui, J. Ratiometric and Highly Selective Fluorescent Sensor for Cadmium under Physiological PH Range: A New Strategy to Discriminate Cadmium from Zinc. J. Org. Chem. 2007, 72, 3554–3557. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Das, M.; Pal, K.; Jana, S.; Dutta, B.; Ray, P.P.; Jana, K.; Sinha, C. Three-Dimensional-Coordination Polymer of Zn(II)-Carboxylate: Structural Elucidation, Photoelectrical Conductivity, and Biological Activity. ACS Omega 2019, 4, 17649–17661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, B.; Dey, A.; Sinha, C.; Pratim Ray, P.; Mir, M.H. Photochemical Structural Transformation of a Linear 1D Coordination Polymer Impacts the Electrical Conductivity. Inorg. Chem. 2018, 57, 8029–8032. [Google Scholar] [CrossRef]
- Wu, S.; Zheng, Y.; Liu, Q.; Li, R.; Peng, T. Low Cost and Solution-Processable Zinc Phthalocyanine as Alternative Hole Transport Material for Perovskite Solar Cells. RSC Adv. 2016, 6, 107723–107731. [Google Scholar] [CrossRef]
- Kumar, G.; Kumar, G.; Gupta, R. Effect of Pyridyl Donors from Organic Ligands: Versus Metalloligands on Material Design. Inorg. Chem. Front. 2021, 8, 1334–1373. [Google Scholar] [CrossRef]
- Hancock, R.D. The Pyridyl Group in Ligand Design for Selective Metal Ion Complexation and Sensing. Chem. Soc. Rev. 2013, 42, 1500–1524. [Google Scholar] [CrossRef]
- Barbieri, A.; Accorsi, G.; Armaroli, N. Luminescent Complexes beyond the Platinum Group: The D10 Avenue. Chem. Commun. 2008, 19, 2185–2193. [Google Scholar] [CrossRef]
- Kokina, T.E.; Rakhmanova, M.I.; Shekhovtsov, N.A.; Glinskaya, L.A.; Komarov, V.Y.; Agafontsev, A.M.; Baranov, A.Y.; Plyusnin, P.E.; Sheludyakova, L.A.; Tkachev, A.V.; et al. Luminescent Zn(Ii) and Cd(Ii) Complexes with Chiral 2,2′-Bipyridine Ligands Bearing Natural Monoterpene Groups: Synthesis, Speciation in Solution and Photophysics. Dalton Trans. 2020, 49, 7552–7563. [Google Scholar] [CrossRef]
- Dumur, F.; Contal, E.; Wantz, G.; Gigmes, D. Photoluminescence of Zinc Complexes: Easily Tunable Optical Properties by Variation of the Bridge Between the Imido Groups of Schiff Base Ligands. Eur. J. Inorg. Chem. 2014, 4186–4198. [Google Scholar] [CrossRef]
- Vickers, S.M.; Frischmann, P.D.; Maclachlan, M.J. Family of Cadmium Acetate Coordination Networks With Structurally Diverse [Cd4(OAc)9(μ3-OH)]2− Secondary Building Units. Inorg. Chem. 2011, 50, 2957–2965. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, J.; Pérez-yáñez, S.; Rodríguez-diéguez, A.; García, J.Á.; Rojas, S. Towards Correlating Dimensionality and Topology in Luminescent MOFs Based on Terephthalato and Bispyridyl-like Ligands. Dalton Trans. 2021, 50, 9269–9282. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, M.; Vázquez, S.; Ayllón, J.A.; Calvet, T.; Font-Bardia, M.; Pons, J. Zn(II) and Cd(II) Coordination Dimers Based on Mixed Benzodioxole-Carboxylate and N-Donor Ligands: Synthesis, Characterization, Crystal Structures and Photoluminescence Properties. ChemistrySelect 2017, 2, 632–639. [Google Scholar] [CrossRef]
- Moreno-Gómez, L.; Sánchez-Férez, F.; Calvet, T.; Font-Bardia, M.; Pons, J. Zn(II) and Cd(II) Monomer, Dimer and Polymer Compounds Coordinated by Benzoic Acid and 4-Acetylpyridine: Synthesis and Crystal Structures. Inorg. Chim. Acta 2020, 506, 119561. [Google Scholar] [CrossRef]
- Sánchez-Férez, F.; Rius-Bartra, J.M.; Calvet, T.; Font-Bardia, M.; Pons, J. Steric and Electronic Effects on the Structure and Photophysical Properties of Hg(II) Complexes. Inorg. Chem. 2021, 60, 3851–3870. [Google Scholar] [CrossRef]
- Deacon, G.B.; Phillips, R.J. Relationships between the Carbon-Oxygen Stretching Frequencies of Carboxylato Complexes and the Type of Carboxylate Coordination. Coord. Chem. Rev. 1980, 33, 227–250. [Google Scholar] [CrossRef]
- Nandi, C.K.; Hazra, M.K.; Chakraborty, T. Vibrational Coupling in Carboxylic Acid Dimers. J. Chem. Phys. 2005, 123, 124310. [Google Scholar] [CrossRef]
- Williams, D.H.; Fleming, I. Spectroscopic Methods in Organic Chemistry, 7th ed.; Springer Nature: Cham, Switzerland, 2008. [Google Scholar]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds Part A: Theory and Applications, 6th ed.; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Pinsky, M.; Avnir, D. Continuous Symmetry Measures. 5. The Classical Polyhedra. Inorg. Chem. 1998, 37, 5575–5582. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen-Sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[1,7-Bis(N-Methylbenzimidazol-2′-Yl)-2,6-Dithiaheptane]Copper(II) Perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Nishio, M. The CH/π Hydrogen Bond in Chemistry. Conformation, Supramolecules, Optical Resolution and Interactions Involving Carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873–13900. [Google Scholar] [CrossRef]
- Singh, J.; Hundal, G.; Gupta, R. Mononuclear and Dinuclear NiII and CuII Complexes with a Pyrrolecarboxamide Ligand: Core Conversions and Unusual Presence of a Dimer and Two Monomers in the Same Unit Cell. Eur. J. Inorg. Chem. 2009, 3259–3265. [Google Scholar] [CrossRef]
- Morse, P.M.; Girolami, G.S. Are d0ML6 Complexes Always Octahedral? The X-Ray Structure of Trigonal-Prismatic [Li(Tmed)]2[ZrMe6]. J. Am. Chem. Soc. 1989, 111, 4114–4116. [Google Scholar] [CrossRef]
- Friese, J.C.; Krol, A.; Puke, C.; Kirschbaum, K.; Giolando, D.M. Trigonal Prismatic vs Octahedral Coordination Geometry: Syntheses and Structural Characterization of Hexakis(Arylthiolato) Zirconate Complexes. Inorg. Chem. 2000, 39, 1496–1500. [Google Scholar] [CrossRef]
- Llunell, M.; Casanova, D.; Cirera, J.; Bofill, J.M.; Alemany, P.; Alvarez, S.; Pinsky, M.; Avnir, D. SHAPE Version 2.1: Program for the Stereochemical Analysis of Molecular Fragments by means of Continuous Shape Measures and Associated Tools; Universitat de Barcelona: Barcelona, Spain, 2013. [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomie Distances in Halides and Chaleogenides. Acta Crystallogr. Sect. A Found. Crystallogr. 1976, 32, 751–767. [Google Scholar]
- Alessi, P.J.; Carter, E.C.; Fairchild, M.D.; Hunt, R.W.G.; Kránicz, B.; Moore, J.R.; Morren, L.; Nobbs, J.H.; Ohno, Y.; Pointer, M.R.; et al. CIE 15: Technical Report: Colorimetry, 3rd ed.; Carter, E.C., Ohno, Y., Pointer, M.R., Robertson, A.R., Sève, R., Schanda, J.D., Witt, K., Eds.; International Commission on Illumination: Washington, DC, USA, 2004. [Google Scholar]
- Würth, C.; Grabolle, M.; Pauli, J.; Spieles, M.; Resch-Genger, U. Relative and Absolute Determination of Fluorescence Quantum Yields of Transparent Samples. Nat. Protoc. 2013, 8, 1535–1550. [Google Scholar] [CrossRef]
- Chen, R.F. Fluorescence Quantum Yields of Tryptophan and Tyrosine. Anal. Lett. 1967, 1, 35–42. [Google Scholar] [CrossRef]
- Dawson, W.R.; Windsor, M.W. Fluorescence Yields of Aromatic Compounds. J. Phys. Chem. 1968, 72, 3251–3260. [Google Scholar] [CrossRef]
- Hale, G.M.; Querry, M.R. Optical Constants of Water in the 200-Nm to 200-Μm Wavelength Region. Appl. Opt. 1973, 12, 555. [Google Scholar] [CrossRef]
- Shakerizadeh-Shirazi, F.; Hemmateenejad, B.; Mehranpour, A.M. Determination of the Empirical Solvent Polarity Parameter ET(30) by Multivariate Image Analysis. Anal. Methods 2013, 5, 891–896. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The Calculations of Excited-State Properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persistence of Vision Pty. Ltd. Persistence of Vision; Persistence of Vision Pty. Ltd.: Williamstown, VIC, Australia, 2004. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, version D.01; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Chai, J.D.; Head-Gordon, M. Systematic Optimization of Long-Range Corrected Hybrid Density Functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaJohn, L.A.; Christiansen, P.A.; Ross, R.B.; Atashroo, T.; Ermler, W.C. Ab Initio Relativistic Effective Potentials with Spin-Orbit Operators. III. Rb through Xe. J. Chem. Phys. 1987, 87, 2812–2824. [Google Scholar] [CrossRef]
- Improta, R.; Barone, V.; Scalmani, G.; Frisch, M.J. A State-Specific Polarizable Continuum Model Time Dependent Density Functional Theory Method for Excited State Calculations in Solution. J. Chem. Phys. 2006, 125, 054103. [Google Scholar] [CrossRef]
- Improta, R.; Scalmani, G.; Frisch, M.J.; Barone, V. Toward Effective and Reliable Fluorescence Energies in Solution by a New State Specific Polarizable Continuum Model Time Dependent Density Functional Theory Approach. J. Chem. Phys. 2007, 127, 074504. [Google Scholar] [CrossRef]
- Martin, R.L. Natural Transition Orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths | ||||||
|---|---|---|---|---|---|---|
| Zn(1)-O(5) | 2.0372(12) | Zn(1)-O(1)#1 | 2.0489(12) | |||
| Zn(1)-O(2) | 2.0388(11) | Zn(1)-N(1) | 2.0338(14) | |||
| Bond angles | ||||||
| N(1)-Zn(1)-O(5) | 101.93(5) | O(2)-Zn(1)-O(6)#1 | 87.85(5) | |||
| N(1)-Zn(1)-O(2) | 100.22(5) | N(1)-Zn(1)-O(1)#1 | 98.73(5) | |||
| O(5)-Zn(1)-O(2) | 89.35(5) | O(5)-Zn(1)-O(1)#1 | 88.98(5) | |||
| N(1)-Zn(1)-O(6)#1 | 97.36(6) | O(2)-Zn(1)-O(1)#1 | 160.91(5) | |||
| O(5)-Zn(1)-O(6)#1 | 160.70(6) | O(6)#1-Zn(1)-O(1)#1 | 87.47(5) | |||
| Intermolecular interactions | H⋯A (Å) | D⋯A (Å) | D-H (Å) | >D-H⋯A (°) | ||
| C18-H18⋯Cg(1) | 3.395 | 4.273(3) | 0.950 | 154.63 | ||
| C18-H18⋯O8 | 2.598(2) | 3.467(3) | 0.950 | 152.31 | ||
| π⋯π interactions | ||||||
| Cg(I)⋯Cg(J) | Cg⋯Cg a | α b | β, γ c | Cg(I)_Perp,Cg(J)_Perp d | Slippage e | |
| Cg(2)⋯Cg(2) | 3.5075(10) | 0 | 16.5, 16.5 | 3.3638(7), 3.3639(7) | 0.994 | |
| Cg(2)⋯Cg(3) | 3.7861(10) | 10.37(8) | 31.1, 20.7 | 3.5410(7), 3.2430(7) | - | |
| Monomer | ||||||
|---|---|---|---|---|---|---|
| Bond lengths | ||||||
| Zn(1A)-O(1A) | 2.001(3) | Zn(1A)-N(3A) | 2.154(3) | |||
| Zn(1A)-O(5A) | 2.052(3) | Zn(1A)-O(9A) | 2.184(3) | |||
| Zn(1A)-N(1A) | 2.140(3) | Zn(1A)-O(10A) | 2.257(3) | |||
| Bond angles | ||||||
| O(1A)-Zn(1A)-O(5A) | 117.34(11) | N(1A)-Zn(1A)-O(9A) | 91.03(11) | |||
| O(1A)-Zn(1A)-N(1A) | 91.70(12) | N(3A)-Zn(1A)-O(9A) | 89.91(10) | |||
| O(5A)-Zn(1A)-N(1A) | 88.62(11) | O(1A)-Zn(1A)-O(10A) | 151.96(10) | |||
| O(1A)-Zn(1A)-N(3A) | 92.36(11) | O(5A)-Zn(1A)-O(10A) | 90.69(10) | |||
| O(5A)-Zn(1A)-N(3A) | 88.51(11) | N(1A)-Zn(1A)-O(10A) | 88.07(11) | |||
| N(1A)-Zn(1A)-N(3A) | 175.79(12) | N(3A)-Zn(1A)-O(10A) | 88.89(10) | |||
| O(1A)-Zn(1A)-O(9A) | 92.89(10) | O(9A)-Zn(1A)-O(10A) | 59.09(9) | |||
| O(5A)-Zn(1A)-O(9A) | 149.77(11) | |||||
| TA value (°) | ATA value (°) | |||||
| O(10A)-Cg(3)-Cg(4)-N(1A) | 46.72 | 58.41 | ||||
| N(3A)-Cg(3)-Cg(4)-O(5A) | 64.48 | |||||
| O(9A)-Cg(3)-Cg(4)-O(1A) | 64.04 | |||||
| Dimer | ||||||
| Bond lengths | ||||||
| Zn(1B)-O(5B) | 2.025(3) | Zn(1B)-N(1B) | 2.153(3) | |||
| Zn(1B)-O(6B)#1 | 2.034(3) | Zn(1B)-O(2B) | 2.271(3) | |||
| Zn(1B)-O(1B) | 2.140(3) | Zn(1B)⋯Zn(1B) | 3.9893(6) | |||
| Zn(1B)-N(3B) | 2.151(3) | |||||
| Bond angles | ||||||
| O(5B)-Zn(1B)-O(6B)#1 | 125.03(11) | O(1B)-Zn(1B)-N(1B) | 93.02(11) | |||
| O(5B)-Zn(1B)-O(1B) | 145.44(11) | N(3B)-Zn(1B)-N(1B) | 176.78(12) | |||
| O(6B)#1-Zn(1B)-O(1B) | 89.46(11) | O(5B)-Zn(1B)-O(2B) | 86.09(10) | |||
| O(5B)-Zn(1B)-N(3B) | 88.48(11) | O(6B)#1-Zn(1B)-O(2B) | 148.82(11) | |||
| O(6B)#1-Zn(1B)-N(3B) | 89.27(12) | O(1B)-Zn(1B)-O(2B) | 59.55(10) | |||
| O(1B)-Zn(1B)-N(3B) | 90.17(11) | N(3B)-Zn(1B)-O(2B) | 93.82(11) | |||
| O(5B)-Zn(1B)-N(1B) | 89.12(11) | N(1B)-Zn(1B)-O(2B) | 88.17(11) | |||
| O(6B)#1-Zn(1B)-N(1B) | 90.34(12) | |||||
| Intramolecular interactions | H⋯A (Å) | D⋯A (Å) | D-H (Å) | >D-H⋯A (°) | ||
| O(6A)-H(6A)⋯O(2A) | 1.723 | 2.503(4) | 0.840 | 153.7 | ||
| π⋯ π interactions | ||||||
| Cg(I)⋯Cg(J) | Cg⋯Cg a | α b | β, γ c | Cg(I)_Perp, Cg(J)_Perp d | Slippage e | |
| Cg(1)⋯Cg(2) | 3.884(2) | 1.7(2) | 15.1, 16.1 | 3.7307(17), 3.7493(17) | - | |
| TA value (°) | ATA value (°) | |||||
| N(3B)-Cg(5)-Cg(6)-O(5B) | 68.38 | 58.81 | ||||
| O(1B)-Cg(5)-Cg(6)-O(6B) | 62.50 | |||||
| O(2B)-Cg(5)-Cg(6)-N(1B) | 45.55 | |||||
| Intermolecular Interactions. | H⋯A (Å) | D⋯A (Å) | D-H (Å) | >D-H⋯A (°) | ||
|---|---|---|---|---|---|---|
| N(2A)-H(2AA)⋯O(14A) | 2.07 | 2.934(4) | 0.88 | 168.4 | ||
| N(2A)-H(2BA)⋯O(9A) | 2.05 | 2.903(4) | 0.88 | 164 | ||
| N(4A)-H(4AA)⋯O(13A) | 2.04 | 2.921(4) | 0.88 | 176.1 | ||
| N(4A)-H(4BA)⋯O(2B) | 2.02 | 2.855(4) | 0.88 | 157 | ||
| N(2B)-H(2BA)⋯O(10B) | 1.99 | 2.858(4) | 0.88 | 166.8 | ||
| N(2B)-H(2BB)⋯O(12A) | 2.46 | 3.268(5) | 0.88 | 153.6 | ||
| N(4B)-H(4BA)⋯O(9B) | 2.05 | 2.913(4) | 0.88 | 165.9 | ||
| N(4B)-H(4BB)⋯O(10A) | 2.01 | 2.868(4) | 0.88 | 164.7 | ||
| O(1W)-H(1WO)⋯O(2A) | 2.10 | 2.900(6) | 0.84 | 160.1 | ||
| π⋯π interactions | ||||||
| Cg(I)⋯Cg(J) | Cg⋯Cg a | α b | β, γ c | Cg(I)_Perp, Cg(J)_Perp d | Slippage e | |
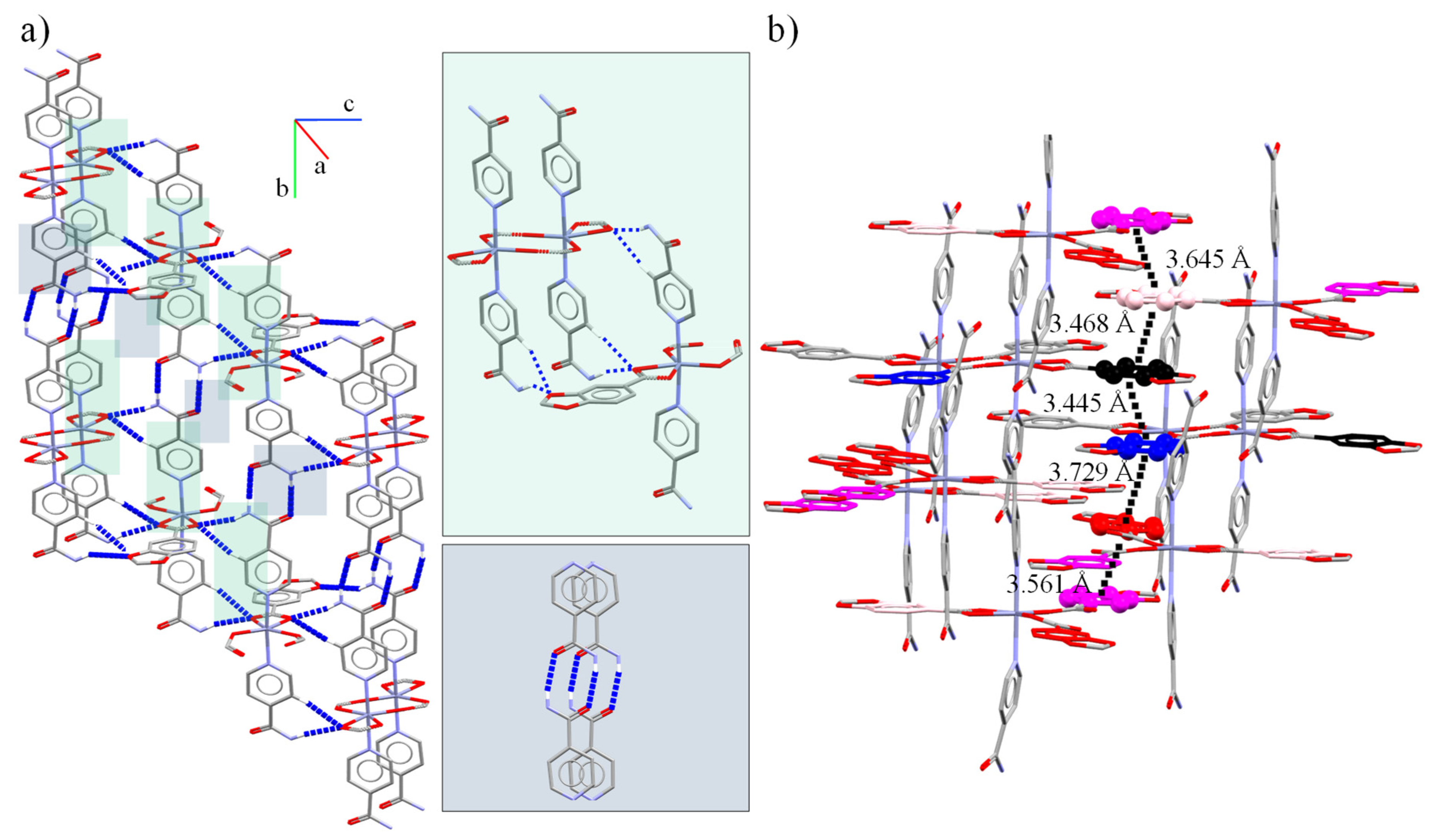
| Cg(7)⋯Cg(6) | 3.445(2) | 1.24(18) | 10.2, 9.0 | 3.4022(16), 3.3905(14) | - | |
| Cg(7)⋯Cg(3) | 3.468(2) | 3.01(18) | 13.6, 15.4 | 3.3432(16), 3.3710(14) | - | |
| Cg(6)⋯Cg(5) | 3.729(2) | 4.64(17) | 25.5, 22.5 | 3.4456(14), 3.3670(15) | - | |
| Cg(5)⋯Cg(4) | 3.561(2) | 2.91(18) | 16.2, 18.6 | 3.3740(15), 3.4202(16) | - | |
| Cg(4)⋯Cg(3) | 3.645(2) | 2.60(18) | 24.2, 21.7 | 3.3852(16), 3.3244(14) | - | |
| Molecule A | |||||
|---|---|---|---|---|---|
| Bond lengths | |||||
| Cd(1A)-O(2A) | 2.285(2) | Cd(1A)-N(2A) | 2.322(3) | ||
| Cd(1A)-O(5A) | 2.330(2) | Cd(1A)-O(6A)#1 | 2.395(2) | ||
| Cd(1A)-O(5A)#1 | 2.482(2) | Cd(1A)-O(1A) | 2.485(2) | ||
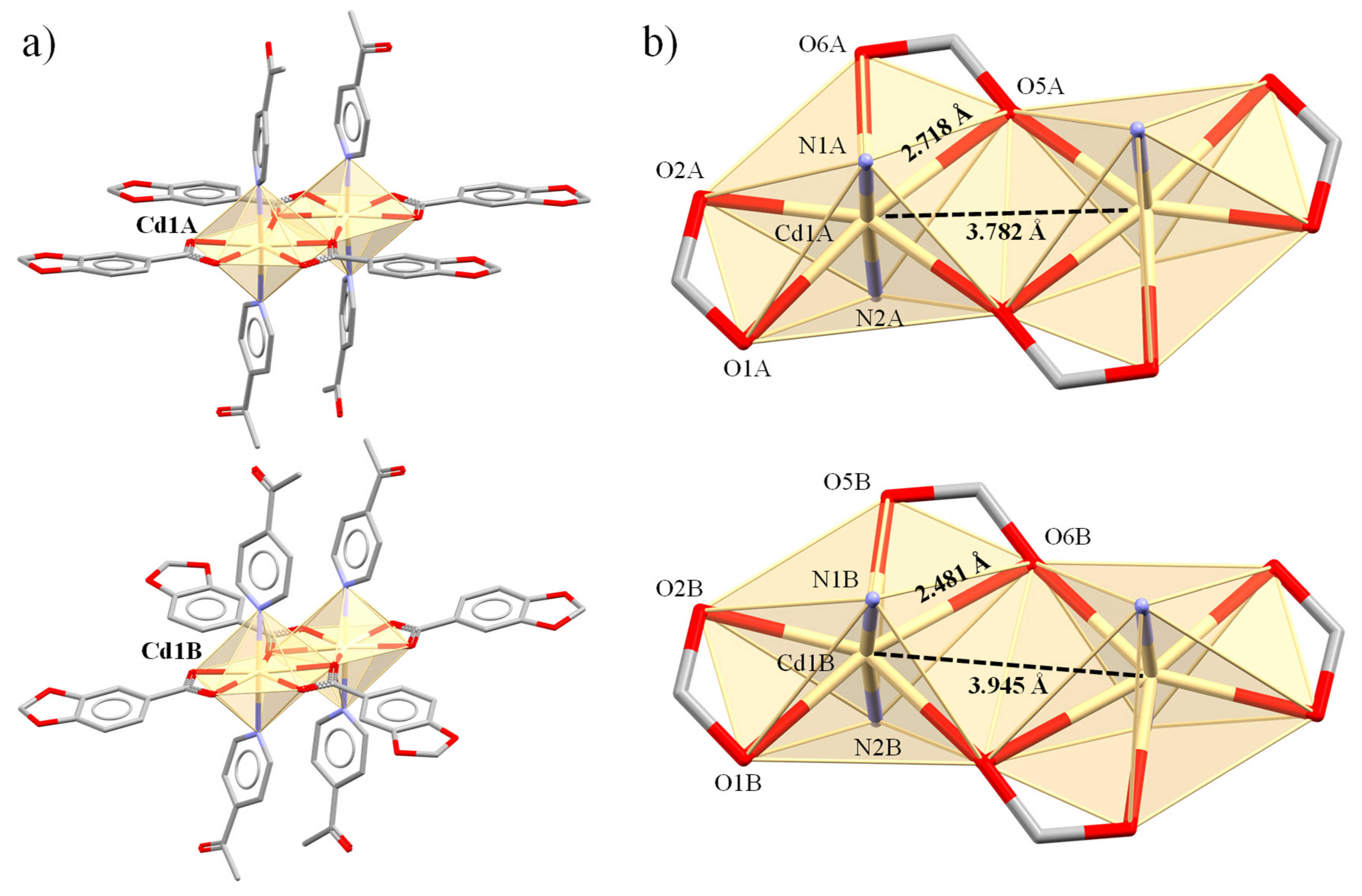
| Cd(1A)-N(1A) | 2.332(3) | Cd(1A)⋯Cd(1A) | 3.7820(5) | ||
| Bond angles | |||||
| O(2A)-Cd(1A)-N(2A) | 93.46(9) | N(2A)-Cd(1A)-O(5A)#1 | 92.29(8) | ||
| O(2A)-Cd(1A)-O(5A) | 149.25(7) | O(5A)-Cd(1A)-O(5A)#1 | 76.42(8) | ||
| N(2A)-Cd(1A)-O(5A) | 87.09(8) | N(1A)-Cd(1A)-O(5A)#1 | 90.58(8) | ||
| O(2A)-Cd(1A)-N(1A) | 89.75(9) | O(6A)#1-Cd(1A)-O(5A)#1 | 53.41(7) | ||
| N(2A)-Cd(1A)-N(1A) | 172.17(9) | O(2A)-Cd(1A)-O(1A) | 54.75(7) | ||
| O(5A)-Cd(1A)-N(1A) | 86.51(8) | N(2A)-Cd(1A)-O(1A) | 87.86(8) | ||
| O(2A)-Cd(1A)-O(6A)#1 | 80.79(7) | O(5A)-Cd(1A)-O(1A) | 94.61(7) | ||
| N(2A)-Cd(1A)-O(6A)#1 | 95.65(8) | N(1A)-Cd(1A)-O(1A) | 88.17(8) | ||
| O(5A)-Cd(1A)-O(6A)#1 | 129.79(7) | O(6A)#1-Cd(1A)-O(1A) | 135.54(7) | ||
| O(2A)-Cd(1A)-O(5A)#1 | 134.18(7) | O(5A)#1-Cd(1A)-O(1A) | 171.00(7) | ||
| Molecule B | |||||
| Bond lengths | |||||
| Cd(1B)-O(5B) | 2.265(2) | Cd(1B)-N(1B) | 2.318(3) | ||
| Cd(1B)-O(6B)#2 | 2.317(2) | Cd(1B)-N(2B) | 2.335(3) | ||
| Cd(1B)-O(1B) | 2.366(2) | Cd(1B)-O(2B) | 2.367(2) | ||
| Cd(1B)-O(6B) | 2.718(2) | Cd(1B)⋯Cd(1B) | 3.9452(6) | ||
| Bond angles | |||||
| O(5B)-Cd(1B)-O(6B)#2 | 127.84(8) | N(1B)-Cd(1B)-O(1B) | 93.92(8) | ||
| O(5B)-Cd(1B)-N(1B) | 96.03(9) | N(2B)-Cd(1B)-O(1B) | 86.04(8) | ||
| O(6B)#2-Cd(1B)-N(1B) | 85.79(8) | O(5B)-Cd(1B)-O(2B) | 85.45(8) | ||
| O(5B)-Cd(1B)-N(2B) | 88.90(8) | O(6B)#2-Cd(1B)-O(2B) | 145.20(8) | ||
| O(6B)#2-Cd(1B)-N(2B) | 86.31(8) | N(1B)-Cd(1B)-O(2B) | 102.18(9) | ||
| N(1B)-Cd(1B)-N(2B) | 172.09(9) | N(2B)-Cd(1B)-O(2B) | 84.33(9) | ||
| O(5B)-Cd(1B)-O(1B) | 141.05(7) | O(1B)-Cd(1B)-O(2B) | 55.62(7) | ||
| O(6B)#2-Cd(1B)-O(1B) | 90.36(8) | ||||
| π⋯ π interactions | |||||
| Cg(I)⋯Cg(J) | Cg⋯Cg a | α b | β, γ c | Cg(I)_Perp, Cg(J)_Perp d | Slippage e |
| Cg(1)⋯Cg(2) | 3.7430 | 16.0 | 25.0, 10.4 | 3.6813, 3.3912 | - |
| Cg(3)⋯Cg(4) | 3.8415 | 13.0 | 21.2, 18.6 | 3.6418, 3.5810 | - |
| Molecule A | |||||
| Bond lengths | |||||
| Cd(1A)-N(3A) | 2.307(3) | Cd(1A)-O(6A)#1 | 2.360(2) | ||
| Cd(1A)-N(1A) | 2.331(3) | Cd(1A)-O(1A) | 2.427(3) | ||
| Cd(1A)-O(5A) | 2.337(2) | Cd(1A)-O(5A)#1 | 2.524(2) | ||
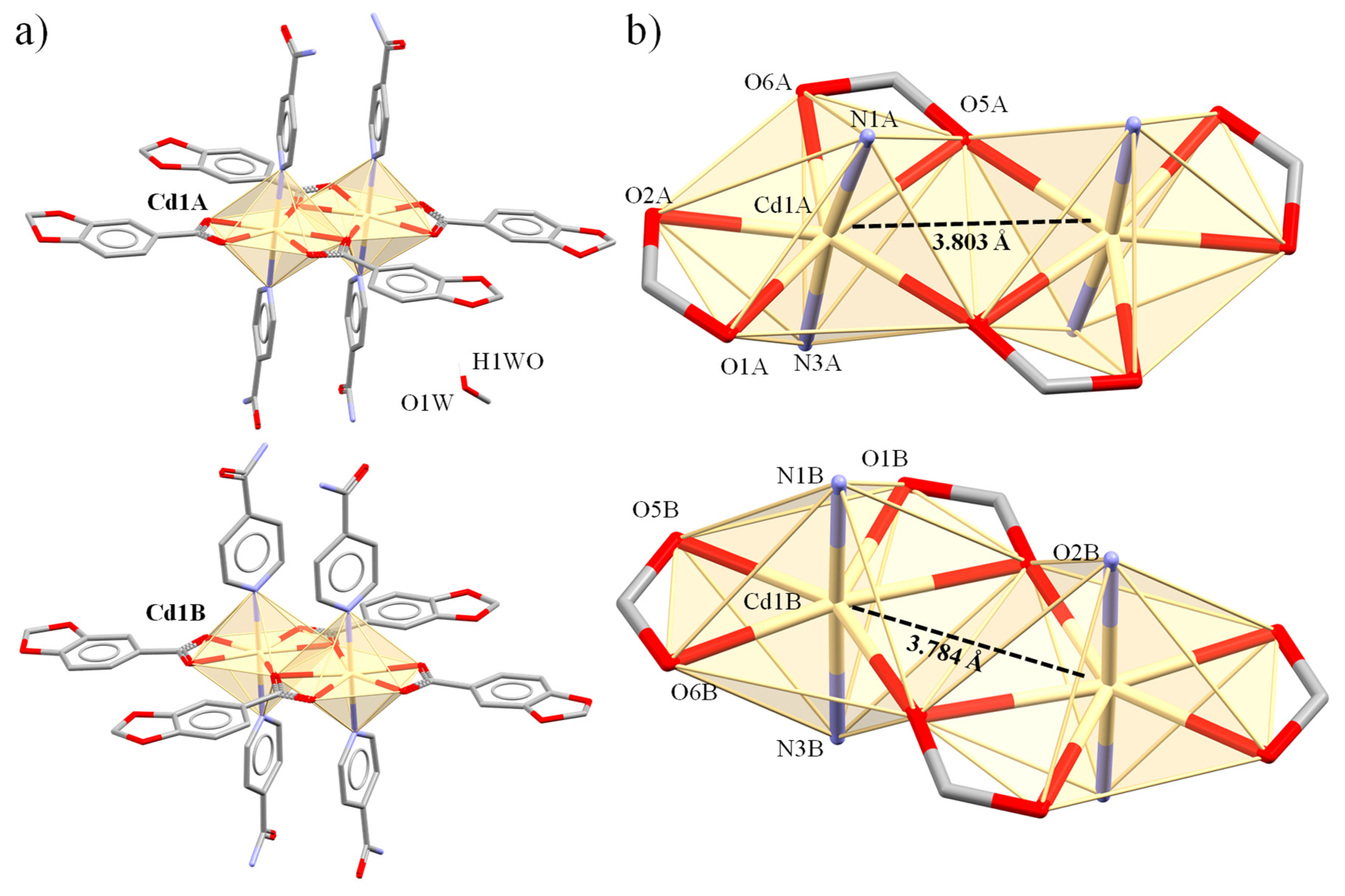
| Cd(1A)-O(2A) | 2.354(2) | Cd(1A)⋯Cd(1A) | 3.8026(6) | ||
| Bond angles | |||||
| N(3A)-Cd(1A)-N(1A) | 173.41(9) | N(1A)-Cd(1A)-O(1A) | 85.03(9) | ||
| N(3A)-Cd(1A)-O(5A) | 87.98(8) | O(5A)-Cd(1A)-O(1A) | 92.34(8) | ||
| N(1A)-Cd(1A)-O(5A) | 85.46(8) | O(2A)-Cd(1A)-O(1A) | 54.32(8) | ||
| N(3A)-Cd(1A)-O(2A) | 93.79(8) | O(6A)#1-Cd(1A)-O(1A) | 136.98(8) | ||
| N(1A)-Cd(1A)-O(2A) | 91.36(9) | N(3A)-Cd(1A)-O(5A)#1 | 90.99(8) | ||
| O(5A)-Cd(1A)-O(2A) | 146.66(8) | N(1A)-Cd(1A)-O(5A)#1 | 88.15(8) | ||
| N(3A)-Cd(1A)-O(6A)#1 | 91.19(8) | O(5A)-Cd(1A)-O(5A)#1 | 77.08(8) | ||
| N(1A)-Cd(1A)-O(6A)#1 | 93.53(8) | O(2A)-Cd(1A)-O(5A)#1 | 136.07(7) | ||
| O(5A)-Cd(1A)-O(6A)#1 | 130.48(8) | O(6A)#1-Cd(1A)-O(5A)#1 | 53.42(7) | ||
| O(2A)-Cd(1A)-O(6A)#1 | 82.81(8) | O(1A)-Cd(1A)-O(5A)#1 | 167.86(8) | ||
| N(3A)-Cd(1A)-O(1A) | 94.64(9) | ||||
| Molecule B | |||||
| Bond lengths | |||||
| Cd(1B)-O(2B)#2 | 2.306(2) | Cd(1B)-O(5B) | 2.362(2) | ||
| Cd(1B)-N(3B) | 2.321(2) | Cd(1B)-O(6B) | 2.411(2) | ||
| Cd(1B)-O(1B) | 2.328(2) | Cd(1B)-O(2B) | 2.572(2) | ||
| Cd(1B)-N(1B) | 2.329(3) | Cd(1B)⋯Cd(1B) | 3.7838(6) | ||
| Bond angles | |||||
| O(2B)#2-Cd(1B)-N(3B) | 86.45(8) | N(3B)-Cd(1B)-O(6B) | 86.89(8) | ||
| O(2B)#2-Cd(1B)-O(1B) | 131.21(8) | O(1B)-Cd(1B)-O(6B) | 137.68(8) | ||
| N(3B)-Cd(1B)-O(1B) | 92.87(9) | N(1B)-Cd(1B)-O(6B) | 91.31(8) | ||
| O(2B)#2-Cd(1B)-N(1B) | 86.99(8) | O(5B)-Cd(1B)-O(6B) | 54.62(7) | ||
| N(3B)-Cd(1B)-N(1B) | 173.17(9) | O(2B)#2-Cd(1B)-O(2B) | 78.31(8) | ||
| O(1B)-Cd(1B)-N(1B) | 92.89(9) | N(3B)-Cd(1B)-O(2B) | 91.92(8) | ||
| O(2B)#2-Cd(1B)-O(5B) | 145.66(8) | O(1B)-Cd(1B)-O(2B) | 52.93(8) | ||
| N(3B)-Cd(1B)-O(5B) | 91.89(8) | N(1B)-Cd(1B)-O(2B) | 88.63(8) | ||
| O(1B)-Cd(1B)-O(5B) | 83.13(8) | O(5B)-Cd(1B)-O(2B) | 136.03(7) | ||
| N(1B)-Cd(1B)-O(5B) | 92.44(8) | O(6B)-Cd(1B)-O(2B) | 169.35(7) | ||
| O(2B)#2-Cd(1B)-O(6B) | 91.05(8) | ||||
| π⋯ π interactions | |||||
| Cg(I)⋯Cg(J) | Cg⋯Cg a | α b | β, γ c | Cg(I)_Perp, Cg(J)_Perp d | Slippage e |
| Cg(1)⋯Cg(1) | 3.6090(19) | 1 | 14.9 | 3.4870(14) | - |
| Cg(2)⋯Cg(2) | 3.6435(18) | 0 | 18.3 | 3.4597(13) | 1.142 |
| Cg(3)⋯Cg(3) | 3.7740(19) | 1 | 24.4 | 3.4372(14) | - |
| Cg(4)⋯Cg(4) | 3.606(2) | 3 | 2.5 | 3.6021(16), 3.6023(16) | - |
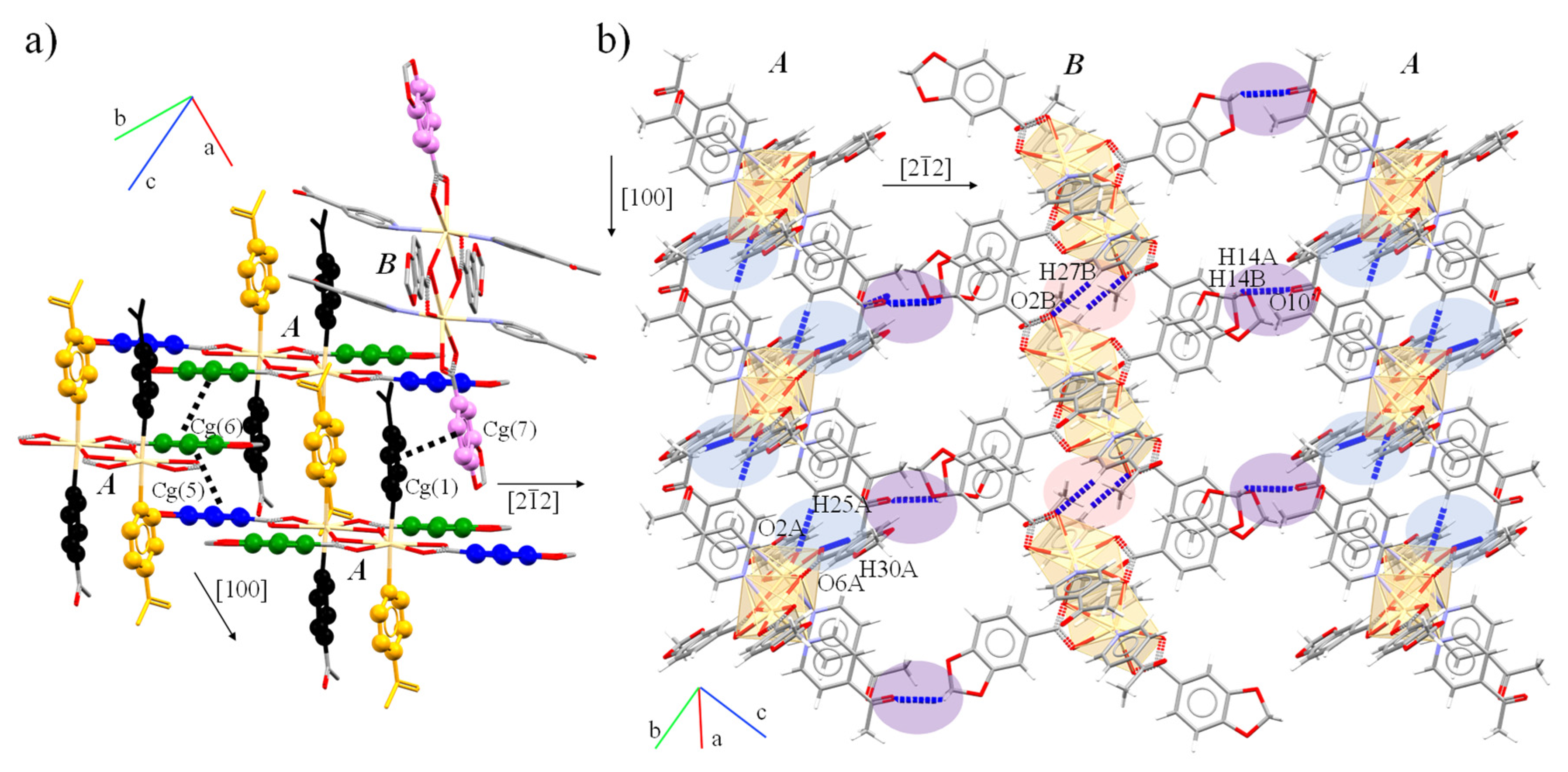
| Intermolecular Interactions | H⋯A (Å) | D⋯A (Å) | D-H (Å) | >D-H⋯A (°) | ||
|---|---|---|---|---|---|---|
| C6B-H6BB⋯O10A | 2.45 | 3.24(1) | 0.99 | 135.9 | ||
| C14B-H14B⋯O10A | 2.497 | 3.06(1) | 0.99 | 115.7 | ||
| C25A-H25A⋯O2A | 2.241 | 3.190(5) | 0.95 | 177.2 | ||
| C27B-H27B⋯O2B | 2.365 | 3.228(5) | 0.95 | 150.8 | ||
| C30A-H30A⋯O6A | 2.371 | 3.317(5) | 0.98 | 161.9 | ||
| C30B-H30E⋯O5B | 2.480 | 3.423(6) | 0.98 | 161.3 | ||
| π⋯ π interactions | ||||||
| Cg(I)⋯Cg(J) | Cg⋯Cga | α b | β, γ c | Cg(I)_Perp, Cg(J)_Perp d | Slippage e | |
| Cg(1)⋯Cg(7) | 3.8236 | 9.0 | 25.6, 17.1 | 3.6555, 3.4478 | - | |
| Cg(5)⋯Cg(6) | 3.7876 | 1.0 | 29.0, 28.4 | 3.3325, 3.3129 | - | |
| Cg(6)⋯Cg(6) | 3.5058 | 0 | 18.3, 18.3 | 3.3287, 3.3287 | 1.100 | |
| Intermolecular Interactions. | H⋯A (Å) | D⋯A (Å) | D-H (Å) | >D-H⋯A (°) | ||
|---|---|---|---|---|---|---|
| N2A-H2AA⋯O10A | 1.997 | 2.873(3) | 0.88 | 174.1 | ||
| N2A-H2AB⋯O4A | 2.161 | 2.909(4) | 0.88 | 142.6 | ||
| N4A-H4AA⋯O9A | 2.046 | 2.899(3) | 0.88 | 163.1 | ||
| N4A-H4AB⋯O5B | 2.015 | 2.849(3) | 0.88 | 157.8 | ||
| N4B-H4BA⋯O9B | 2.050 | 2.928(4) | 0.88 | 175.7 | ||
| N4B-H4BB⋯O1W | 2.172 | 2.980(8) | 0.88 | 152.7 | ||
| N2B-H2BA⋯O10B | 2.011 | 2.874(4) | 0.88 | 166.2 | ||
| N2B-H2BB⋯O2A | 2.022 | 2.832(3) | 0.88 | 152.5 | ||
| C26A-H26A⋯O5B | 2.418 | 3.315(4) | 0.95 | 157.3 | ||
| π⋯π interactions | ||||||
| Cg(I)⋯Cg(J) | Cg⋯Cg a | α b | β, γ c | Cg(I)_Perp, Cg(J)_Perp d | Slippage e | |
| Cg(8)⋯Cg(7) | 3.560(2) | 6.19(18) | 8.2, 13.4 | 3.4625(14), 3.5233(17) | - | |
| Cg(5)⋯Cg(6) | 3.824(2) | 2.39(16) | 26.2, 27.7 | 3.3857(14), 3.4303(13) | - | |
| Cg(6)⋯Cg(7) | 3.534(2) | 1.95(17) | 17.3, 15.6 | 3.4046(13), 3.3746(17) | - | |
| Sample | λex | λmax-em | Δλ | ϕs |
|---|---|---|---|---|
| 1 | 263 | 354 | 9774 | 0.74 |
| 2 | 225 225 * 251 * | 344 344 * 318 * | 15,375 15,375 8,394 | 0.01 0.03 0.02 |
| 3 | 315 | 355 | 3,577 | 0.47 |
| 4 | 226 | 347 | 15,429 | 0.09 |
| 1 | 2 | |
|---|---|---|
| Empirical formula | C46H34ZnN2O18 | C130H108Zn4N16O50 |
| Formula weight | 1033.49 | 2955.80 |
| T (K) | 100(2) | 100(2) |
| Wavelength (Å) | 0.71073 | 0.71073 |
| System, space group | Triclinic, P-1 | Triclinic, P-1 |
| Unit cell dimensions | ||
| a (Å) | 8.2648(5) | 10.8798(6) |
| b (Å) | 11.5381(6) | 16.9461(8) |
| c (Å) | 11.7350(7) | 17.3401(9) |
| α (°) | 103.454(2) | 84.455(2) |
| β (°) | 90.849(2) | 80.089(2) |
| γ (°) | 105.143(2) | 77.457(2) |
| V (Å3) | 1047.14(11) | 3068.3(3) |
| Z | 1 | 1 |
| Dcalc (g cm3) | 1.639 | 1.600 |
| µ (mm−1) | 1.231 | 0.878 |
| F (000) | 528 | 1520 |
| Crystal size (mm3) | 0.180 × 0.120 × 0.090 | 0.100 × 0.080 × 0.040 |
| hkl ranges | −13 ≤ h ≤ 13 −18 ≤ k ≤ 18 −18 ≤ l ≤ 18 | −15 ≤ h ≤ 15 −24 ≤ k ≤ 24 −24 ≤ l ≤ 24 |
| θ range (°) | 1.886 to 34.487 | 2.076 to 30.569 |
| Reflections collected/ unique/[Rint] | 47884/8790/ [Rint] = 0.0343 | 96200/18771/ [R(int) = 0.01031] |
| Completeness to θ (%) | 99.7 | 99.9 |
| Absorption Correction | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max. and min. transmis. | 0.7468 and 0.6880 | 0.7461 and 0.6818 |
| Refinement method | Full matrix least-squares on |F|2 | Full-matrix least-squares on |F|2 |
| Data/restrains/parameters | 8790/0/308 | 18771/1/902 |
| Goodness of fit (GOF) on |F|2 | 1.077 | 1.074 |
| Final R indices [I>2σ(I)] | R1 = 0.0384, wR2 = 0.1025 | R1 = 0.0628, wR2 = 0.1442 |
| R indices (all data) | R1 = 0.0469 wR2 = 0.1102 | R1 = 0.1404, wR2 = 0.1947 |
| Extinction coefficient | n/a | n/a |
| Largest. Diff. Peak and hole (e Å−3) | 1.540 and −0.544 | 2.970 and −1.091 |
| 3 | 4 | |
|---|---|---|
| Empirical formula | C60H48Cd2N4O20 | C113H92Cd4N16O41 |
| Formula weight | 1369.82 | 2779.62 |
| T (K) | 100(2) | 100(2) |
| Wavelength (Å) | 0.71073 | 0.71073 |
| System, space group | Triclinic, P-1 | Monoclinic, P2/c |
| Unit cell dimensions | ||
| a (Å) | 10.2552(6) | 15.4163(10) |
| b (Å) | 11.9963(7) | 17.3028(12) |
| c (Å) | 24.7728(15) | 22.9004(17) |
| α (°) | 82.798(2) | 90 |
| β (°) | 89.674(2) | 105.565(2) |
| γ (°) | 65.1870(10) | 90 |
| V (Å3) | 2740.8(3) | 5884.55 |
| Z | 2 | 2 |
| Dcalc (g cm3) | 1.660 | 1.569 |
| µ (mm−1) | 0.862 | 0.806 |
| F (000) | 1384 | 2804 |
| Crystal size (mm3) | 0.131 × 0.092 × 0.042 | 0.400 × 0.120 × 0.040 |
| hkl ranges | −13 ≤ h ≤ 13 −15 ≤ k ≤ 15 −32 ≤ l ≤ 32 | −22 ≤ h ≤ 21 0 ≤ k ≤ 25 0 ≤ l ≤ 33 |
| θ range (°) | 2.191 to 27.526 | 2.190 to 31.067 |
| Reflections collected/ unique/[Rint] | 75664/12559/ [Rint] = 0.0750 | 18818/18818/ [R(int) = 0.0751] |
| Completeness to θ (%) | 99.9 | 99.8 |
| Absorption Correction | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max. and min. transmis. | 0.7455 and 0.6770 | 0.7454 and 0.6631 |
| Refinement method | Full matrix least-squares on |F|2 | Full-matrix least-squares on |F|2 |
| Data/restrains/parameters | 12559/0/788 | 18818/10/776 |
| Goodness of fit (GOF) on |F|2 | 1.082 | 1.052 |
| Final R indices [I > 2σ(I)] | R1 = 0.0404, wR2 = 0.0814 | R1 = 0.0428, wR2 = 0.1115 |
| R indices (all data) | R1 = 0.0634, wR2 = 0.0888 | R1 = 0.0833, wR2 = 0.1439 |
| Extinction coefficient | n/a | n/a |
| Largest. Diff. Peak and hole (e Å−3) | 1.087 and −1.028 | 2.986 and −1.223 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Férez, F.; Rius-Bartra, J.M.; Ayllón, J.A.; Calvet, T.; Font-Bardia, M.; Pons, J. Tuning Photophysical Properties by p-Functional Groups in Zn(II) and Cd(II) Complexes with Piperonylic Acid. Molecules 2022, 27, 1365. https://doi.org/10.3390/molecules27041365
Sánchez-Férez F, Rius-Bartra JM, Ayllón JA, Calvet T, Font-Bardia M, Pons J. Tuning Photophysical Properties by p-Functional Groups in Zn(II) and Cd(II) Complexes with Piperonylic Acid. Molecules. 2022; 27(4):1365. https://doi.org/10.3390/molecules27041365
Chicago/Turabian StyleSánchez-Férez, Francisco, Joaquim Mª Rius-Bartra, José A. Ayllón, Teresa Calvet, Mercè Font-Bardia, and Josefina Pons. 2022. "Tuning Photophysical Properties by p-Functional Groups in Zn(II) and Cd(II) Complexes with Piperonylic Acid" Molecules 27, no. 4: 1365. https://doi.org/10.3390/molecules27041365
APA StyleSánchez-Férez, F., Rius-Bartra, J. M., Ayllón, J. A., Calvet, T., Font-Bardia, M., & Pons, J. (2022). Tuning Photophysical Properties by p-Functional Groups in Zn(II) and Cd(II) Complexes with Piperonylic Acid. Molecules, 27(4), 1365. https://doi.org/10.3390/molecules27041365