
Complementary, Cooperative Ditopic Halogen Bonding and Electron Donor-Acceptor π-π Complexation in the Formation of Cocrystals


Abstract
:1. Introduction
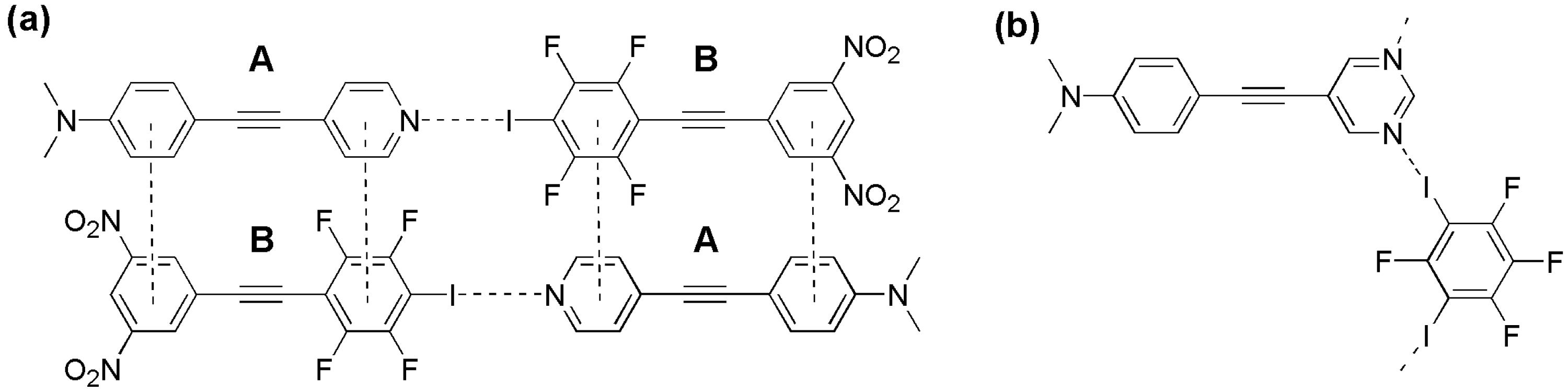
2. Results
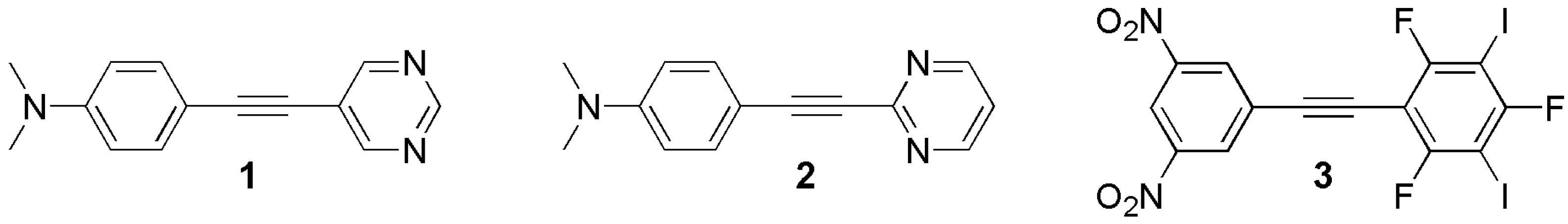
2.1. Materials
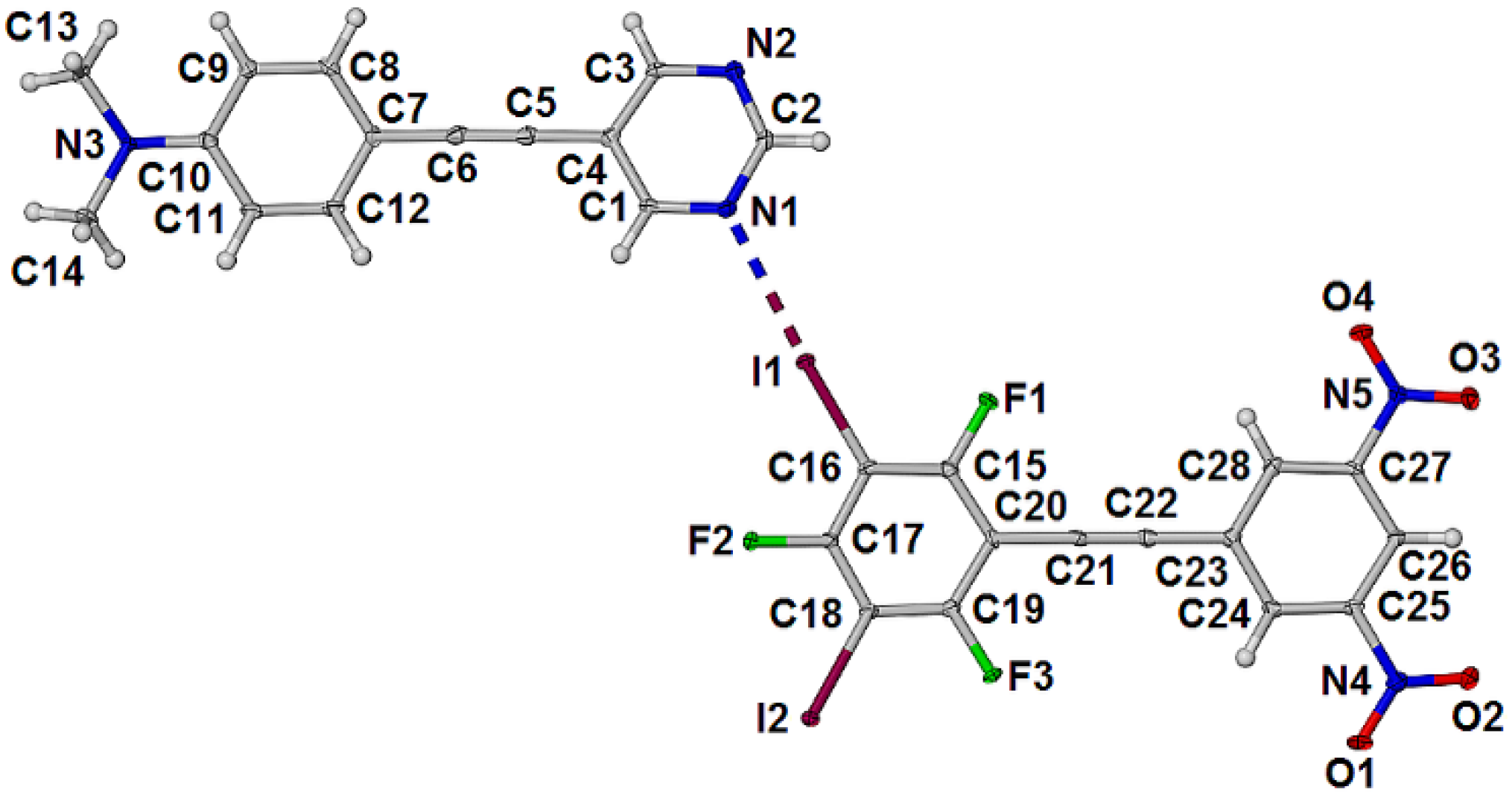
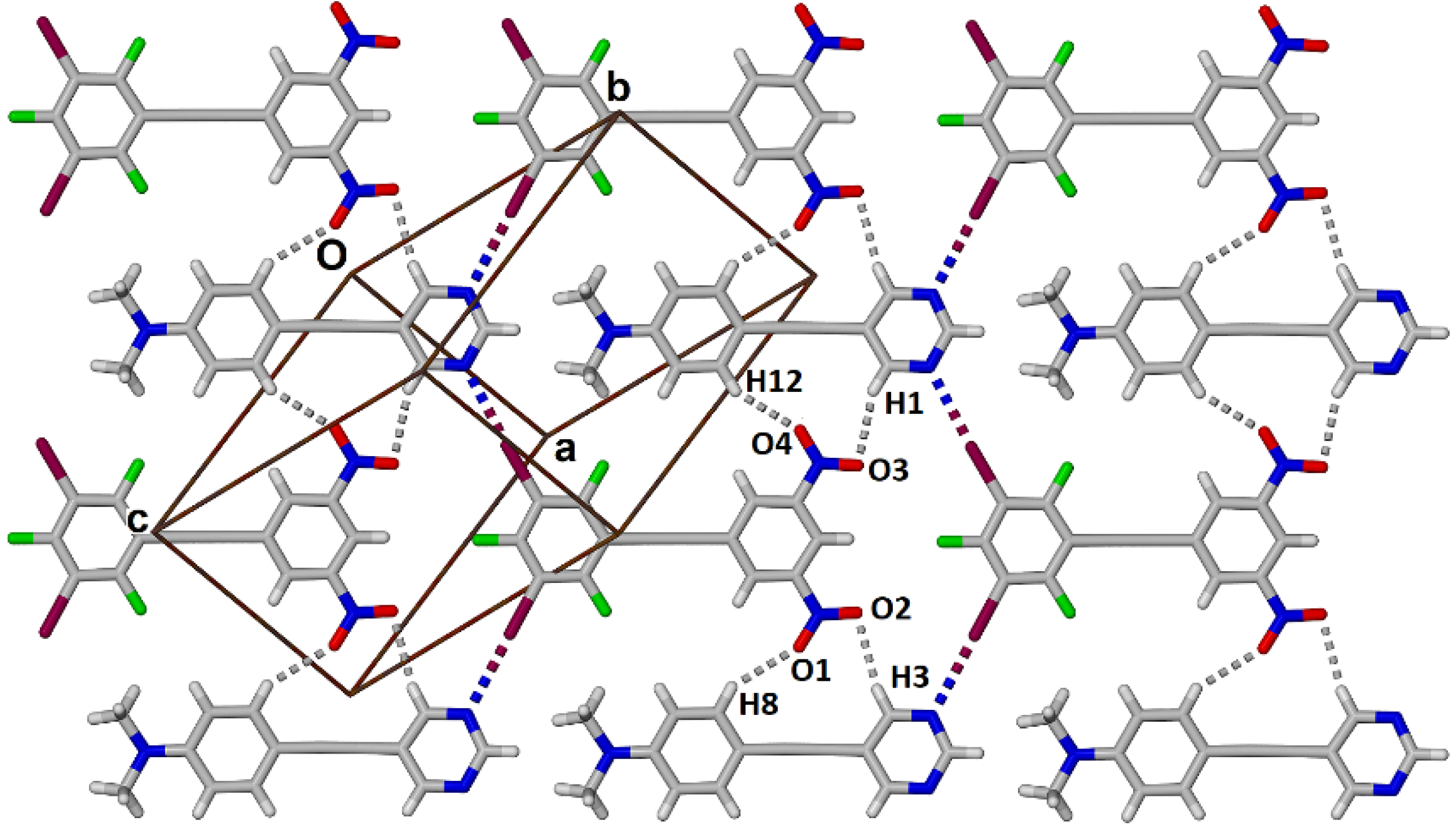
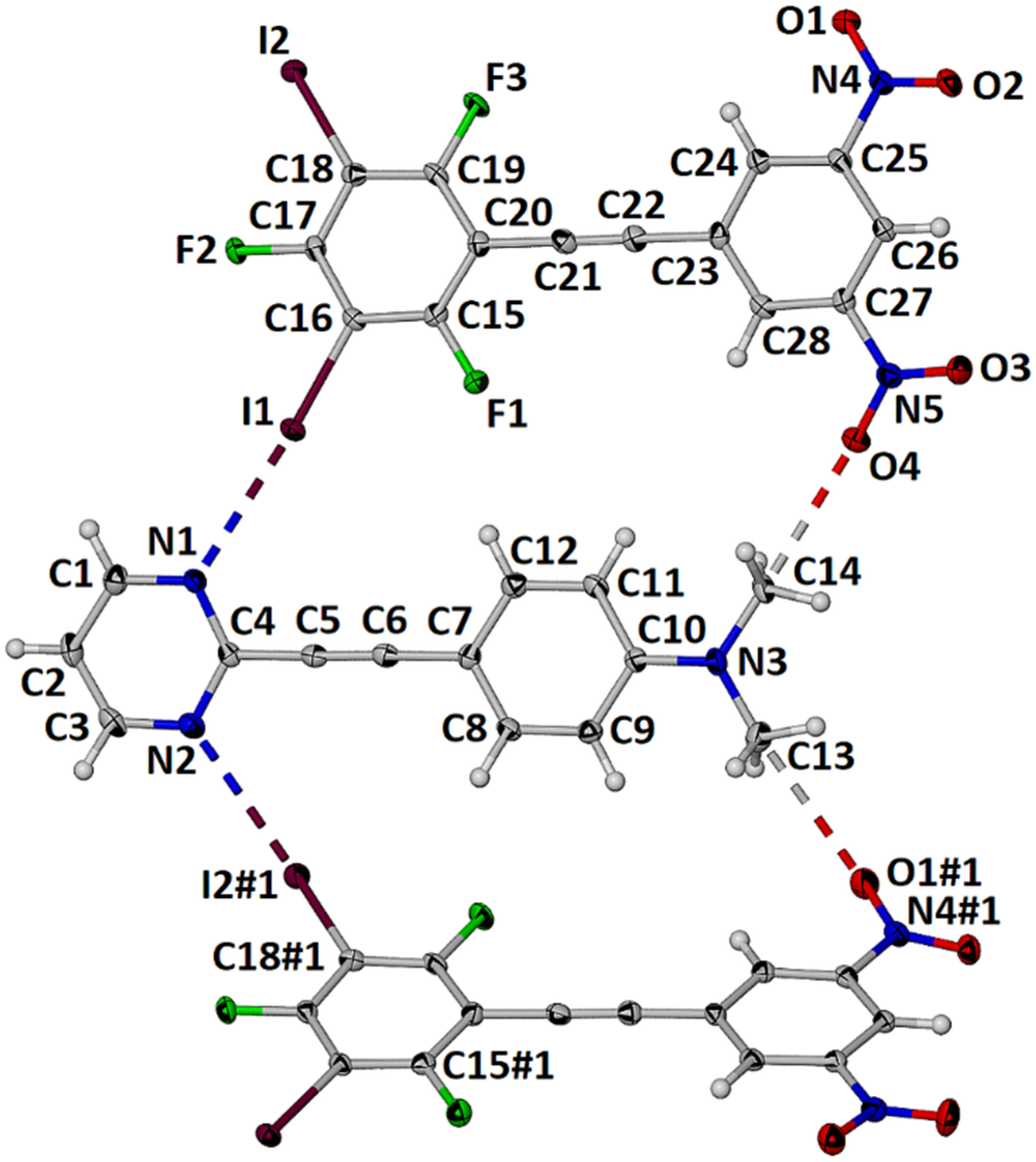
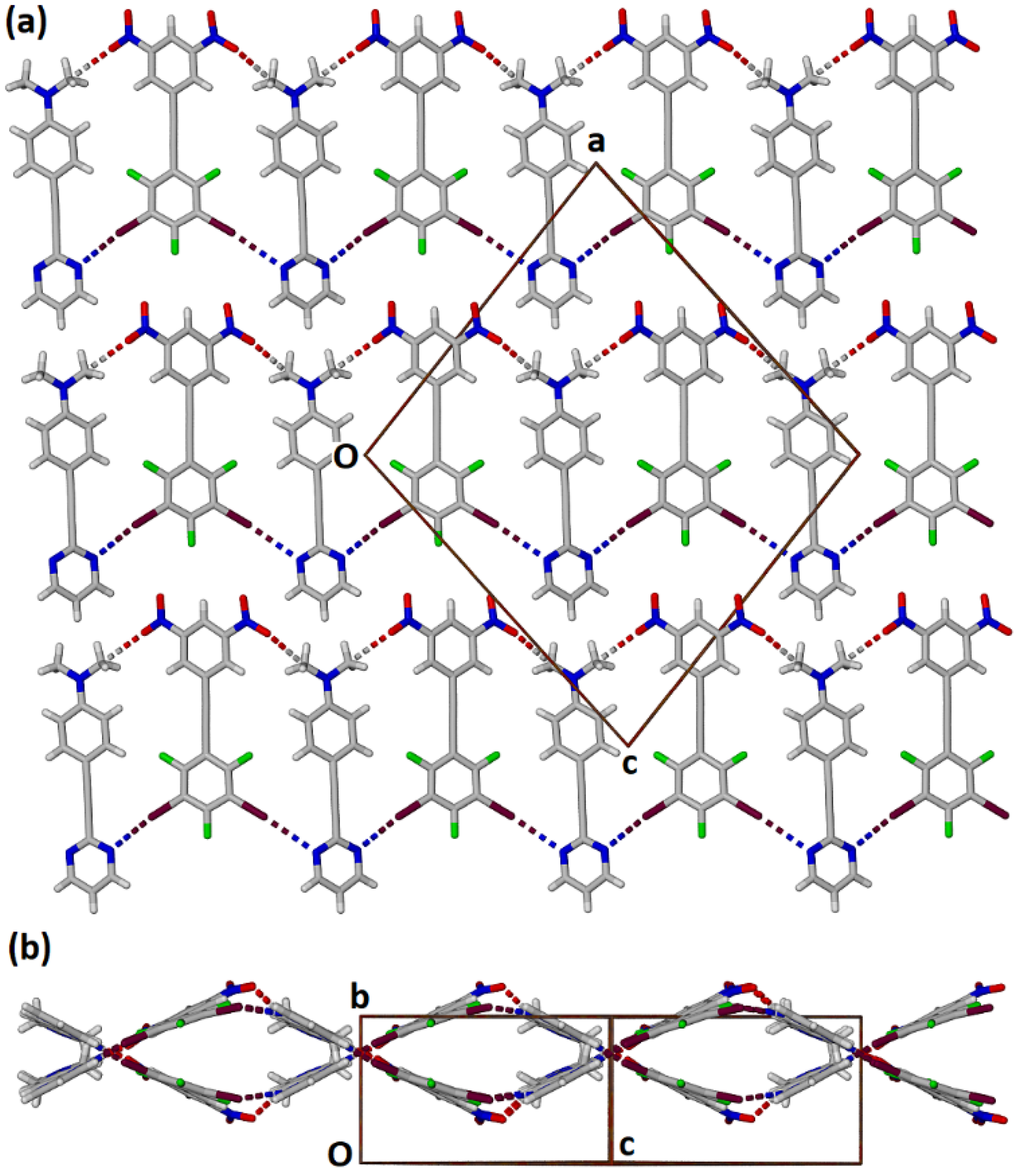
2.2. Formation and Analysis of Cocrystals
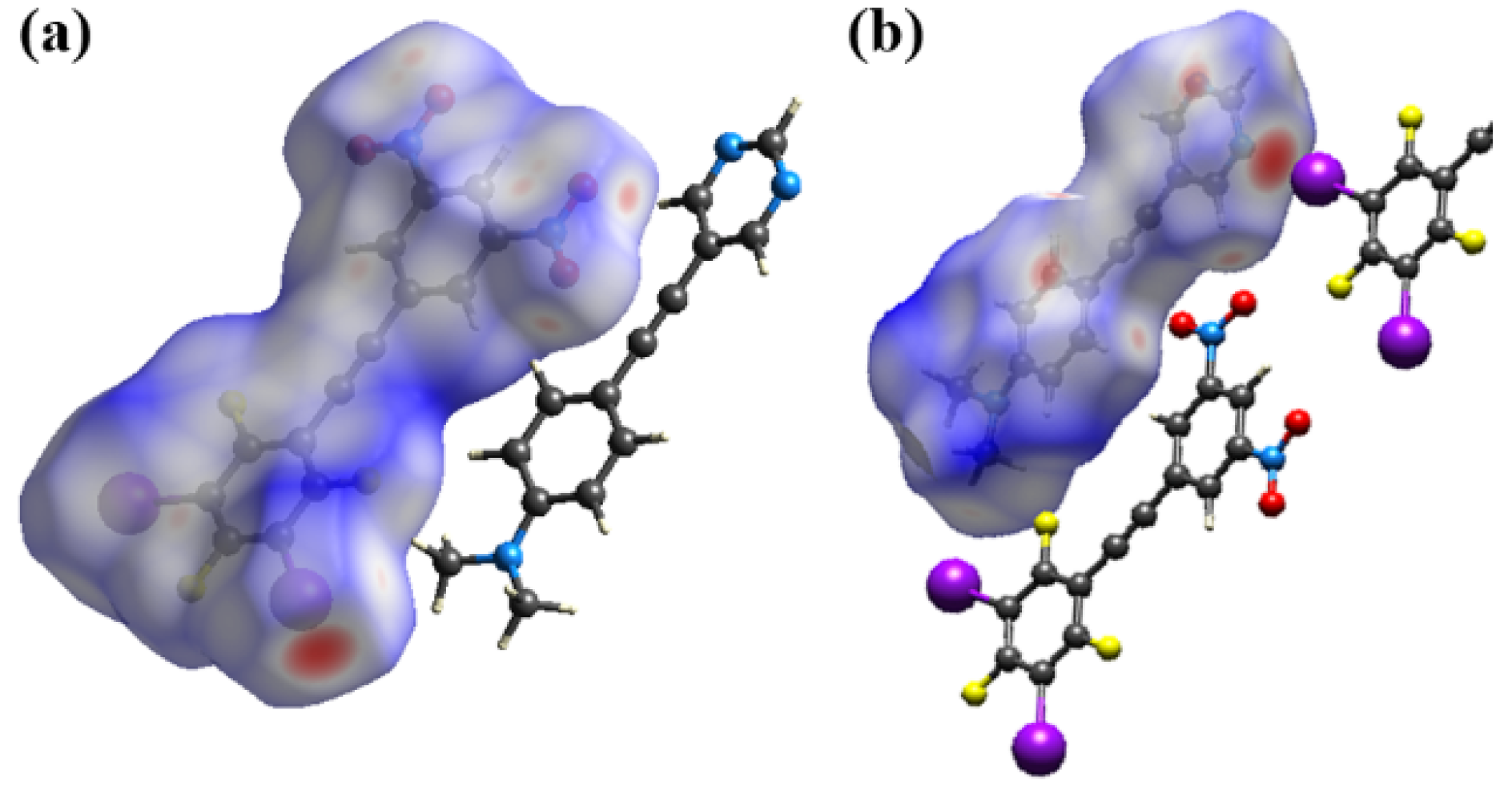
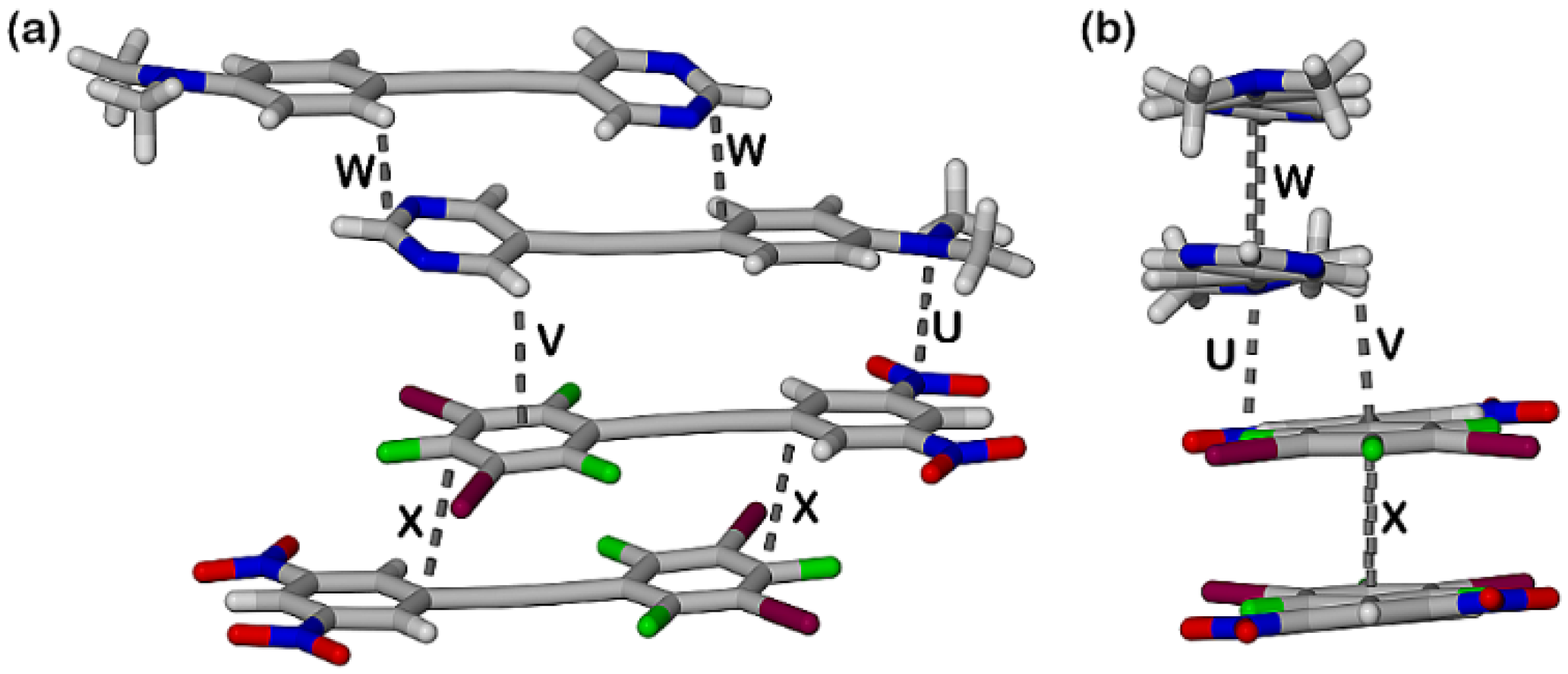
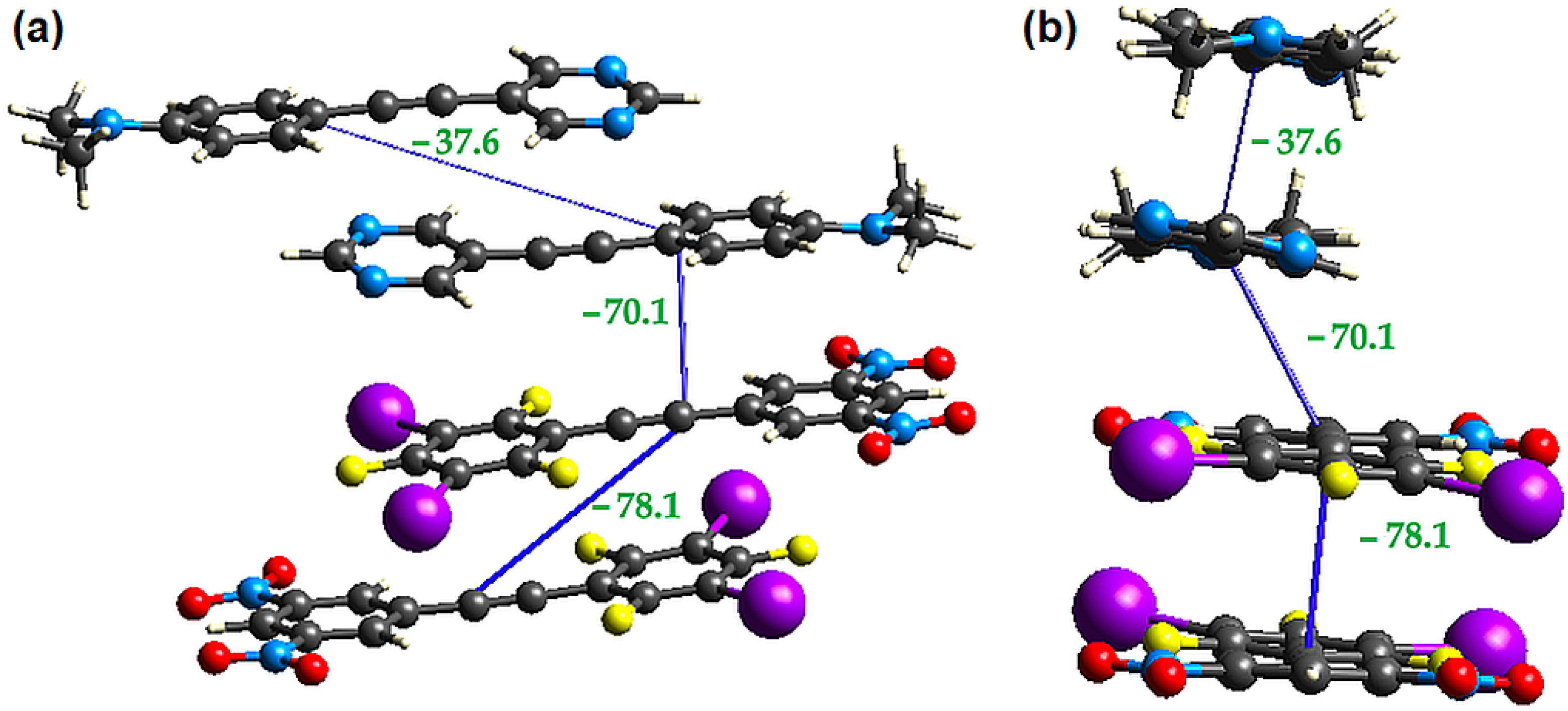
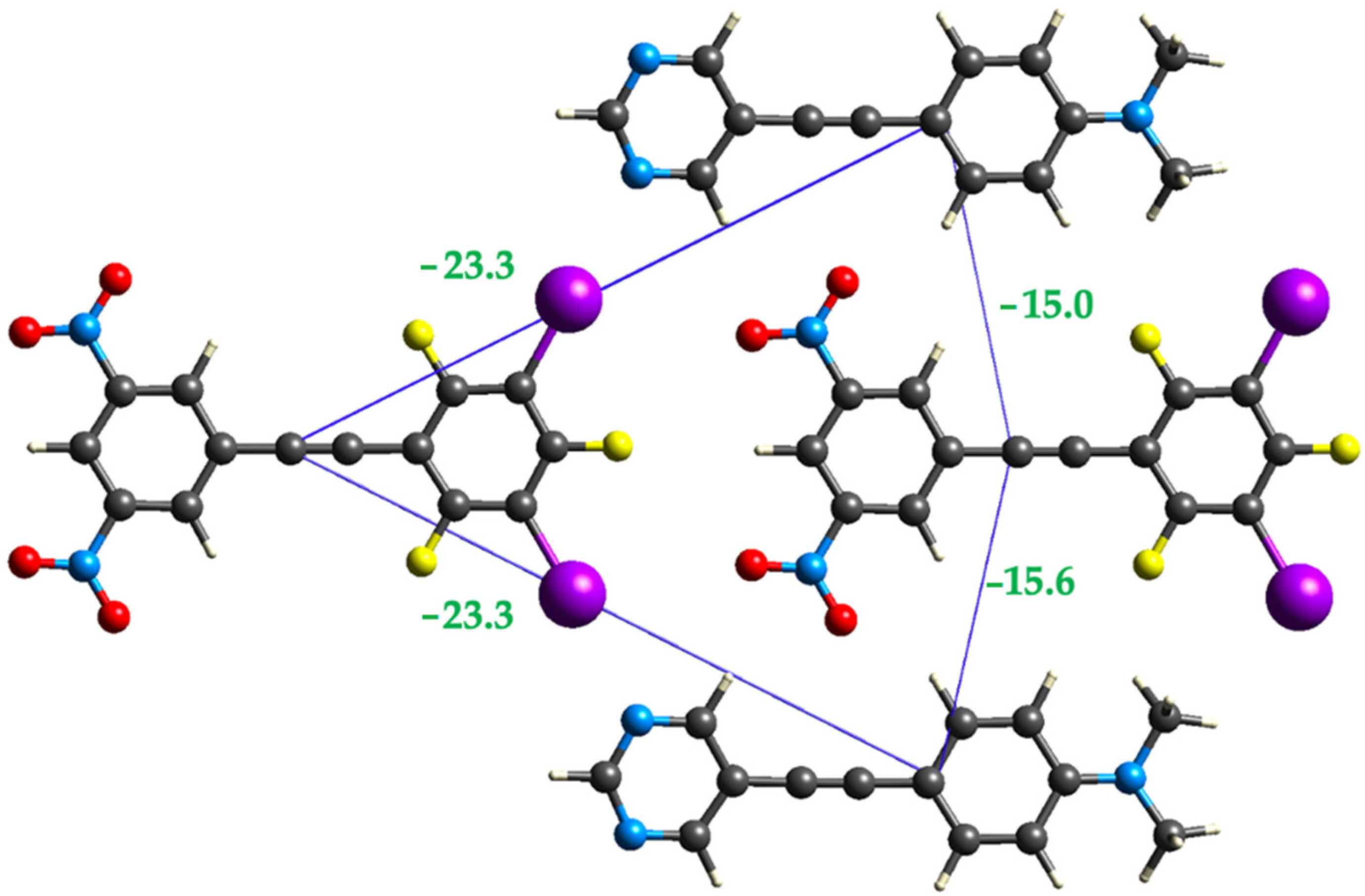
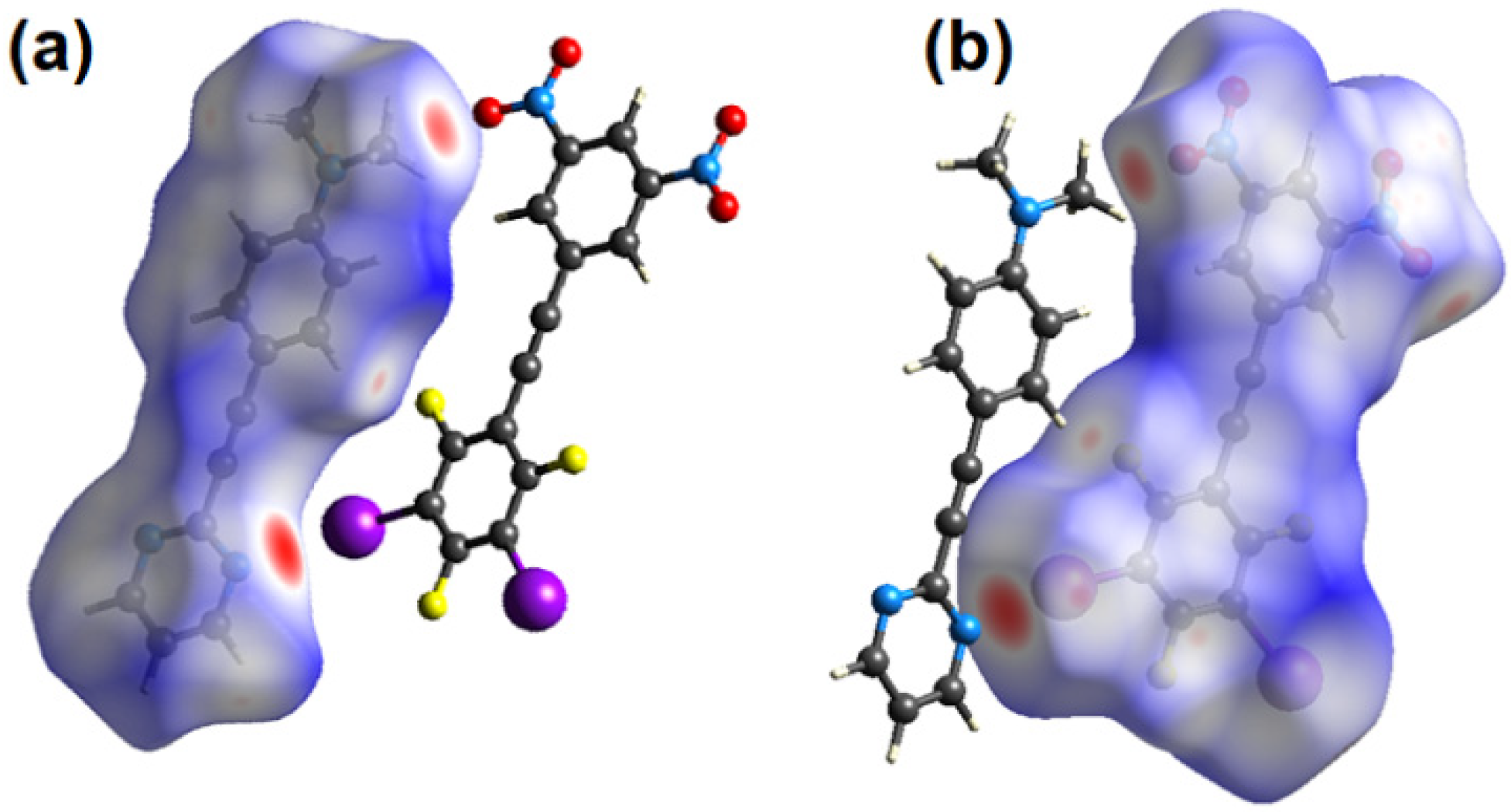
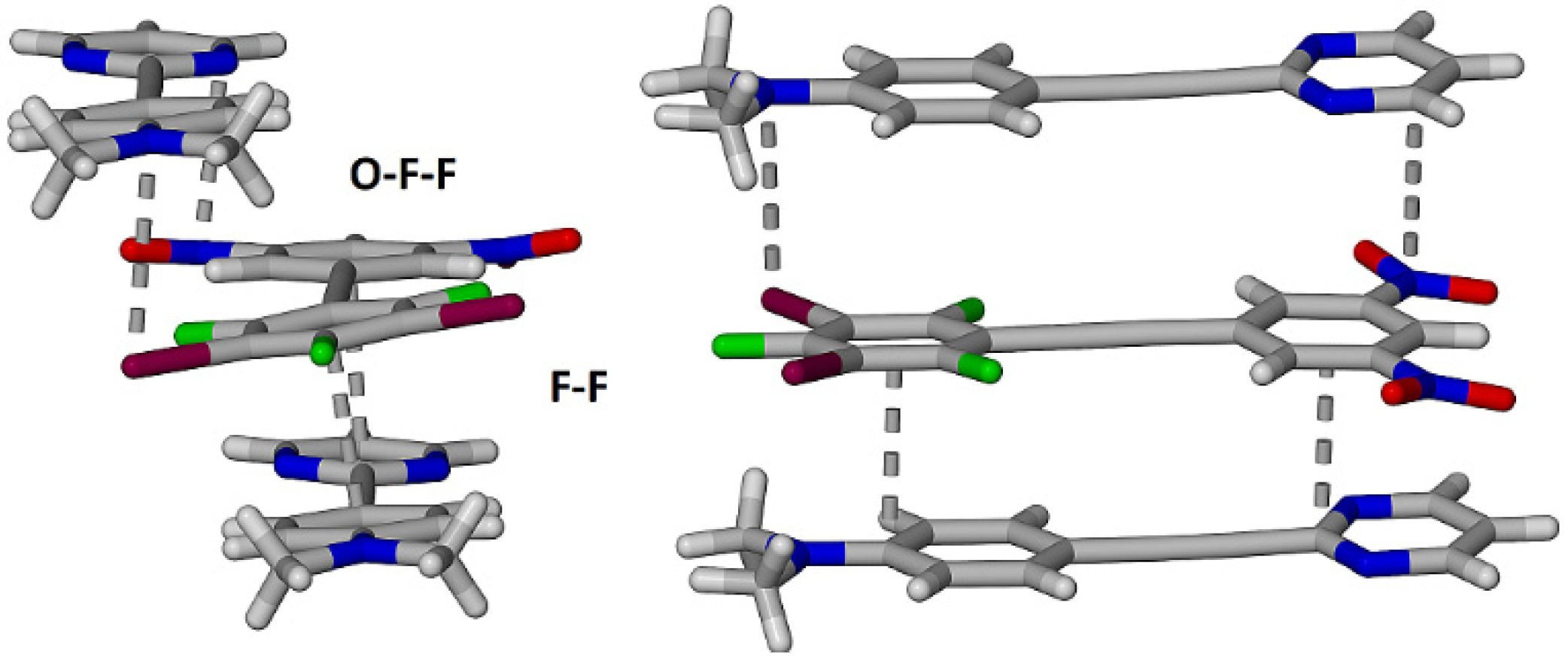
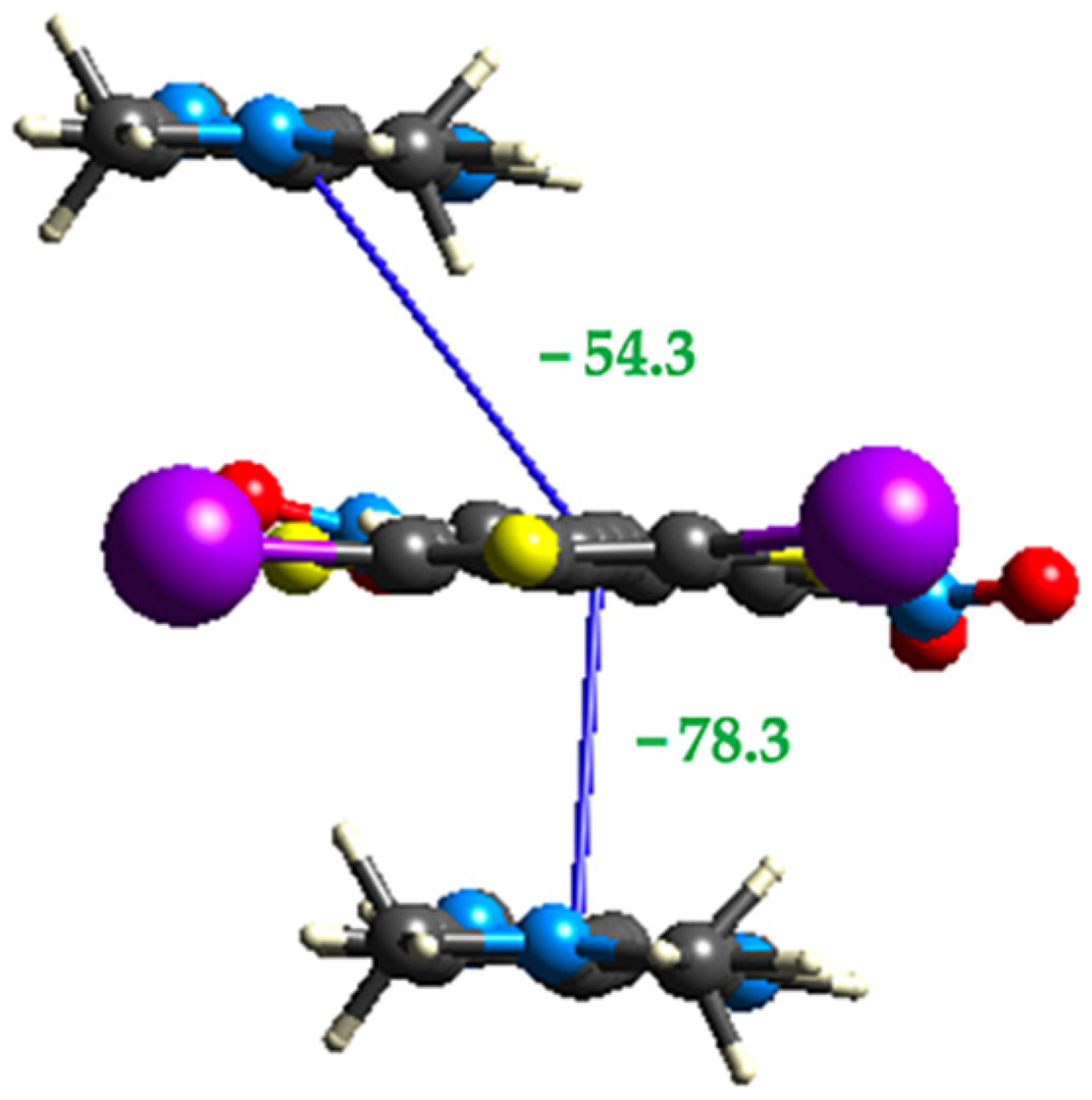
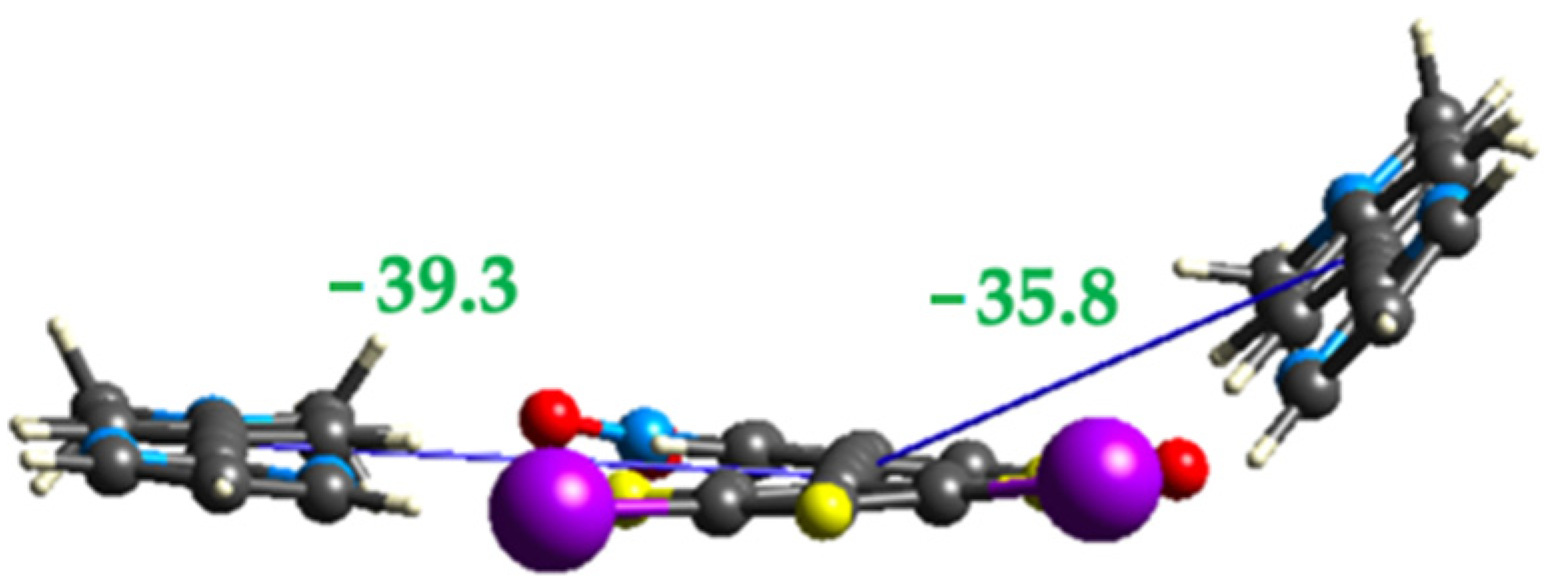
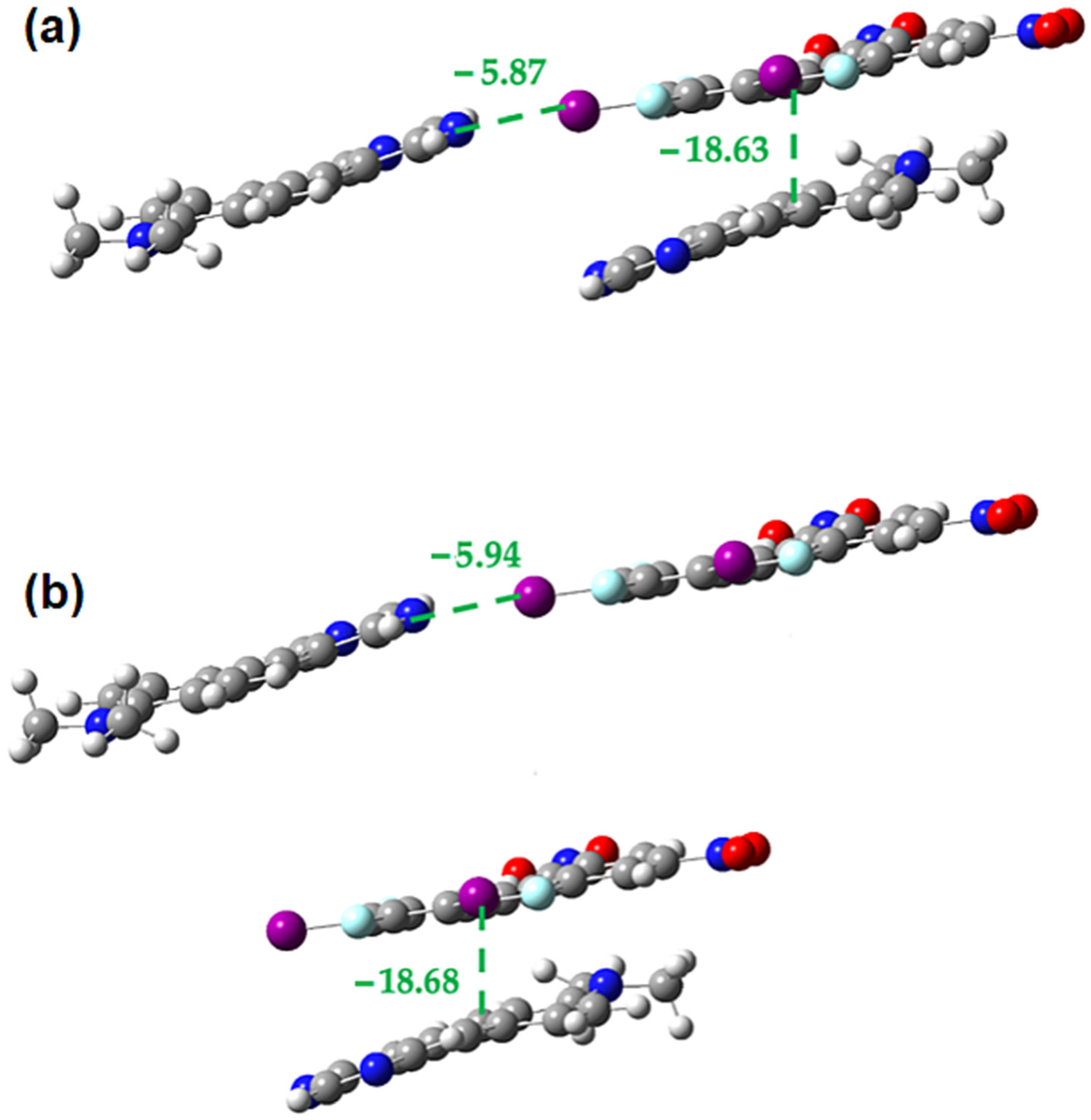
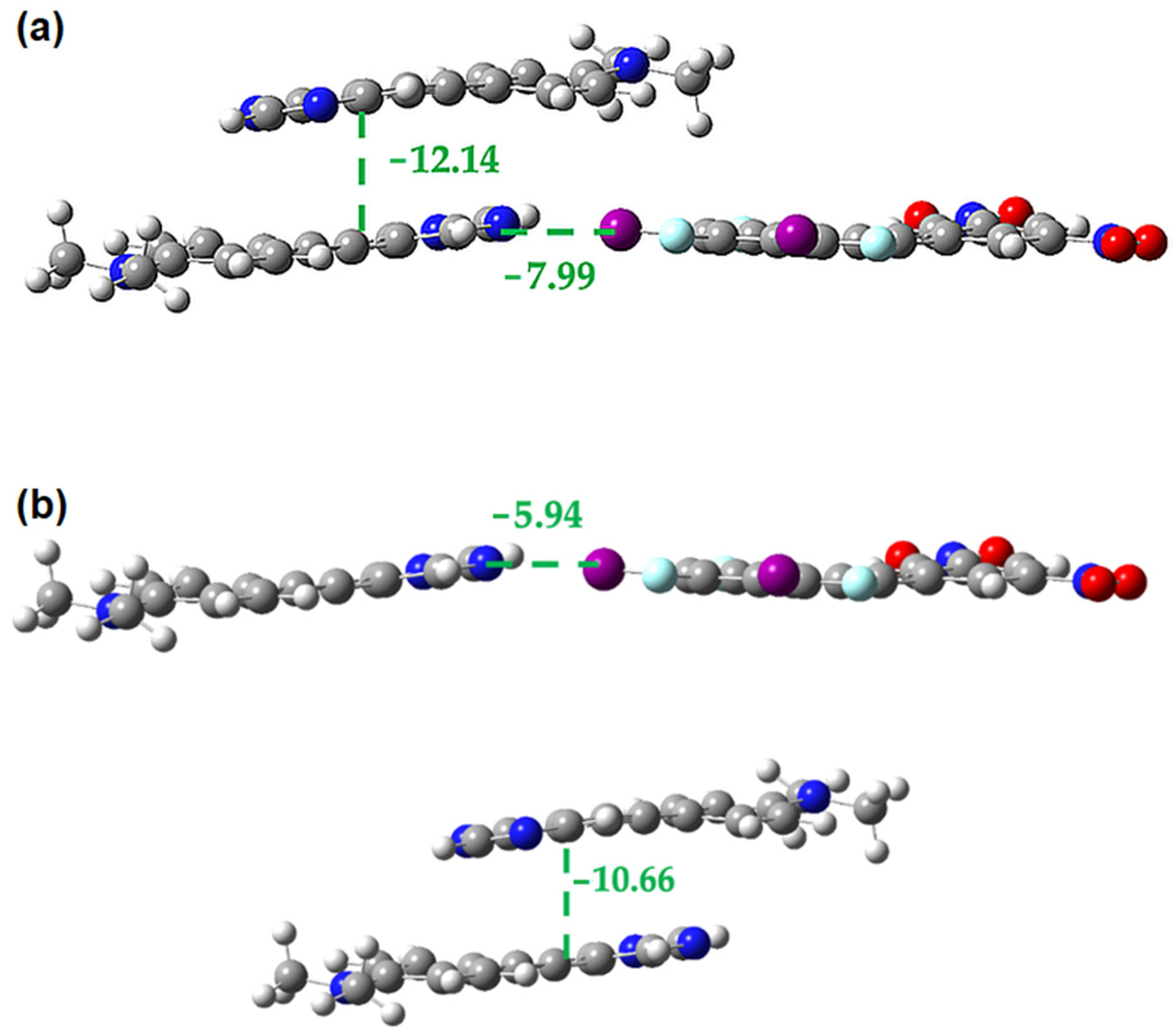
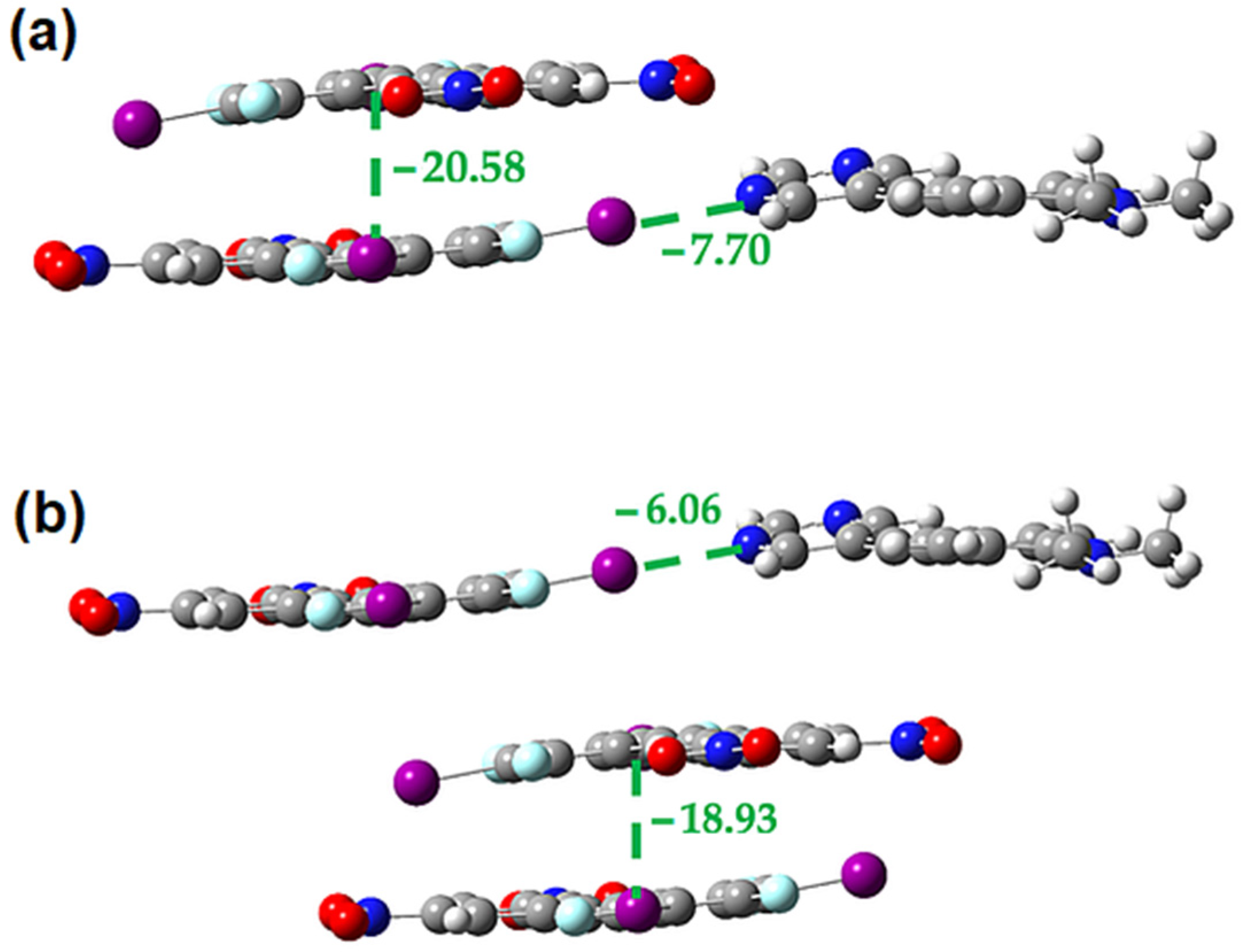
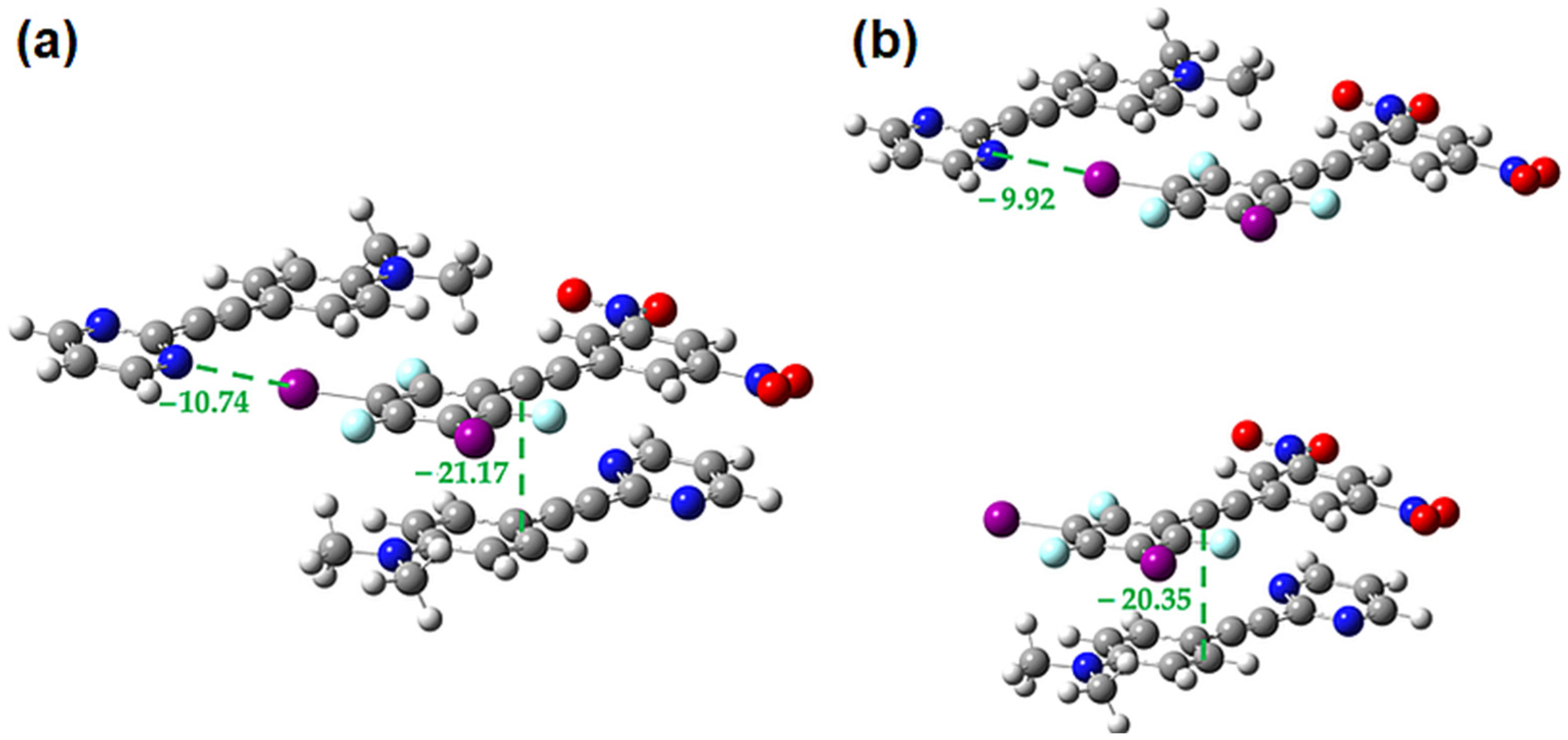
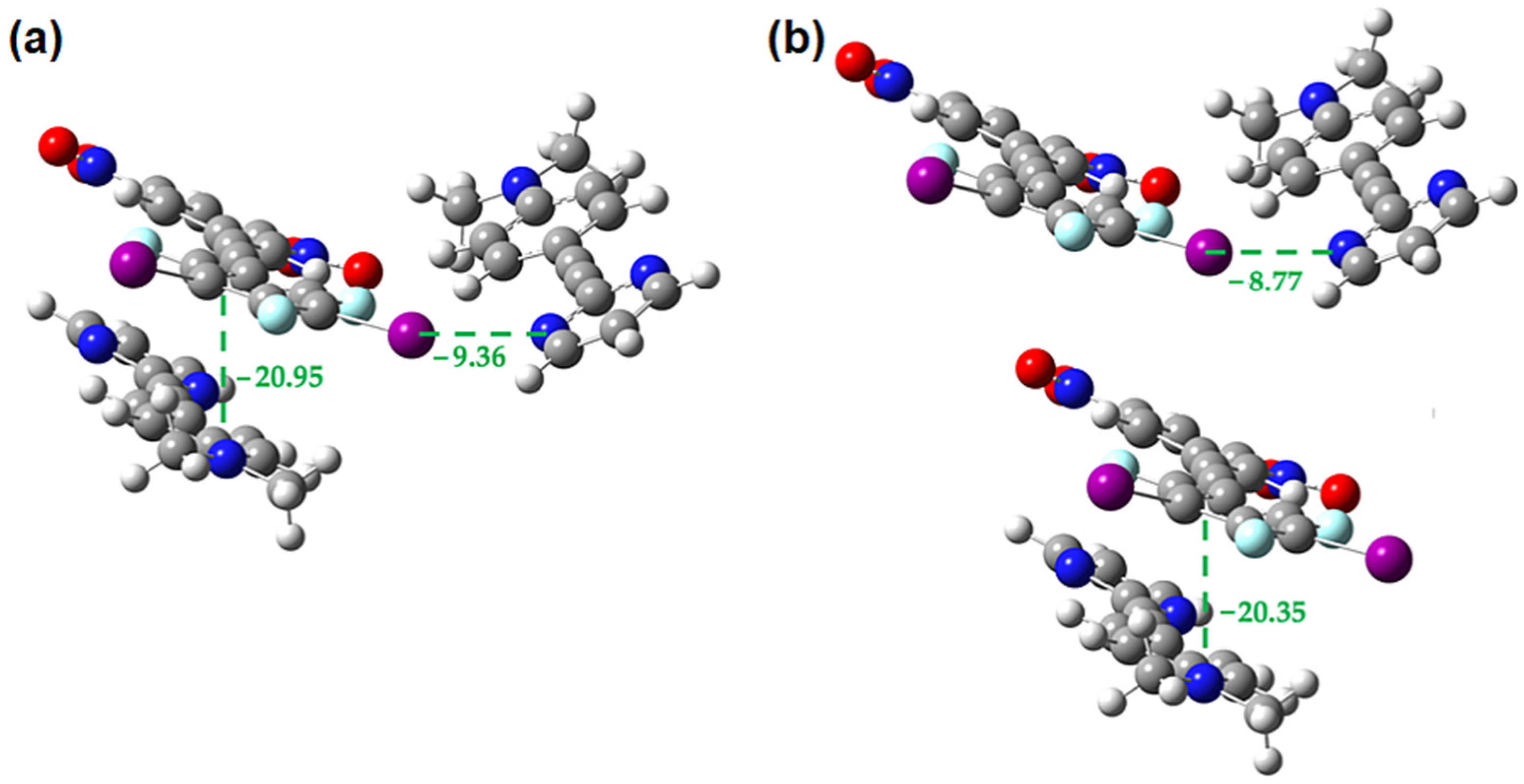
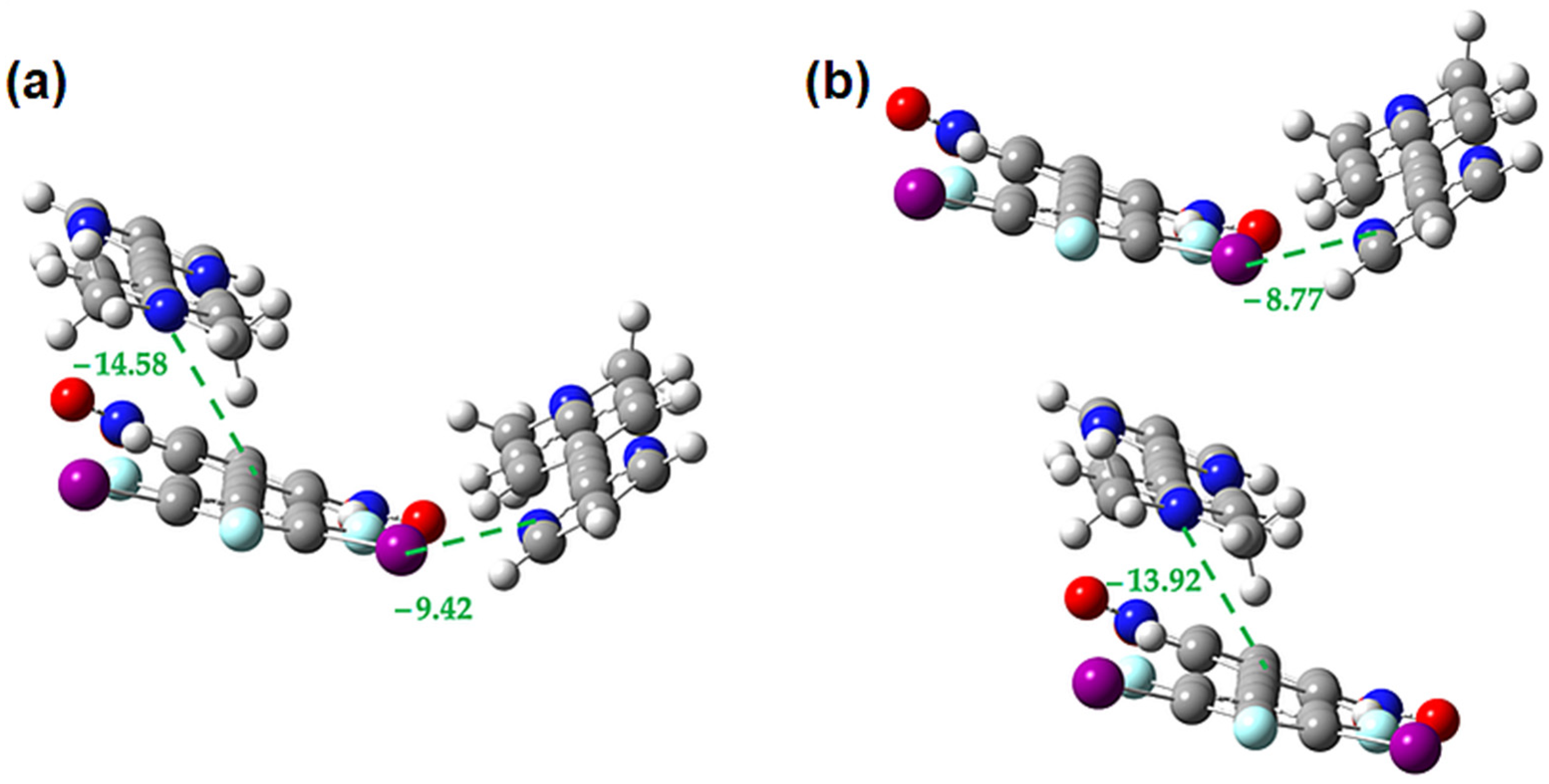
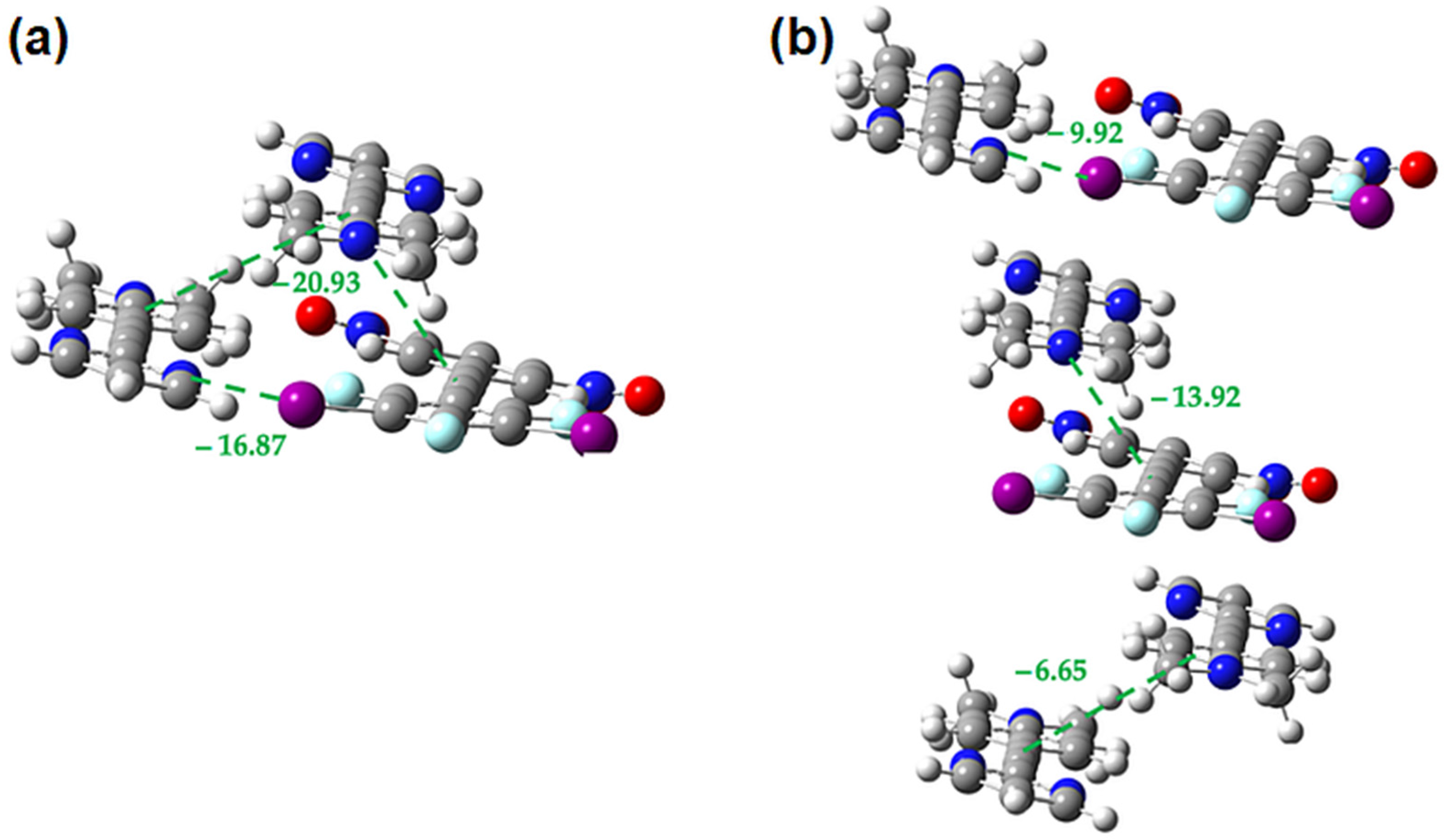
2.3. Computational Analysis of the Interplay between Halogen Bonding and π-Stacking
3. Materials and Methods
3.1. Preparation of Materials
3.2. Cocrystallization
3.3. Structure Refinement
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Rest, C.; Kandanelli, R.; Fernandez, G. Strategies to create hierarchical self-assembled structures via cooperative non-covalent interactions. Chem. Soc. Rev. 2015, 44, 2543–2572. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Sastry, G.N. Cooperative or Anticooperative: How Noncovalent Interactions Influence Each Other. J. Phys. Chem. B 2015, 119, 11121–11135. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zhou, Y.; Zhu, X. Supramolecular Dendritic Polymers: From Synthesis to Applications. Acc. Chem. Res. 2014, 47, 2006–2016. [Google Scholar] [PubMed]
- Li, F.; Yager, K.G.; Dawson, N.M.; Jiang, Y.-B.; Malloy, K.J.; Qin, Y. Stable and Controllable Polymer/Fullerene Composite Nanofibers through Cooperative Noncovalent Interactions for Organic Photovoltaics. Chem. Mater. 2014, 26, 3747–3756. [Google Scholar] [CrossRef]
- Huang, Z.; Qin, K.; Deng, G.; Wu, G.; Bai, Y.; Xu, J.; Wang, Z.; Yu, Z.; Scherman, O.A.; Zhang, X. Supramolecular Chemistry of Cucurbiturils: Tuning Cooperativity with Multiple Noncovalent Interactions from Positive to Negative. Langmuir 2016, 32, 12352–12360. [Google Scholar] [CrossRef] [Green Version]
- Loh, C.C.J. Exploiting non-covalent interactions in selective carbohydrate synthesis. Nat. Rev. Chem. 2021, 5, 792–815. [Google Scholar] [CrossRef]
- Motloch, P.; Bols, P.S.; Anderson, H.L.; Hunter, C.A. Cooperative assembly of H-bonded rosettes inside a porphyrin nanoring. Chem. Sci. 2021, 12, 1427–1432. [Google Scholar] [CrossRef]
- Quentin, J.; Swenson, D.C.; MacGillivray, L.R. Supramolecular Sandwiches: Halogen-Bonded Coformers Direct [2+2] Photoreactivity in Two-Component Cocrystals. Molecules 2020, 25, 907. [Google Scholar]
- Majumdar, P.; Tharammal, F.; Gierschner, J.; Varghese, S. Tuning Solid-State Luminescence in Conjugated Organic Materials: Control of Excitonic and Excimeric Contributions through π Stacking and Halogen Bond Driven Self-Assembly. ChemPhysChem 2020, 21, 616–624. [Google Scholar] [CrossRef]
- Liu, Y.H.; Dadvand, A.; Titi, H.M.; Hamzehpoor, E.; Perepichka, D.H. Halogen bonding vs. π-stacking interactions in new bis(acenaphthylene)dione semiconductors. CrystEngComm 2021, 23, 8255–8259. [Google Scholar] [CrossRef]
- Zhu, W.; Zheng, R.; Zhen, Y.; Yu, Z.; Dong, H.; Fu, H.; Shi, Q.; Hu, W. Rational Design of Charge-Transfer Interactions in Halogen-Bonded Co-crystals toward Versatile Solid-State Optoelectronics. J. Am. Chem. Soc. 2015, 137, 11038–11046. [Google Scholar] [CrossRef] [PubMed]
- Nwachukwu, C.I.; Kehoe, Z.R.; Bowling, N.P.; Speetzen, E.D.; Bosch, E. Cooperative halogen bonding and polarized π-stacking in the formation of colored mixed-stack charge-transfer co-crystals. New J. Chem. 2018, 42, 10615–10622. [Google Scholar] [CrossRef]
- Nwachukwu, C.I.; Patton, L.J.; Bowling, N.P.; Bosch, E. Ditopic halogen bonding with bipyrimidines and activated pyrimidines. Acta Cryst. 2020, C76, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Gromwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer 17.5; University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dey, S.K.; Ojha, B.; Das, G. A subtle interplay of C–H hydrogen bonds in complexation of anions of varied dimensionality by a nitro functionalized tripodal podand. CrystEngComm 2011, 13, 269–278. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Veluthaparam, R.V.P.; Saha, A.; Saha, B.K. The Effects of Electronegativity of X and Hybridization of C on the X−C···O Interactions: A Statistical Analysis on Tetrel Bonding. ChemPlusChem 2021, 86, 1123–1127. [Google Scholar] [CrossRef]
- Thalladi, V.R.; Weiss, H.-C.; Blaser, D.; Boese, R.; Nangia, A.; Desiraju, G.R. C−H···F Interactions in the Crystal Structures of Some Fluorobenzenes. J. Am. Chem. Soc. 1998, 120, 8702. [Google Scholar] [CrossRef]
- Bowling, N.P.; Speetzen, E.D.; Bosch, E. Arylethynyl Helices Supported by π-Stacking and Halogen Bonding. ChemPlusChem 2021, 86, 745. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Wu, J.Y.; Dai, G.L. Study on the Halogen Bond and π-π-Stacking Interaction between Fluoro Substituted Iodobenzene and Pyrazine. J. Mol. Model. 2020, 26, 333. [Google Scholar] [CrossRef]
- Kautny, P.; Kriegner, H.; Bader, D.; Dusek, M.; Reider, G.A.; Frohlich, J.; Stoge, B. Ethyne-Linked Push–Pull Chromophores: Implications of Crystal Structure and Molecular Electronics on the Quadric Nonlinear Activity. Cryst. Growth Des. 2017, 17, 4124–4136. [Google Scholar] [CrossRef]
- Bosch, E. CCDC 2129575: Experimental Crystal Structure Determination. 2021. Available online: https://doi.org/10.5517/ccdc.csd.cc29gzzr (accessed on 14 February 2022).
- Apex 2; Bruker AXS: Madison, WI, USA, 2014.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Barbour, L.J. X-Seed—A software tool for supramolecular crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer Model Energies and Energy Frameworks: Extension to Metal Coordination Compounds, Organic Salts, Solvates and Open-Shell Systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5642–5648. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-Type Basis Sets for Local Spin Density Functional Calculations. Part I. Boron through Neon, Optimization Technique and Validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef] [Green Version]
- Sosa, C.; Andzelm, J.; Elkin, B.C.; Wimmer, E.; Dobbs, K.D.; Dixon, D.A. A Local Density Functional Study of the Structure and Vibrational Frequencies of Molecular Transition-Metal Compounds. J. Phys. Chem. 1992, 96, 6630–6636. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Non-Covalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.E.; Krieg, J. A Consistent and Accurate Ab Initio Parameterization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boys, S.F.; Bernardi, F. Calculation of Small Molecular Interactions by Differences of Separate Total Energies—Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How Does Basis Set Superposition Error Change the Potential Surfaces for Hydrogen Bonded Dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef] [Green Version]























{kind=link}
{kind=link}
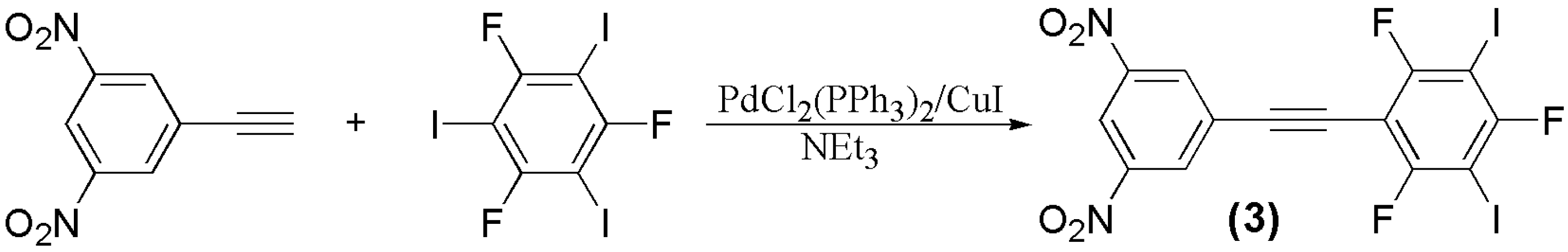{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| π-Dimer | Eele | Epol | Edis | Erep | Etotal |
|---|---|---|---|---|---|
| D-D | −53.0 | −3.4 | −109.9 | 123.3 | −78.1 |
| D-A | −48.9 | −4.8 | −88.3 | 100.6 | −70.1 |
| A-A | −34.7 | −2.3 | −59.0 | 84.4 | −37.6 |
| Interaction Type | R 1 | Eele | Epol | Edis | Erep | Etotal |
|---|---|---|---|---|---|---|
| Halogen bond | 12.21 | −46.8 | −5.6 | −9.5 | 62.5 | −23.3 |
| Halogen bond | 12.25 | −42.3 | −5.1 | −9.3 | 53.9 | −23.3 |
| Side-by-side | 8.41 | −13.3 | −2.5 | −27.2 | 39.8 | −15.0 |
| Side-by-side | 8.44 | −11.5 | −2.5 | −26.0 | 34.0 | −15.6 |
| π-Dimer | Eele | Epol | Edis | Erep | Etotal |
|---|---|---|---|---|---|
| O-F-F | −35.5 | −5.2 | −72.9 | 81.9 | −54.3 |
| F-F | −54.7 | −6.5 | −96.7 | 110.0 | −78.4 |
| rXB (Å) | C-I-N Angle (Degrees) | rTB (Å) | Eele | Epol | Edis | Erep | Etotal |
| 3.022(3) | 171.77(9) | 2.905(3) | −49.9 | −7.5 | −27.3 | 69.4 | −39.3 |
| 3.261(3) | 162.93(10) | 3.044(4) | −34.3 | −5.3 | −31.4 | 51.3 | −35.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Speetzen, E.D.; Nwachukwu, C.I.; Bowling, N.P.; Bosch, E. Complementary, Cooperative Ditopic Halogen Bonding and Electron Donor-Acceptor π-π Complexation in the Formation of Cocrystals. Molecules 2022, 27, 1527. https://doi.org/10.3390/molecules27051527
Speetzen ED, Nwachukwu CI, Bowling NP, Bosch E. Complementary, Cooperative Ditopic Halogen Bonding and Electron Donor-Acceptor π-π Complexation in the Formation of Cocrystals. Molecules. 2022; 27(5):1527. https://doi.org/10.3390/molecules27051527
Chicago/Turabian StyleSpeetzen, Erin D., Chideraa I. Nwachukwu, Nathan P. Bowling, and Eric Bosch. 2022. "Complementary, Cooperative Ditopic Halogen Bonding and Electron Donor-Acceptor π-π Complexation in the Formation of Cocrystals" Molecules 27, no. 5: 1527. https://doi.org/10.3390/molecules27051527
APA StyleSpeetzen, E. D., Nwachukwu, C. I., Bowling, N. P., & Bosch, E. (2022). Complementary, Cooperative Ditopic Halogen Bonding and Electron Donor-Acceptor π-π Complexation in the Formation of Cocrystals. Molecules, 27(5), 1527. https://doi.org/10.3390/molecules27051527