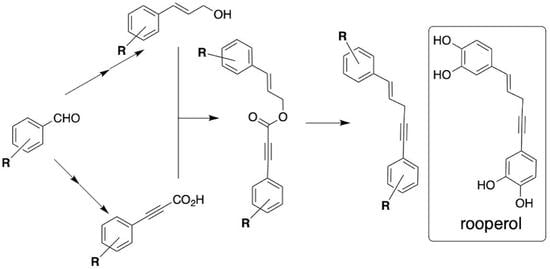
4. Materials and Methods
All reactions were carried out under argon in oven-dried glassware with magnetic stirring. Unless otherwise noted, all materials were obtained from commercial suppliers and were used without further purification. THF was distilled from sodium/benzophenone prior to use. Flash chromatography was performed with EM Reagent silica gel (230–400 mesh) using the mobile phase indicated. Melting points (open capillary) are uncorrected. Unless otherwise noted,
1H and
13C NMR spectra were determined in CDCl
3 on a spectrometer operating at 400 and 100 MHz, respectively, and are reported in ppm using solvent as internal standard (7.26 ppm for
1H and 77.0 ppm for
13C in CDCl
3). Mass spectra were obtained by atmospheric pressure chemical ionization (APCI), chemical ionization using methane as the ionizing gas (CI), or by electrospray ionization (ESI). Copies of all NMR and MS spectra are available in the
Supporting Information.
General Procedure-Silyl Protection of Benzaldehydes: 1,2-bis-((
tert-butyldimethyl-silyl)
oxy)
benzaldehyde (
8A) [
17]: To solution of 3,4-dihydroxybenzaldehyde (4 g, 29 mmol) in dichloromethane (120 mL) under argon was added imidazole (7.9 g, 4 equivalent). The reaction mixture was cooled in an ice bath and tert-butyldimethyl-silyl chloride (12 g, 2.5 equivalent) was added. The reaction mixture was allowed to stir overnight, then diluted with dichloromethane and washed twice with 1 N HCl, once with saturated aqueous NaHCO
3, once with brine, and then, the organic layer was dried over Na
2SO
4. After filtration, the solution was evaporated under vacuum and the residue subjected to flash chromatography purification (SiO
2, 10% EtOAc/ hexanes) to yield the product (9.7 g, 91%) as a viscous oil, which crystallized to a pale yellow solid. Mp: 42–43 °C (lit: 39–41 °C [
18]); IR (neat, ATR): cm
−1 2953, 2926, 2856, 1694, 1569, 1504, 1298, 1284, 1269, 1248, 1213, 1157, 1104, 973, 897, 825, 781, 731;
1H NMR (400 MHz, CDCl
3): δ 9.81 (s, 1H), 7.38–7.34 (m, 2H), 6.94 (d, 1H,
J = 8.0 Hz), 0.99 (s, 18H), 0.25 (s, 6H), 0.23 (s, 6H);
13C{
1H} NMR (100 MHz, CDCl
3): δ 190.7, 153.3, 147.6, 130.7, 125.2, 120.7, 120.5, 25.8, 25.7, 18.4, 18.2, −4.0, −4.2; MS (APCI, pos)
m/z (%): 367(67) [M + H]
+.
4-((
tert-butyldimethylsilyl)
oxy)
benzaldehyde (
8B) [
19]: Prepared from 4-hydroxybenzaldehyde following the general procedure described for
8A, which afforded a quantitative yield (9.69 g) of a colorless oil. IR (neat, ATR): cm
−1 2955, 2930, 2858, 1697, 1506, 1255, 1154, 902;
1H NMR (500 MHz, CDCl
3): δ 9.89 (s, 1H), 7.79 (d, 2H,
J = 8.6 Hz), 6.94 (d, 2H, J = 8.6 Hz), 0.99 (s, 9H), 0.25 (s, 6H);
13C{
1H} NMR (125 MHz, CDCl
3): δ 190.8, 161.5, 131.9, 130.4, 120.5, 25.5, 18.2, −4.3; MS (ESI, pos)
m/z (%): 237(55) [M + H]
+.
3,5-bis((
tert-butyldimethylsilyl)
oxy)
benzaldehyde (
8C) [
20]: Prepared from 3,5-dihydroxybenzaldehyde following the general procedures described above for
8A, which afforded a 93% yield (1.28 g) of a slightly yellow oil that slowly crystalized in the freezer. Mp = 28–29 °C; IR (neat, ATR, cm
−1) 2955, 2930, 2857, 1702, 1587, 1330, 1253, 1165, 826;
1H NMR (500 MHz, CDCl
3): δ 9.86 (s, 1H), 6.95 (d, 2H,
J = 2.3 Hz), 6.59 (t, 1H,
J = 2.3 Hz), 0.99 (s, 18H), 0.22 (s, 12H);
13C{
1H} NMR (125 MHz, CDCl
3): δ 191.7, 157.3, 138.4, 118.4, 114.3, 25.6, 18.2, −4.4; MS (APCI, pos)
m/z (%): 367(17) [M + H]
+.
2,2-dimethyl-4H-benzo[d][1,3]dioxine-6-carbaldehyde 8(
F): Prepared as previously reported [
21] from 4-bromosalicyl alcohol isopropylidene acetal to afford 11.17 g (93% yield) of colorless oil that slowly recrystallized in the freezer to afford off-white crystals. Mp = 55–58 °C (lit. 56–58 °C [
21]); IR (neat, ATR): cm
−1 2992, 2869, 1690, 1496, 1384, 1269;
1H NMR (500 MHz, CDCl
3): δ 9.85 (s, 1H), 7.70 (dd, 1H,
J = 8.5, 1.0 Hz), 7.55 (t, 1H,
J = 1.0 Hz), 6.93 (d, 1H,
J = 8.5 Hz), 4.90 (s, 2H), 1.57 (s, 6H);
13C{
1H} NMR (125 MHz, CDCl
3): δ 190.7, 156.8, 130.5, 129.4, 126.6, 119.7, 117.7, 100.8, 60.6, 24.8; MS (ESI, pos)
m/z (%): 215(14) [M + Na]
+, 191(15), 161(11).
General Procedure—Wittig reaction: Methyl (
E)
-3-(
3,4-bis((
tert-butyldimethylsilyl)
oxy)
phenyl)
acrylate (
4A) [
22]: To a solution of methyl (triphenylphosphoranylidene)acetate (18.4 g, 55 mmol) in 200 mL of DCM at room temperature was added a ca. 1 M solution of 3,4-bis(
tert-butyldimethylsilyloxy)benzaldehyde (
8A, 6.33 g, 45.8 mmol) in DCM dropwise over 5 min. Upon completion of the addition, the mixture was stirred for an additional 18 h at room-temperature, and the solvent was removed by evaporation. The resulting pasty oil was diluted with 40 mL of hexanes, and the Ph
3PO that precipitated was removed by filtration, and the filter cake was washed with two 40 mL portions of hexanes. The combined organic layers were concentrated under reduced pressure. Flash chromatography on silica gel (11:1 hexanes/EtOAc) of the residue afforded 4.17 g (91%) of 4′A0 as a white solid. Mp = 63.0–64.0 °C; IR (ATR, neat): cm
−1 2947, 2930, 2857, 1721, 1631, 1506, 1472, 1422, 1289, 1249, 1161, 1126, 911, 838, 813, 777;
1H NMR (500 MHz, CDCl
3): δ 6.72 (d, 2H,
J = 2.1 Hz), 7.64 (d, 1H,
J = 15.9 Hz), 6.46 (t, 1H,
J = 2.1 Hz), 6.44 (d, 1H,
J = 15.9 Hz), 3.88 (s, 3H), 0.99 (s, 18H), 0.19 (s, 12H);
13C{
1H} NMR (125 MHz, CDCl
3): δ 167.7, 149.4, 147.1, 144.8, 128.0, 122.2, 121.1, 120.4, 115.4, 51.5, 25.9, 25.8, 18.5, 18.4, −4.0, −4.1; MS (APCI, pos)
m/z (%): 423(66) [M + H]
+.
Methyl (
E)
-3-(
4-((
tert-butyldimethylsilyl)
oxy)
phenyl)
acrylate (
4B) [
23]: Prepared from aldehyde
8B (2.22 g, 9.4 mmol) following the general procedure described for
4A, which afforded a
4B as a white solid (2.73 g, 91% yield). Mp = 34.0–36.0 °C; IR (ATR, neat): cm
−1 2953, 2928, 2856, 1710, 1635, 1598, 1508, 1436, 1324, 1251, 1192, 1166, 988, 908, 834, 780;
1H NMR (400 MHz, CDCl
3): δ 7.64 (d, 1H,
J = 15.9), 7.41 (d, 2H,
J = 8.6), 6.84 (d, 3H,
J = 8.6), 6.30 (d, 1H,
J = 15.9), 3.79 (s, 3H), 0.99 (s, 9H), 0.21 (s, 6H);
13C{
1H} NMR (100 MHz, CDCl
3): δ 167.6, 157.8, 144.5, 129.6, 127.6, 120.4, 120.4, 115.4, 51.5, 25.6, 18.2, −4.4; MS (ESI, pos)
m/z (%): 293(24) [M + H]
+.
Methyl (
E)
-3-(
3,5-bis((
tert-butyldimethylsilyl)
oxy)
phenyl)
acrylate (
4C) [
24]: Prepared from aldehyde
8C following the general procedure described for
4A, which afforded
4C as a colorless oil, (3.93 g, 95% yield). IR (ATR, neat) cm
−1 2951, 2928, 2857, 1716, 1638, 1578, 1437, 1281, 1154, 1002, 828, 778;
1H NMR (500 MHz, CDCl
3): δ 7.55 (d, 1H,
J = 15.9 Hz), 6.62 (dd, 2H,
J = 2.2, 0.4 Hz), 6.36 (t, 1H,
J = 2.2 Hz), 6.34 (d, 1H,
J = 15.9 Hz), 3.80 (s, 3H), 0.98 (s, 18H), 0.20 (s, 12H);
13C{
1H} NMR (125 MHz, CDCl
3): δ 167.4, 156.9, 144.8, 136.1, 117.9, 114.2, 113.2, 51.7, 25.6, 18.2, −4.4; MS (APCI, pos)
m/z (%): 423(58) [M + H]
+.
Methyl (
E)
-3-(
3,4-difluorophenyl)
acrylate (
4D) [
25]: Prepared from 3,4-difluorobenzaldehyde following the general procedure described for
4A, which afforded
4D as a white solid (3.17 g, 91%). Mp = 76.9–77.9 °C; IR (ATR, neat): cm
−1 3057, 2957, 2920, 1704, 1640, 1514, 1494, 1435, 1330, 1270, 1250, 1220, 1189, 1175, 1144, 1111, 987, 870, 814, 790;
1H NMR (400 MHz, CDCl
3): δ 7.57 (d, 1H,
J = 16.0 Hz), 7.32 (ddd, 1H,
J = 11.0, 7.6, 2.1 Hz), 7.25–7.21 (m, 1H), 7.20–7.12 (m, 1H), 6.33 (dd, 1H,
J = 16.0, 0.3 Hz), 3.79 (s, 3H);
13C{
1H} NMR (100 MHz, CDCl
3): δ 166.8, 151.5 (dd,
J = 253, 13 Hz), 153.4 (dd,
J = 249, 13 Hz), 142.4, 131.6 (dd,
J = 5, 5 Hz), 124.7 (dd,
J = 6, 3 Hz), 118.9 (d,
J = 2 Hz), 117.8 (d,
J = 17 Hz), 116.3 (d,
J = 17 Hz), 51.7;
19F{
1H} NMR (376 MHz, CDCl
3): δ −134.2 (d,
J = 21 Hz), −136.6 (d,
J = 21 Hz); MS (APCI, pos)
m/z (%): 199(5) [M + H]
+.
Methyl (
E)
-3-(
benzo[d][1,3]
dioxol-5-yl)
acrylate (
4E) [
26]: Prepared from piperonal following the general procedure described for
4A, which afforded
4E as a white solid (5.4 g, 96%). Mp: 133–135 °C; IR (ATR, neat): cm
−1 2950, 2901, 1699, 1622, 1597, 1594, 1495, 1454, 1438, 1367, 1304, 1255, 1201, 1169, 1124, 1105, 1035, 1004, 931, 916, 821;
1H NMR (400 MHz, CDCl
3): δ 7.53 (d, 1H,
J = 16.0 Hz), 6.98–7.04 (m, 2H), 6.82 (d, 1H,
J = 8.4 Hz), 6.26 (d, 1H, J = 16.0 Hz), 6.01 (s, 2H), 3.79 (s, 3H);
13C{
1H} NMR (100 MHz, CDCl
3): δ 167.6, 149.6, 148.3, 144.5, 128.8, 124.4, 115.7, 108.5, 106.4, 101.5, 51.6; MS (ESI, pos)
m/z (%): 207(78) [M + H]
+.
Methyl (E)-3-(2,2-dimethyl-4H-benzo[d][1,3]dioxin-6-yl)acrylate (4F): Prepared from 2,2-dimethyl-4H-benzo(d)(1,3)dioxine-6-carbaldehyde (8F) following the general procedure described for 4A, which afforded 4F as a white, waxy solid (249 mg, 99%). IR (ATR, neat): cm−1 2992, 2947, 2860, 1716, 1627, 1609, 1582, 1497, 1448, 1463, 1384, 1374, 1267, 1191, 1166, 1114, 1062, 953, 863, 830, 796; 1H NMR (400 MHz, CDCl3): δ 7.60 (1H, dd, J = 16.0, 3.0 Hz), 7.33–7.35 (m, 1H), 7.13 (s, 1H), 6.81 (dd, 1H, J = 8.4, 3.3 Hz), 6.26–6.29 (m, 1H), 4.83 (s, 2H), 3.77 (s, 3H), 1.53 (s, 6H); 13C{1H} NMR (100 MHz, CDCl3): δ 167.6, 153.3, 144.4, 128.0, 126.7, 124.9, 119.7, 117.6, 115.4, 100.1, 60.6, 51.5, 24.7; MS (ESI, pos) m/z (%): 271(10) [M + Na]+, 304(71) [M + Na + CH3OH]+; HRMS (ESI, pos): m/z calculated for C14H16O4 [M + H]+ 249.1121, found 249.1124.
General Procedure–DIBAL-H Reduction: (E)-3-(3,4-bis((tert-butyldimethylsilyl)oxy)phenyl)prop-2-en-1-ol (5A): To a solution of 4A (3.0 g, 7.1 mmol) in 70 mL of dry DCM in a 250 mL rb flask at −78 °C under Ar was added dropwise a solution of DIBAL-H (1.2 M solution in hexane, 18 mL, 21.6 mmol). Upon completion of the addition, the reaction mixture was allowed to warm to room temperature over 2 h and then carefully added dropwise to a stirred mixture of 2 M HCl (100 mL) and ice. After stirring for 30 min, the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure to afford a colorless oil that was subjected to flash chromatography purification (SiO2, 0–10% EtOAc/hexanes) to yield the product (2.25 g, 80%) as a colorless oil. IR (ATR, neat): cm−1 3360(br), 2929, 2886, 2857, 1598, 1511, 1471, 1419, 1301, 1254, 1124, 989, 905, 840, 781; 1H NMR (400 MHz, CDCl3): δ 6.88 (d, 1H, J = 2.2 Hz), 6.85 (dd, 1H, J = 8.3, 2.2 Hz), 6.77 (d, 1H, J = 8.2 Hz), 6.48 (dt, 1H, J = 15.8, 1.3 Hz), 6.18 (dt, 1H, J = 15.8, 5.9 Hz), 4.30–4.27 (m, 1H), 1.38 (t, 1H, J = 5.7 Hz), 0.99 (s. 9H), 0.98 (s, 9H), 0.20 (s, 6H), 0.19 (s, 6H); 13C{1H} NMR (100 MHz, CDCl3): δ 146.9, 131.2, 130.2, 126.3, 121.0, 119.9, 119.0, 63.9, 25.9, 25.8, 18.5, 18.4, −4.1; MS (ESI, pos) m/z (%): 417(25) [M + Na]+, 377(15); HRMS (ESI, pos): m/z calculated for C21H38NaO3Si2 [M + Na]+ 417.2257, found 417.2253.
(
E)
-3-(
4-((
tert-butyldimethylsilyl)
oxy)
phenyl)
prop-2-en-1-ol (
5B) [
23]: Prepared from
4B following the general procedure described for
5A, which afforded
5B as a colorless oil, 1.3 g (85%). IR (ATR, neat): cm
−1 3325(br), 2954, 2928, 2857, 2602, 1507, 1252, 1168, 908, 835, 799, 778;
1H NMR (400 MHz, CDCl
3): δ 7.26 (d, 2H,
J = 8.5 Hz), 6.79 (d, 2H,
J = 8.5 Hz), 6.55 (d, 1H,
J = 15.9 Hz), 6.24 (dt, 1H,
J = 15.5, 5.9 Hz), 4.39 (dd, 2H,
J = 5.9, 1.2 Hz), 2.36 (d, 1H,
J = 0.3 Hz), 0.98 (s, 9H), 0.20 (s, 6H);
13C{
1H} NMR (100 MHz, CDCl
3): δ 155.5, 131.0, 129.9, 127.6, 126.4, 120.2, 63.9, 25.7, 18.2, -4.4; MS (APCI, pos)
m/z (%): 264(5) [M]
+, 247(67).
(
E)
-3-(
3,5-bis((
tert-butyldimethylsilyl)
oxy)
phenyl)
prop-2-en-1-ol (
5C) [
24]. Prepared from
4C following the general procedure described for
5A, which afforded
5C as a colorless oil, 914 mg (95%). IR (neat, ATR): cm
−1 3292(br), 2928, 2857, 1581, 1435, 1251, 1164, 1021, 827, 777;
1H NMR (400 MHz, CDCl
3): δ 6.50–6.46 (m, 3H), 6.29 (dt, 1H,
J = 10.0, 5.0 Hz), 6.25 (t, 1H,
J= 2.1 Hz), 4.29 (dd, 2H,
J = 5.6, 1.3Hz), 0.98 (s, 18H), 0.19 (s, 12H);
13C{
1H} NMR (100 MHz, CDCl
3): δ 156.6, 138.4, 131.0, 128.6, 111.7, 111.6, 63.7, 25.7, 18.2, −4.4.
(E)-3-(3,4-difluorophenyl)prop-2-en-1-ol (5D). Prepared from 4D following the general procedure described for 5A, which afforded 5D as a colorless oil, 1.18 g (92%). IR (neat, ATR): cm−1 3271(br), 2987, 2845, 1603, 1514, 1289, 1270, 1090, 1018, 967; 1H NMR (400 MHz, CDCl3): δ 7.21–7.16 (m, 1H), 7.13–7.06 (m, 2H), 6.53 (d, 1H, J = 15.9 Hz), 6.27 (dt, 1H, J = 15.9, 5.5 Hz), 4.32 (dd, 2H, J = 5.5, 1.4 Hz); 13C{1H} NMR (100 MHz, CDCl3): δ 150.5 (dd, J = 247, 13 Hz), 149,8 (dd, J = 249, 13 Hz), 133.9 (dd, J = 6, 4 Hz), 129.6 (d, J = 2 Hz), 128.8, 122.6 (dd, J = 6, 3 Hz) 117.3 (d, J = 18 Hz), 114.7 (d, J = 8 Hz), 63.2; 19F NMR (376 MHz, CDCl3) δ: −137.9 (d, J = 21 Hz), −139.0 (d, J = 21 Hz); MS (ESI, pos) m/z (%): 171 [M + H]+(9), 157(12); HRMS (EI, pos): m/z calculated for C9H8OF2 [M]+ 170.0543, found 170.0539.
(E)-3-(benzo[d][1,3]dioxol-5-yl)prop-2-en-1-ol (5E). Prepared from 4E following the general procedure described for 5A, which afforded 5E as a white solid, 1.94 g (90%). Mp: 79.5–80.0 °C; IR (neat, ATR): cm−1 3350, 2920, 2895, 2851, 1499, 1441, 1243, 1083, 1034, 1003, 965, 920, 909; 1H NMR (500 MHz, CDCl3): δ 6.93 (d, 1H, J = 1.5 Hz), 6.81 (1H, dd, J= 8.0, 1.5 Hz), 6.75 (d, 1H, J = 8.0 Hz), 6.52 (dd, 1H, J = 15.8, 1.3 Hz) 6.20 (dd, 1H, J = 15.8, 5.9 Hz), 5.96 (s, 2H), 4.29 (td, 2H, J = 5.8, 1.3 Hz), 1.44 (t, 1H, J = 5.9 Hz); 13C{1H} NMR (100 MHz, CDCl3): δ 148.0, 147.3, 131.1, 131.0, 126.6, 121.1, 108.2, 105.7, 101.1, 63.7; MS (ESI, pos) m/z (%): 177(20) [M-H]+.
(E)-3-(2,2-dimethyl-4H-benzo[d][1,3]dioxin-6-yl)prop-2-en-1-ol (5F). Prepared from 4′C3 following the general procedure described for 4A0, which afforded a white solid, 248 mg (quant.). Mp: 130.0–130.9 °C; IR (neat, ATR): cm−1 3355(br), 2992, 2854, 1498, 1383, 1260, 1200, 1142, 1115, 1056, 1004, 975, 958, 868, 831; 1H NMR (500 MHz, CDCl3): δ 7.20 (dd, 1H, J = 8.5, 1.8 Hz), 6.98 (d, 1H, J = 2.0 Hz), 6.77 (d, 1H, J = 8.5 Hz), 6.49–6.52 (m, 1H), 6.21 (dt, 1H, J = 15.8, 5.9 Hz), 4.82 (s, 2H), 4.28 (dd, 2H, J = 5.9, 1.5 Hz), 1.54 (s, 6H); 13C{1H} NMR (125 MHz, CDCl3): δ 150.9, 130.8, 129.1, 125.5, 126.2, 122.7, 119.4, 117.3, 99.7, 63.8, 60.8, 24.7; MS (APCI, pos) m/z (%): 203(87) [M − H2O + H]+; HRMS (CI, pos) calculated for C13H16O3 [M]+ 220.1099, found 220.1101.
General Procedure–Vinyl Dibromide Formation: ((
4-(
2,2-dibromovinyl)
-1,2-phenylene)
bis(
oxy))
bis(
tert-butyldimethylsilane) (
6A) [
27]. To a 250 mL round-bottom flask with stirbar under Ar was placed 8.92 g (26.9 mmole) of CBr
4. DCM (30 mL) was added to the flask via syringe, and the reaction flask was cooled in an ice bath. Triphenylphosphine (14.09 g, 53.7 mmole) was added the flask in portions, and the resulting mixture was stirred under Ar for 5 min. A solution of 4.94 g (13.5 mmole) of the aldehyde
8A in 10 mL DCM was added to the reaction mixture, and the ice bath was removed. The mixture was allowed to stir at room temperature for 30 min, when TLC monitoring indicated the reaction was complete. Saturated sodium bicarbonate solution was carefully added until the aqueous layer was neutral by pH paper. The aqueous layer was then extracted with three 80 mL portions of DCM. The combined organic extracts were washed with saturated brine and then dried over anhydrous sodium sulfate. The dried solution was filtered and then evaporated under reduced pressure, and the residue subjected to chromatography (0–5% EtOAc/hex) to afford 6.36 g (91 % yield) of
6A as a slightly yellow oil. IR (ATR, neat): cm
−1 2952, 2928, 2857, 1503, 1471, 1294, 1251, 1128, 881, 903, 835, 777;
1H NMR (400 MHz, CDCl
3): δ 7.38 (s, 1H), 7.28 (d, 1H,
J = 2.3 Hz), 6.99 (ddd, 1H,
J = 8.3, 2.3, 0.6 Hz), 6.85 (d, 1H,
J = 8.3 Hz), 1.05 (s, 9H), 1.04 (s, 9H), 0.28 (s, 6H), 0.27 (s, 6H);
13C{
1H} NMR (100 MHz, CHCl
3): δ 147.5, 145.6, 136.4, 128.4, 122.5, 120.7, 120.6, 86.8, 25.9, 25.8, 18.5, 18.4, −4.1; MS (ESI, neg)
m/z (%): 521(2), 519(1), 523(1) [M − H]
–.
tert-butyl(4-(2,2-dibromovinyl)phenoxy)dimethylsilane (6B): Prepared from 8B following the general procedure described for 6A to afford 2.26 g (58% yield) of 8B as a colorless oil: IR (ATR, neat): cm−1 2953, 2928, 2856, 1601, 1505, 1462, 1252, 1171, 908, 870, 836, 775, 691; 1H NMR (500 MHz, CDCl3): δ 7.44–7.46 (m, 2H), 7.40 (s, 1H), 6.82–6.83 (m, 2H), 0.99 (s, 9H), 0.22 (s, 6H); 13C{1H} NMR (125 MHz, CHCl3): δ 156.0, 136.4, 129.8, 128.3, 119.9, 87.2, 25.6, 18.2, −4.4; MS (ESI, neg) m/z (%):393(8), 391(15), 389(7) [M − H] –; HRMS (EI, pos): m/z calculated for C14H20OSiBr2 [M]+ 391.9630, found 391.9636.
((5-(2,2-dibromovinyl)-1,3-phenylene)bis(oxy))bis(tert-butyldimethylsilane) (6C): Prepared from 8C following the general procedure described for 6A to afford 3.83 g (89% yield) of 6C as a pale yellow oil. IR (ATR, neat): cm−1 2854, 2929, 2857, 1580, 1431, 1334, 1252, 1163, 1030, 827, 810, 338; 1H NMR (400 MHz, CDCl3) δ: 7.53 (2, 1H), 6.65 (dd, 2H, J = 2.1, 0.4 Hz), 6.33 (t, 1H, J= 2.1 Hz), 0.98 (s, 18H), 0.20 (s, 12H); 13C{1H} NMR (100 MHz, CDCl3) δ 156.5, 136.7, 136.6, 113.4, 112.7, 89.4, 25.7, 18.2, −4.38; MS (ESI, neg) m/z (%): 521(6) [M − H]-; HRMS (EI, pos): m/z calculated for C20H34O2Si2Br2 (M)+ 522.0444, found 522.0441.
4-(
2,2-dibromovinyl)
-1,2-difluorobenzene (
6D) [
28] Prepared from 3,4-difluorobenzaldehyde following the general procedure described for
6A to afford 10.13 g (97% yield) of a
6D as a colorless oil. IR (neat, ATR): cm
−1 3019, 1604, 1513, 1432, 1418, 1287, 1214, 1115, 882;
1H NMR (400 MHz, CDCl
3): δ 7.22–7.15 (m, 1H), 7.39 (d, 1H,
J = 0.27), 7.39–7.48 (m, 2H);
13C{1H} NMR (100 MHz, CDCl
3): δ 91.4 (d,
J = 2.52), 117.1 (d, J = 16.2 Hz), 117.3 (d,
J = 14.8 Hz), 125.5 (dd,
J = 6.5, 3.6), 132.1 (dd,
J = 6.5, 4.3), 135.1 (d,
J = 1.6), 150.0 (dd,
J = 247.9, 11.7 Hz), 150.1 (dd,
J = 249.9, 10.7 Hz);
19F{
1H} NMR (376 MHz, CDCl
3) δ: 136.2 (d, J = 20.7 Hz), 136.8 (d, J = 20.7 Hz).
5-(
2,2-dibromovinyl)
benzo(d)(1,3)dioxole (
6E) [
29]: Prepared from piperonal following the general procedure described for
6A to afford 3.87 g (95% yield) of
6E as a crystalline yellow solid. Mp = 50.9–51.7 °C; IR (ATR, neat): 2962, 2907, 1500, 1486, 1443, 1305, 1255, 1192, 1098, 1032, 924, 864, 840, 798, 752;
1H NMR (400 MHz, CDCl3) δ: 5.98 (2H, s), 6.80 (1H, d,
J = 8.1 Hz), 6.95 (1H, ddd,
J = 8.1, 1.7, 0.7 Hz), 7.18 (1H, dd,
J = 1.7, 0.4), 7.37 (1H, s);
13C{
1H} NMR 100 MHz, CDCl
3) δ: 87.9, 101.4, 108.1, 108.2, 123.4, 129.2, 136.3, 147.6, 147.8; MS (APCI, pos)
m/z (%): 309(1), 307(2), 305(1) [M + H]
+.
General Procedure–Corey-Fuchs Elimination with Trapping by CO2: 3-(
3,4-bis((
tert-butyldimethylsilyl)
oxy)
phenyl)
propiolic acid (
7A): To a solution of 5.0 g (9.57 mmol) of the vinyl dibromide
6A in 40 mL of THF under argon at −78 °C was added dropwise 8.8 mL of a 2.6 M solution (2.4 equivalent) of n-butyllithium in hexane. The reaction mixture was stirred at −78 °C for 1h, the ice bath was removed, and the reaction mixture was allowed to warm to room temperature. Chips of clean dry ice (ca. 5 g, large excess) were carefully added to the reaction mixture and allowed to fully dissolve/sublime while the mixture returned to room temperature. Water (20 mL) was added to the reaction mixture, and the THF was removed under reduced pressure. The residue was transferred to a separatory funnel, and ice-cold 1 M HCl (75 mL) and DCM (100 mL) were added. The aqueous phase was acidified with 6N HCl until the organic phase was no longer cloudy. The organic phase was collected. The aqueous phase was extracted twice more with 50 mL DCM, and extracts were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and evaporated. Recrystallization (EtOAc/hex) of the residue afford 2.77g (78%) of
7A as a white solid. Mp = 143–145 °C. IR (KBr): cm
−1 3100, 2955, 2932, 2859, 2208, 1676, 1507;
1H NMR (400 MHz, CDCl
3): δ 7.13 (1H, dd,
J = 8.3, 2.0 Hz), 7.07 (1H, d,
J = 2.0 Hz), 6.81 (1H, d,
J = 8.3 Hz), 0.99 (9H, s), 0.98 (9H, s), 0.22 (6H, s), 0.21 (6H, s).
13C{
1H} NMR (100 MHz, CDCl
3): δ 158.5, 150.8, 147.0, 127.9, 125.7, 121.2, 111.4, 90.1, 79.3, 25.8, 18.5, 18.4, −4.1, −4.2; MS (ESI, neg)
m/z (%): 405(18) [M − H]
–; Matches lit [
14].
3-(4-((tert-butyldimethylsilyl)oxy)phenyl)propiolic acid (7B). Prepared from 6B following the general procedure described for 7A to afford 560 mg (62% yield) of 7B as a tan solid. Mp = 104.2–106.3 °C; IR (ATR, neat): 2951, 2928, 2857, 2197, 1667, 1595, 1507, 1255, 1208m 1163, 903, 835, 781 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.51 (d, 2H, J = 8.7 Hz), 6.83 (d, 2H, J = 8.7 Hz), 0.98 (s, 9H), 0.22 (s, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 158.8, 158.6, 135.3, 120.5, 111.5, 90.0, 79.8, 25.5, 18.2, −4.4; MS (ESI, neg) m/z (%): 275(15) [M − H]–, 231(46), 160(6), 135(16), 117(7); HRMS (ESI, neg): m/z calculated for C15H20O3Si [M − H]– 275.1109, found 275.1112.
3-(3,5-bis((tert-butyldimethylsilyl)oxy)phenyl)propiolic acid (7C): Prepared from 6C following the general procedure described for 7A to afford 1.6 g (quant. yield) of 7C as a white semi-solid. IR (ATR, neat): cm−1 2954, 2928, 2857, 2223, 1677, 1578, 1427, 1251, 1167, 1028, 825, 776; 1H-NMR (CDCl3, 500 MHz): δ 6.71 (2H, d, J = 2.2 Hz), 6.45 (1H, t, J = 2.2 Hz), 0.97 (18H, s), 0.21 (12H, s); 13C{1H} NMR (100 MHz, CHCl3): δ 158.2, 156.7, 120.0, 118.1, 116.1, 88.9, 79.4, 25.6, 18.2, −4.4; MS (ESI, neg) m/z (%): 405(11) (M-H), 361(49), 247(13),101(8); HRMS (ESI, neg): m/z calculated for C9H4F2O2 [M − H]– 181.0107, found 181,0110.
3-(3,4-difluorophenyl)propiolic acid (7D). Prepared from 6D following the general procedure described for 7A to afford 193 mg (23% yield) of 7D as a pale yellow solid. Mp 169.0–171.6 °C; IR (ATR, neat): cm−1 3078, 2901, 2851, 2221, 1795, 1685, 1629, 1601, 1511, 1438, 1277, 1251, 1207, 1042, 916, 865; 1H-NMR (CDCl3, 400 MHz): δ 10.18 (s(br), 1H), 7.46–7.42 (m, 1H), 7.40–7.39 (m, 1H), 7.38 (q, 1H, J = 9.0 Hz); 13C{1H} NMR (100 MHz, CHCl3): δ 167.7, 151.6 (dd, J = 253, 12 Hz), 149.8 (dd, J = 248, 13 Hz), 130.6, 121.8 (d, J = 19 Hz), 118.6 (d, J = 18 Hz), 116.7 (d, J = 18 Hz), 116.3 (dd, J = 4, 3 Hz), 82.1 (d, J = 3 Hz), 82.0; MS (ESI, neg) m/z (%): 181(21) (M-H); HRMS (ESI, neg): m/z calculated for C9H4F2O2 [M − H]– 181.0107, found 181.0110.
3-(
benzo(d)(1,3)dioxol-5-yl)
propiolic acid (
7E) [
30]. Prepared from
6E following the general procedure described for
7A to afford 1.9 g (81% yield) of
7E as an orange solid. Mp = 162.8–165.6 °C (dec); IR (ATR, neat): cm
−1 2982, 2915, 2871, 2206, 1667, 1489, 1446, 1410, 1305, 1243, 1193, 1098, 1035, 923, 860, 811 cm
−1;
1H NMR (400 MHz, CD3OD) δ: 6.02 (2H, s); 6.86 (1H, dd, J = 8.06, 0.36), 7.01 (1H, dd, J = 1.61, 0.35), 7.15 (1H, dd, J = 8.06, 1.64);
13C NMR (400 MHz, CD3OD) δ: 80.7, 87.2, 103.3, 109.8, 113.1, 113.8, 129.7, 149.3, 151.6, 156.8; MS (ESI, neg)
m/z (%): 189(9) [M − H]
–, 145(14), 75(54).
3-(
2,2-dimethyl-4H-benzo(d)(1,3)dioxin-6-yl)
propiolic acid (
7F): A solution of 300 mg (1.6 mmole) of 6-ethynyl-2,2-dimethyl-4
H-benzo(
d)(1,3)dioxine [
16] in 1 mL of THF was cooled to −78 °C. To this solution was added dropwise 637 µL (1.6 mmole) of 2.5 M BuLi in hexanes. The solution was warmed to −20 °C, stirred at −20 °C for 30 min, and then, CO
2 was bubbled through the reaction mixture over 4 h during which the reaction was allowed to warm to rt. The reaction was quenched by addition of a small amount of water, the solvent was evaporated, and the residue was subjected to flash chromatography (10% EtOAc/Hex + 1% AcOH) to afford 229 mg (62%) of
7F as a white solid. Mp = 119.5–121.0 °C; IR (ATR, neat): cm
−1 2989, 2938, 2866, 2205, 1659, 1608, 1495, 1327, 1273, 1252, 1218, 1120, 884;
1H-NMR (CDCl
3, 500 MHz): δ 7.32 (1H, dd,
J = 8.8, 2.0 Hz), 7.28 (1H, d,
J = 2.0 Hz), 6.81 (1H, d,
J = 8.5 Hz), 4.83 (2H, s), 1.55 (6H, s);
13C{
1H} NMR (125 MHz, CHCl
3): δ 157.6, 154.0, 144.6, 130.4, 119.9, 117.8, 110.6, 100.6, 89.6, 79.5, 60.4, 24.8; MS (ESI, neg)
m/z (%): 231(4) [M − H]
–, 187(38), 129(11), 75(4), 69(14), 59(20); HRMS (ESI, neg):
m/z calculated for C
13H
12O
4 [M − H]
- 231.0663, found 231.0665.
General Procedure – DCC Coupling: (E)-3-(3,4-bis((tert-butyldimethylsilyl)oxy)phenyl)allyl 3-(3,4-bis((tert-butyldimethylsilyl)oxy)phenyl)propiolate (2AA): The carboxylic acid (7A) (2.413 g, 5.93 mmol) and DMAP (112 mg, 0.92 mmol) were placed in a round-bottomed flask that was then purged with argon. To the flask was added 40 mL of dry DCM and ca. 1 g of 4Å molecular sieves. A solution of the alcohol (4A) (1.815 g, 4.60 mmol) in 15 mL DCM was added via syringe. The flask was placed in an ice bath, and a solution of 1.43g (6.90 mmol) of DCC in 3 mL DCM was added dropwise via syringe. The ice bath was removed, and the reaction was allowed to stir at room temperature for 2 h. The crude reaction mixture was filtered through a plug of silica gel, which was washed with 50 mL of 30% ethyl acetate/hexanes. The filtrate was evaporated and purified by flash chromatography (0–2% ethyl acetate/hexanes) to afford 2.91 g (81% yield) of ester 2AA as a pale yellow oil. IR (ATR, neat): cm−1 2955, 2930, 2216, 1711, 1511; 1H NMR (400 MHz, CDCl3): δ 7.10 (1H, dd, J = 8.3, 2.0 Hz), 7.04 (1H, d, J = 2.0 Hz), 6.88 (2H, m), 6.78 (2H, m), 6.59 (1H, d, J = 15.8 Hz), 6.14 (1H, dt, J = 15.8, 6.8 Hz), 4.84 (2H, dd, 6.8, 1.1 Hz), 0.99 (9H, s), 0.97 (18H, s), 0.96 (9H, s), 0.22 (6H, s), 0.21 (6H, s), 0.20 (6H, s), 0.19 (6H, s); 13C{1H} NMR (100 MHz, CD3COCD3): δ 154.1, 151.1, 148.0, 147.9, 147.7, 135.6, 131.1, 128.3, 126.0, 122.4, 122.0, 121.5, 121.3, 120.1, 112.9, 86.9, 80.5, 67.1, 26.4, 26.3, 26.2, 19.2, 19.09, 19.07, 19.04, −3.81, −3.84, −3.9; HRMS (ESI, pos) calc. for C42H70NaO6Si4 [M + Na]+ 805.4142, found 805.4148.
(E)-3-(4-((tert-butyldimethylsilyl)oxy)phenyl)allyl 3-(4-((tert-butyldimethylsilyl)oxy)phenyl)propiolate (2BB). Prepared from 4B and 7B following the general procedure described for 2AA to afford 64 mg (42% yield) of 2BB as a colorless oil. IR(ATR, neat): cm−1 2914, 1848, 2236, 1734, 1623, 1568, 1559, 1309, 1268, 1241, 1178, 1086, 640; 1H-NMR (CDCl3, 500 MHz): δ 7.48 (d, 2H, J = 8.8 Hz), 7.28 (d, 2H, J = 8.5 Hz), 6.83–6.79 (m, 4H), 6.65 (d, 1H, J = 15.8 Hz), 6.19 (dt, 1H, J = 15.8, 6.7 Hz), 4.85 (dd, 2H, J= 6.7, 1.1 Hz), 0.99 (s, 9H), 0.98 (s, 9H), 0.21 (s, 6H), 0.20 (s, 6H); 13C{1H} NMR (125 MHz, CDCl3): δ 158.1, 155.9, 154.1, 135.1, 134.9, 129.3, 127.9, 120.4, 120.2, 120.0, 112.0, 87.3, 80.1, 66.6, 25.6, 25.5, 18.2, −4.4; MS (ESI, pos) m/z (%): 523(3) [M + H]+.
(E)-3-(3,5-bis((tert-butyldimethylsilyl)oxy)phenyl)allyl 3-(3,5-bis((tert-butyldimethylsilyl)oxy)phenyl)propiolate (2CC): Prepared from 4C and 7C following the general procedure described for 2AA to afford 349 mg (70% yield) of 2CC as off-white needles. Mp = 77.4–80.7 °C; IR (ATR, neat): cm−1 2951, 1919, 1857, 2219, 1706, 1579, 1437, 1427, 1250, 1233, 1163, 1153, 1026, 825, 811, 777; 1H NMR (400 MHz, CD3Cl): δ 6.69 (d, 2H, J = 2.2 Hz), 6.59 (d, 1H, J = 15.8 Hz), 6.51 (d, 2H, J = 2.2 Hz), 6.43 (t, 1H, J = 2.2 Hz), 6.27–6.20 (m, 2H), 4.86 (dd, 1H, J = 6.6, 1.0 Hz), 0.98 (s, 18H), 0.97 (s, 18H), 0.19 (s, 12H), 0.18 (s, 12H); 13C{1H} NMR (100 MHz, CDCl3): δ 156.7, 156.6, 153.7, 137.7, 135.3, 122.1, 120.4, 177.9, 115.7, 112.2, 112.9, 86.6, 79.8, 66.5, 25.7, 25.6, 18.1, −4.4, −4.5; MS (ESI, pos) m/z (%): 783(9) (M + H), 395(11), 377(14); HRMS (APCI, pos) calculated for C42H70O6Si4 [M + H]+ 783.4322, found 783.4303.
(E)-3-(3,4-difluorophenyl)allyl 3-(3,4-difluorophenyl)propiolate (2DD): Prepared from 4D and 7D following the general procedure described for 2AA to afford 154 mg (76% yield) of 2DD as an amber oil. IR (ATR, neat): cm−1 2949, 2938, 2856, 2215, 1724, 1644, 1603, 1513, 1435, 1293, 1269, 1242, 1165, 1142, 965, 811, 785; 1H NMR (400 MHz, CDCl3): δ 7.42–7.33 (2H, m), 7.23–7.09 (4H, m), 6.63 (1H, d, J = 15.87), 6.23 (1H, dt, J = 15.84, 6.46), 4.87 (2H, dd, J = 6.47, 1.23); 13C{1H} NMR (100 MHz, CDCl3): δ 153.2, 152.1 (dd, J = 256, 13 Hz), 150.4 (dd, J = 251, 16 Hz), 150.2 (dd, J = 158, 23 Hz), 150.0 (dd, J = 251 13 Hz), 133.2 (dd, J = 6, 4 Hz), 133.1, 130.0 (dd, J = 7, 4 Hz), 123.1 (d, J = 2Hz), 123.0 (dd, J = 7, 4 Hz), 121.9 (d, J = 19 Hz), 118.0 (d, J = 17 Hz), 117.4 (d, J = 17 Hz), 116.2 (dd, J = 7, 4 Hz), 115.0 (d, J = 17 Hz), 84.1 (d, J = 2 Hz), 80.5, 66.1; 19F{1H} NMR (376 MHz, CDCl3) δ: −137.0 (d, J = 20 Hz), −139.3 (d, J = 21 Hz), −140.1 (d, J = 20 Hz), −140.9 (d, J = 21 Hz); MS (APCI, pos) m/z (%): 389(13) (M + MeOH + Na+); HRMS (CI, pos) calculated for C18H12F4O2 [M]+ 336.0773, found 336.0771.
(
E)
-3-(
benzo(d)(1,3)dioxol-5-yl)
allyl 3-(
benzo(d)(1,3)dioxol-5-yl)
propiolate (
2EE) [
31]: Prepared from
4E and
7E following the general procedure described for
2AA to afford 1.58 g (86% yield) of
2EE as an off-white, chalky solid. 86%. Mp = 104.9–106.5 °C; IR(ATR, neat): cm
−1 2916, 2901, 2851, 2207, 1692, 1618, 1490, 1444, 1303, 1236, 1185, 1096, 1034, 931, 804;
1H NMR (400MHz, CDCl
3): δ 7.15 (1H, dd,
J = 8.1, 1.6), 7.00–6.93 (2H, m), 6.84 (1H, dd,
J = 8.2, 1.6), 6.80–6.75 (2H, m), 6.62 (1H, d,
J = 15.8), 6.15 (1H, d,
J = 15.8), 5.95 (2, 2H), 5.94 (s, 2H), 4.83 (2H, dd, J = 6.7, 1.2);
13C{
1H} NMR (100 MHz, CDCl
3): δ 153.8, 150.0, 148.0, 147.7, 147.5, 135.0, 130.3, 128.8, 121.6, 120.2, 112.4, 108.7, 108.2, 105.8, 101.7, 101.1, 87.0, 79.4, 66.4. MS (ESI, pos)
m/z (%): 373(3) [M + Na]
+.
(E)-3-(2,2-dimethyl-4H-benzo(d)(1,3)dioxin-6-yl)allyl 3-(2,2-dimethyl-4H-benzo(d)(1,3)dioxin-6-yl)propiolate (2FF): Prepared from 4F and 7F following the general procedure described for 2AA to afford 15 mg (50% yield) of 2FF as a colorless oil. IR(ATR, CHCl3): cm−1 2993, 2940, 2856, 2211, 1704, 1612, 1580, 1497, 1384, 1375, 1312, 1290, 1270, 1254, 1234, 1198, 1144, 1115, 956, 873; 1H NMR (400 MHz, CDCl3): δ 7.39 (dd, 1H, J = 8.5, 1.9 Hz), 7.24–7.21 (m, 2H), 7.02 (s, 1H), 6.79 (t, 2H, J = 8.1 Hz), 6.62 (d, 1H, J = 15.8 Hz), 6.17 (dt, 1H, J = 15.8, 6.7), 4.85–4.81 (m, 4H), 4.80 (s, 2H), 1.54 (s, 12H); 13C{1H} NMR (100 MHz, CDCl3): δ 154.0, 153.6, 151.4, 134.9, 133.3, 130.1, 128.5, 126.6, 123.1, 120.2, 119.8, 119.4, 117.6, 117.3, 111.0, 100.4, 99.8, 87.1, 79.8, 66.6, 60.8, 60.4, 24.8, 24.7; MS (ESI, pos) m/z (%): 435(3) [M + H]+; HRMS (APCI, pos) calculated for C26H26O6 [M + H]+ 435.1802, found 435.1788.
(E)-3-(benzo(d)(1,3)dioxol-5-yl)allyl 3-(3,4-bis((tert-butyldimethylsilyl)oxy)phenyl)propiolate (2EA): Prepared from 4E and 7A following the general procedure described for 2AA to afford 139 mg (59% yield) of 2EA as slightly yellow needles. Mp 95.3–96.7 °C; IR(ATR, neat): cm−1 2927, 2855, 2208, 1556, 1493, 1437, 1411, 1318, 1275, 1238, 1155, 1119, 1035, 935, 896, 839, 783; 1H NMR (500MHz, CDCl3): δ 7.10 (dd, 1H, J = 8.3, 2.1 Hz), 7.05 (d, 1H, J = 2.1 Hz), 6.94 (d, 1H, J = 1.7 Hz), 6.84 (dd, 1H, J = 8.0, 1.7 Hz), 6.79 (d, 1H, J = 8.3 Hz), 6.76 (8.0 Hz), 6.63 (d, 1H, J = 15.8 Hz), 6.16 (dt, 1H, J = 15.8, 6.7 Hz), 5.96 (s, 2H), 4.84 (dd, 2H, J = 6.7, 1.2 Hz), 0.99 (s, 9H), 0.98 (s, 9H), 0.22 (s, 6H), 0.21 (s, 6H);13C{1H} NMR (125 MHz, CDCl3): δ 154.1, 150.2, 148.1, 147.7, 146.9, 135.1, 130.4, 127.4, 125.4, 121.6, 121.1, 120.3, 111.9, 108.2, 105.9, 101.1, 87.5, 79.6, 66.4, 25.8, 18.5, 18.4, −4.1, −4.2; MS (ESI, pos) m/z (%): 567(21) [M + H]+; HRMS (APCI, pos) calculated for C31H42O6Si2 [M + H]+ 567.2593, found 567.2586.
(
E)
-3-(
benzo(d)(1,3)dioxol-5-yl)
allyl 3-phenylpropiolate (
2EG) [
30]: Prepared from
4E and phenylpropiolic acid following the general procedure described for
2AA to afford 179 mg (51% yield) of
2EG as a white solid. Mp = 55.7–57.2 °C. IR(ATR, neat) cm
−1: 2996, 2896, 2210, 1697, 1501, 1490, 1444, 1299, 1280, 1251, 1182, 1170, 1125, 1036, 953, 930, 911, 752;
1H NMR (400MHz, CDCl
3): δ 7.60–7.58 (m, 2H), 7.45 (t, 1H,
J = 7.5 Hz), 7.37 (t, 2H,
J = 7.6 Hz), 6.95 (d, 1H,
J = 1.4 Hz), 6.85 (dd, 1H,
J = 8.0, 1.4 Hz), 6.77 (d, 1H,
J = 8.0 Hz), 6.63 (d, 1H,
J = 15.8 Hz), 6.16 (dt, 1H,
J = 15.8, 6.7 Hz), 5.96 (s, 2H), 4.86 (d, 2H,
J = 6.7 Hz);
13C{
1H} NMR (100 MHz, CDCl
3): δ 153.8, 148.0, 147.8, 135.2, 132.9, 130.6, 128.5, 121.7, 120.2, 119.5, 108.3, 105.8, 101.1, 86.5, 80.5, 66.6; MS (ESI, pos)
m/z (%): 329(6) [M + Na]
+.
General Procedure—Pd-Catalyzed Decarboxylative Coupling: (
E)
-((
pent-1-en-4-yne-1,5-diylbis(
benzene-4,1,2-triyl))
tetrakis(
oxy))
tetrakis(
tert-butyldimethylsilane) (
1′AA) [
14]: A solution of 134 mg (0.171 mmol) of ester
2AA in 2.5 mL of freshly distilled THF was transferred under argon to a reaction tube containing 10 mg (0.0086 mmol) of Pd(P(Ph)
3)
4, and the tube was sealed. After heating for 4 h in an 80 °C oil bath, the contents of the tube were transferred to a round-bottomed flask with EtOAc, the solvent evaporated, and the residue subjected to flash chromatography (0–1% EtOAc/hexanes) to afford
1′AA as a yellow oil (105 mg, 0.142 mmol, 83%).
1H NMR and IR matched, which was previously reported [
14].
13C{
1H} NMR (100 MHz, CDCl
3): δ 147.3, 146.8, 146.6, 146.4, 130.9, 125.3, 124.3, 122.4, 121.0, 120.9, 119.6, 118.9, 116.5, 105.0, 85.2, 82.4, 26.0, 25.9, 22.9, 18.5, 18.4, −4.1. HRMS (ESI
+) calculated for C
41H
71O
4Si
4 [M + H]
+ 739.4424, found 739.4422.
(E)-((pent-1-en-4-yne-1,5-diylbis(4,1-phenylene))bis(oxy))bis(tert-butyldimethylsilane) (1′BB): Prepared from 2BB following the general procedure described for 1′AA to afford 12 mg (22% yield) of a yellow oil. IR(ATR, neat): cm−1 2954, 2929, 2857, 2220, 1588, 1252, 1164, 906, 835, 779; 1H NMR (500 MHz, CD3Cl): δ 7.33–7.31 (m, 2H), 7.26–7.24 (m, 2H), 6.79–6.76 (m, 4H), 6.62 (d, 1H, J = 15.7 Hz), 6.12–6.07 (m, 1H), 3.32 (dd, 2H, J = 5.6, 1.6 H), 0.99 (s, 9H), 0.98 (s, 9H), 0.19 (s, 12H); 13C{1H} NMR (125 MHz, CDCl3): δ 155.5, 155.1, 132,9, 130.7, 130.5, 127.3, 122.4, 120.1, 120.0, 116.5, 85.5, 82.4, 25.7, 25.6, 22.9, 18.2, –4.4; MS (APCI, pos) m/z (%): 477(16) [M + H]+; HRMS (ESI+) calculated for C29H40O2Si2 [M + H]+ 477.2640, found 477.2640.
(E)-((pent-1-en-4-yne-1,5-diylbis(benzene-5,1,3-triyl))tetrakis(oxy))tetrakis(tert-butyldimethylsilane) (1′CC): Prepared from 2CC following the general procedure described for 1′AA to afford 54 mg (56% yield) of a colorless oil. IR(ATR, neat): cm−1 2954, 2929, 2857, 1578, 1426, 1343, 1252, 1162, 1026, 827, 778; 1H NMR (500 MHz, CD3Cl): δ 6.57–6.50 (m, 3H), 6.50 (d, 2H, J = 2.2 Hz), 6.30 (t, 1H, J = 2.2 Hz), 6.23 (t, 1H, J = 2.2 Hz), 6.16 (dt, 1H, J = 15.7, 5.6 Hz), 3.32 (dd, 2H, J = 5.6, 1.7 Hz), 0.98 (s, 18H), 0.97 (s, 18H), 0.99 (s, 24H); 13C{1H} NMR (125 MHz, CDCl3): δ 156.6, 156.3, 138.9, 131.3, 124.6, 124.3, 116.8, 112.8, 111.6, 111.3, 86.2, 82,7, 25.7, 25.6, 22.9, 18.2, 18.1, −4.2, −4.4; MS (APCI, pos) m/z (%): 739(34) [M + H]+; HRMS (APCI, pos) calculated for C41H70O4Si4 [M + H]+ 739.4424, found 739.4396.
(E)-4,4’-(pent-1-en-4-yne-1,5-diyl)bis(1,2-difluorobenzene) (1DD): Prepared from 2DD following the general procedure described for 1′AA to afford 50 mg (28% yield) of 1DD as a yellow oil. IR(ATR, neat): cm−1 2930, 2204, 1599, 1513, 1430, 1290, 1216, 1114, 965, 872, 820, 773; 1H NMR (400 MHz, CDCl3): δ 7.25–7.06 (6H, m), 6.59 (1H, d, J = 15.7 Hz), 6.15 (1H, dtd, J = 15.7, 5.6), 3.34 (dd, 2H, J = 5.6, 1.3 Hz); 13C{1H} NMR (100 MHz, CDCl3): δ 150.4 (dd, J = 247.6, 12.9 Hz), 150.3 (dd, J = 250.6, 12.5 Hz), 149.9 (dd, J = 248.8, 12.9 Hz), 149.7 (dd, J = 248.8, 12.8 Hz), 134.2 (dd, J = 5.7, 4.1 Hz), 129.7, 128.2 (dd, J = 6.2, 3.6 Hz), 124.9 (d, J = 1.9 Hz), 122.4 (dd, J = 6.1, 3.4 Hz), 120.5 (d, J = 18.3 Hz), 120.3 (dd, J = 7.6, 4.2 Hz), 117.3 (d, J = 17.7 Hz), 117.2 (d, J = 17.3 Hz), 114.6 (d, J = 17.6 Hz), 86.9 (d, J = 1.3 Hz Hz), 81.0, 22.7; MS (ESI, pos) m/z (%): 289(8) [M − H]+; HRMS (ESI, pos) calculated for C17H10F4 [M]+ 290.0719, found 290.0714.
(E)-5,5’-(pent-1-en-4-yne-1,5-diyl)bis(benzo(d)(1,3)dioxole) (1EE): Prepared from 2EE following the general procedure described for 1′AA to afford 350 mg (80% yield) of 1EE as thin yellow needles. Mp = 58.2–60.0 °C; IR(ATR, neat): cm−1 2908, 2854, 1486, 1440, 1246, 1207, 1097, 1036, 967, 925, 813; 1H NMR (400 MHz, CD3COCD3): δ 7.03 (d, 1H, J = 1.7 Hz), 6.98 (dd, 1H, J = 8.0, 1.6 Hz), 6.91–6.90 (m, 1H), 6.88 (dd, 1H, J = 8.0, 1.7 Hz), 6.82 (dd, 1H, J = 8.0, 0.4 Hz), 6,79 (d, 1H, J = 8.0 Hz), 6.65 (dt, 1H, J = 15.7, 1.8 Hz), 6.18 (dt, 1H, J = 15.7, 5.7), 6.02 (s, 2H), 5.98 (s, 2H), 3.32 (dd, 2H J = 5.7, 1.8); 13C{1H} NMR (100 MHz, CD3COCD3): δ 149.0, 148.6, 148.4,148.0, 132.6, 131.6, 126.7, 123.5, 121.7, 117.9, 112.0, 109.1, 108.9, 106.3, 102.3, 102.0, 85.8, 82.2, 23.0; MS (APCI, pos) m/z (%): 307(29) [M + H]+; HRMS (CI+) calculated for C19H14O4 [M]+ 306.0892, found 306.0886.
(E)-6,6’-(pent-1-en-4-yne-1,5-diyl)bis(2,2-dimethyl-4H-benzo(d)(1,3)dioxine) (1FF): Prepared from 2FF following the general procedure described for 1′AA to afford 4 mg (40% yield) of 1FF as a yellow oil: IR(ATR, CHCl3): cm−1 2988, 2923, 2220, 1613, 1582, 1496, 1384, 1265, 1202, 1142, 1117, 1065, 956; 1H NMR (500 MHz, CD3Cl): δ 7.24 (dd, 1H, J = 8.5, 1.9 Hz), 7.20 (dd, 1H, J = 8.5, 1.9 Hz), 7.08 (s, 1H), 6.99 (s, 1H), 6.76 (t, 2H, J = 9.0 Hz), 6.59 (d, 1H, J = 15.7 Hz), 6.08 (dt, 1H, J = 15.7, 5.7 Hz), 4.83 (s, 2H), 4.81 (s, 2H), 3.30 (dd, 2H, J = 5.7, 1.7 Hz), 1.54 (s, 12H); 13C{1H} NMR (125 MHz, CD3COCD3): δ 151.0, 150.7, 131.6, 130.7, 129.7, 128.0, 126.0, 122.5, 122.4, 119.4, 119.3, 117.2, 117.2, 115.5, 99.9, 99.6, 85.3, 82.4, 60.9, 60.6, 29.7, 24.7, 22.9; MS (APCI, pos) m/z (%): 391(1) [M + H]+, 423(5) [M + MeOH + H]+; HRMS (APCI, pos) calculated for C25H26O4 [M + H]+ 391.1904, found 391.1898.
(E)-((4-(5-(benzo(d)(1,3)dioxol-5-yl)pent-4-en-1-yn-1-yl)-1,2-phenylene)bis(oxy))bis(tert-butyldimethylsilane) (1′EA): Prepared from 2EA following the general procedure described for 1′AA to afford 52 mg (43% yield) of 1′EA as a yellow oil. IR(ATR, neat): cm−1 2929, 2857, 2193, 1593, 1557, 1505, 1488, 1407, 1308, 1250, 1099, 1038, 895, 828, 780; 1H NMR (500 MHz, CDCl3): δ 6.94–6.92 (m, 3H), 6.81 (dd, 1H, J = 8.0, 1.2 Hz), 6.76–6.74 (m, 2H), 6.60 (d, 1H, J = 15.7 Hz), 6.07 (dt, 1H, J = 15.7, 5.7 Hz), 5.95 (s, 2H), 3.31 (dd, 2H, J = 5.7, 1.5 Hz), 0.99 (s, 9H), 0.98 (s, 9H), 0.21 (s, 6H), 0.20 (s, 6H); 13C{1H} NMR (125 MHz, CDCl3): δ 147.9, 147.4, 146.9, 146.6, 131.7, 130.9, 125.3, 124.3, 122.8, 120.9, 120.7, 116.4, 108.2, 105.7, 101.0, 84.9, 82.6, 25.9, 22.9, 18.5, 18.4, −4.0, −4.1; (ESI, pos) m/z (%): 523(9) [M + H]+; HRMS (APCI, pos) calculated for C30H40O4Si2 [M + H]+ 521.2538, found 521.2532.
(
E)
-5-(
5-phenylpent-1-en-4-yn-1-yl)
benzo(d)(1,3)dioxole (
1EG) [
32]: Prepared from
2EG following the general procedure described for
1′AA to afford 12 mg (20% yield) of
1EG as a yellow oil. IR(ATR, neat): cm
−1 2916, 2895, 2212, 1502, 1488, 1444, 1279, 1248, 1167, 1124, 1099, 1035, 952, 927, 756;
1H NMR (400 MHz, CD3Cl): δ 7.46–7.44 (m, 2H), 7.31–7.29 (m, 3H), 6.93 (d, 1H,
J = 1.5 Hz), 6.82 (dd, 1H,
J = 8.0, 1.5 Hz), 6.75 (d, 1H,
J = 8.0 Hz), 6.61 (dd, 1H,
J = 15.6, 1.5 Hz), 6.08 (dt, 1H,
J = 15.6, 5.7 Hz), 5.95 (2, 2H), 3.34 (dd, 2H,
J = 5.7, 1.7 Hz);
13C{
1H} NMR (125 MHz, CDCl
3): δ 148.0, 147.0, 131.6, 131.0, 128.2, 127.8, 123.7, 122.5, 120.8, 108.2, 105.7, 101.0, 86.8, 82.8, 22.9; (ESI, pos)
m/z (%): 263(3) [M + H]
+.
General Procedure—Deprotection for Catechol-Containing Products: Rooperol (
1AA) [
14]: In a round-bottomed flask containing 44 mg of the silylester
1′AA (0.06 mmol) in 1 mL of dry DMF under Ar was placed 14 mg (0.24 mmol, 4 equivalent) of anhydrous KF. The flask was placed in an ice bath, and 0.2 mL of a solution prepared as a 1:100 dilution of 33% HBr/AcOH in DMF was added by syringe. After stirring for 1.5 h in the ice bath, the reaction mixture was diluted with EtOAc and washed with H
2O and brine. The organic layer was dried over Na
2SO
4, filtered, and evaporated. The residue was subjected to flash chromatography 25% EtOAc/Hex + 1% MeOH) to afford 11 mg (65% yield) of catechol rooperol (
1AA) as a yellow oil.
1H NMR (400 MHz,
d6-acetone): δ 6.93 (1H, brs), 6.89 (1H, d,
J = 1.7 Hz), 6.80 (1H, dd,
J = 8.0, 1.8 Hz), 6.74–6.77 (3H, m), 6.55 (1H, dt,
J = 15.7, 1.7 Hz), 6.05 (1H, dt,
J = 15.7, 5.7 Hz), 3.25 (2H, dd,
J = 5.7, 1.8 Hz).
13C NMR (100 MHz,
d6-acetone) δ 145.6, 145.0, 144.8, 130.9, 129.4, 123.7, 121.5, 118.3, 118.1, 155.2, 115.1, 114.9, 112.6, 84.1, 82.6, 22.1. IR (thin film) 3019, 1666, 1514, 1388, 1215, 755. MS (ESI
-)
m/z: 281 [M -H]
–, 100); HRMS (ESI
-) calculated for C
17H
13O
4 [M − H]
- 281.0819, found 281.0819.
(E)-4-(5-(benzo(d)(1,3)dioxol-5-yl)pent-4-en-1-yn-1-yl)benzene-1,2-diol (1EA): Prepared from 1′EA following the general procedure described for 1AA to afford 2 mg (18% yield) of 1EA as a colorless oil. IR(ATR, neat): cm−1 2919, 2850, 2220, 1599, 1518, 1445, 1294, 1249, 1196, 1110, 1033, 962, 920, 817, 768; 1H NMR (500 MHz, CD3Cl): δ 6.96–6.92 (m, 3H), 6.82–6.79 (m, 2H), 6.75 (d, 1H, J = 8.0 Hz), 6.59 (dt, 1H, J = 15.6, 1.6 Hz), 6.06 (dt, 1H, J = 15.6, 5.7 Hz), 5.95 (s, 2H), 5.30 (s, 1H), 5.18 (s, 1H), 3.30 (dd, 2H, J = 5.7, 1.7 Hz); 13C{1H} NMR (125 MHz, CD3COCD3): δ 147.9, 147.0, 143.8, 143.0, 131.7, 130.9, 125.3, 122.7, 120.8, 118.6, 116.3, 115.3, 108.2, 105.6, 101.0, 85.1, 82.3, 22.9; (ESI, pos) m/z (%): 295(4) [M + H]+; HRMS (APCI, pos) calculated for C18H14O4 [M + H]+ 295.0965, found 295.0962.
General Procedure—Deprotection for Phenol- and Resorcinol-Containing Products: (
E)
-4,4’-(
pent-1-en-4-yne-1,5-diyl)
diphenol (
1BB) [
33]: To a solution of 10 mg (0.019 mmol) of
1′BB in 0.2 mL of freshly distilled THF was added 4.7 µL (0.082 mmol) of AcOH. The solution was cooled in an ice bath, and 82 µL (0.082 mmol) of a 1M solution of TBAF in THF was added. After 20 min, EtOAc (5 mL) and water (5 mL) were added to the reaction mixture. The layers separated, and the aqueous layer was extracted 1 x EtOAc (5 mL). The combined organic layers were washed 1 x brine (5 mL). The combined aqueous layers were then back-extracted 1 x DCM (10 mL). The combined organics layers were dried over Na
2SO
4, filtered, evaporated, and subjected to flash chromatography 50% EtOAc/hexanes to afford 4 mg (71%) of
1BB as a colorless oil. IR(ATR, neat): cm
−1 2955, 2851, 2224, 1602, 1506, 1238, 965, 831;
1H NMR (500 MHz, CD3Cl): δ 7.25–7.22 (m, 4H), 6.73–6.71 (m, 4H), 6.58 (d, 1H,
J = 15.7 Hz), 6.07 (dt, 1H,
J = 15.7, 5.7 Hz), 3.26 (dd, 2H,
J = 5.7, 1.7 Hz);
13C{
1H} NMR (125 MHz, CDCl
3): δ 158.5, 158.0, 133.9, 131.9, 130.3, 128.4, 122.7, 116.3, 116.2, 116.0, 85.5, 83.6, 23.3; MS (ESI, neg)
m/z (%): 249(100) [M − H]
–.
(E)-5,5’-(pent-1-en-4-yne-1,5-diyl)bis(benzene-1,3-diol) (1CC): Prepared from 1′CC following the general procedure described for 1BB to afford 10 mg (42% yield) of 1CC as a colorless oil. IR(ATR, neat): cm−1 2960, 2925, 2853, 2220, 1587, 1503, 1441, 1340, 1299, 1142,997, 837; 1H NMR (500 MHz, CD3COCD3): δ 8.34 (s, 2H), 8.14 (s, 2H), 6.58 (dt, 1H, J = 15.7, 1.8 Hz), 6.44 (d, 2H, J = 2.2 Hz), 6.43 (d, 2H, J = 2.2 Hz), 6.35 (t, 1H, J = 2.2 Hz), 6.26 (t, 1H, J = 2.2 Hz), 6.20 (dt, 1H, J = 15.7, 5.6 Hz), 3.31 (dd, 2H, J = 5.6, 1.8 Hz); 13C{1H} NMR (125 MHz, CD3COCD3): δ 159.6, 159.4, 140.1, 132.2, 125.9, 125.0, 110.8, 105.7, 104.0, 102.8, 86.6, 83.7, 23.0; (ESI, pos) m/z (%): 283(6) [M + H]+; HRMS (APCI, pos) calculated for C17H14O4 [M + H]+ 283.0965, found 283.0965.
Cancer Cell Cytotoxicity Assays. HeLa, H460, and A549 cells were obtained from ATCC and grown in RPMI culture medium supplemented with 10% heat inactivated FBS (Life Technologies code 10270106). Cell culture media were supplemented with 4 mM glutamine (Lonza code BE17–605E), 100 μg/mL gentamicin (Lonza code 17-5182), and P/S (200 units/mL and 200 μg/mL) (Lonza code 17-602E) at 37 °C with 5% CO2. The effect of the investigated compounds on cell proliferation was determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. The compounds were dissolved in DMSO at a concentration of either 10 or 50 mM prior to cell treatment. The cells were trypsinized and seeded at various cell concentrations depending on the cell type. The cells were grown for 24–72 h, treated with test compounds at required concentrations, and incubated for 72 h in 100 or 200 μL media depending on the cell line used. Three replicates were performed. Cells treated with 0.1% DMSO were used as a negative control. The GI50 corresponds to the concentration of the compound of interest that reduces by 50% the growth of the cancer cell line of interest after having cultured it for 72 h in the presence of the compound in comparison to the untreated control condition.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}